
Abstract
Diabetic nephropathy (DN) is a critical microvascular complication of diabetes. Increasing evidence suggests that dysregulation of glutamine metabolism contributes to DN pathogenesis. This study aimed to explore alterations in glutamine metabolism-related genes (GMRGs) in DN. Nine differentially expressed GMRGs (DE-GMRGs) were identified by intersecting 103 GMRGs with 2,281 DEGs from the GSE142153 dataset comparing normal and DN groups. Notably, DE-GMRGs located on autosomes were significantly enriched in pathways related to glutamine metabolism and the metabolism of alanine, aspartate, and glutamate. The Least Absolute Shrinkage and Selection Operator (LASSO) and Support Vector Machine-Recursive Feature Elimination (SVM-RFE) methods were employed to pinpoint key genes, SLC7A5 and SLC25A12. SLC7A5 was found to be upregulated in DN, whereas SLC25A12 showed decreased expression. The diagnostic potential of these genes was further validated by assessing the area under the receiver operating characteristic (ROC) curve. Correlation analysis revealed strong associations between these key genes and clinical markers such as glomerular filtration rate (GFR), serum creatinine, and immune cells, including mast cells and effector memory CD8 T cells. Drug prediction and molecular docking analyses indicated that valproic acid might serve as an effective therapeutic agent targeting these genes. Glutamine metabolism-related genes SLC7A5 and SLC25A12 were identified as potential diagnostic and therapeutic targets for DN. These findings offer valuable clinical insights for the diagnosis and management of DN.
Similar content being viewed by others
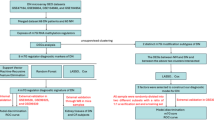


Introduction
Diabetic nephropathy (DN) is a significant complication of both type I and type II diabetes, affecting approximately 20–40% of individuals with diabetes and contributing to the progression to end-stage renal disease (ESRD)1,2,3. With the increasing global prevalence of diabetes across all age groups4, the incidence of DN is also rising, particularly in regions like China5. DN, a microvascular disorder, leads to kidney damage, which is predominantly characterized by glomerulosclerosis and interstitial fibrosis, resulting in proteinuria, reduced filtration efficiency, and impaired glomerular function6,7. The disease’s pathogenesis is multifactorial, involving genetic predisposition, hyperglycemia, dyslipidemia, hypertension, changes in renal hemodynamics, and the metabolism of vasoactive substances8. Currently, the primary treatment strategies for DN focus on controlling blood glucose, lipids, and blood pressure. Although these approaches can slow disease progression, effective therapies remain limited. Recent machine learning-based studies have identified key genes, such as CASP1, MS4A4A, CD53, and GBP2, which may be regulated by macrophages in DN9. Additionally, LASSO-based screening has highlighted CCR2, CX3CR1, and SELP as immune-related biomarkers of DN10. However, the precise molecular mechanisms underlying DN remain unclear, making the identification of novel diagnostic biomarkers and therapeutic targets critical.
Amino acids, as fundamental building blocks of proteins and essential signaling molecules, play a pivotal role in maintaining metabolic and energy balance. The regulation of amino acid metabolism significantly influences kidney function11,12. Glutamine, a prevalent amino acid found in many foods, is a precursor of glutathione and participates in gluconeogenesis, helping to maintain blood glucose levels during starvation13. Recent research has highlighted the protective effects of exogenous glutamine in alleviating DN in type 2 diabetic rats, primarily due to its antioxidant and anti-inflammatory properties14. Additionally, research by Smeeta et al. demonstrated that L-glutamine treatment could mitigate STZ-induced DN by reducing oxidative and nitrosative stress and modulating mRNA expressions of KIM-1, NGAL, TGF-β1, and collagen-1 in animal models15. Despite these promising findings, a comprehensive exploration of glutamine metabolism-related genes (GMRGs) in the context of DN remains underexplored in the literature.
This study utilized DN-related data from public databases and applied advanced bioinformatics methods to identify biomarkers associated with glutamine metabolism in DN. This approach aims to uncover novel insights that could enhance the diagnosis and treatment of DN.
Materials and methods
Data source
The datasets GSE142153 (GPL6480) and GSE99325 (GPL19109 and GPL19184) were sourced from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/). The GSE142153 dataset, serving as the training set, includes 10 normal, 23 DN, and 7 ESRD samples; only the normal and DN samples were used in this study (sample type: peripheral blood mononuclear cells [PBMC]). The GSE99325 dataset, acting as the validation set, contains 175 renal tubule interstitium tissue samples, with 4 normal and 18 DN samples selected for follow-up. Additionally, 103 glutamine metabolism-related genes (GMRGs) were compiled by combining genes obtained from the literature16 and the Molecular Signatures Database (MSigDB).
Identification of DE-GMRGs and functional enrichment analysis
Principal component analysis (PCA) was performed to evaluate the suitability of the GSE142153 dataset, resulting in the removal of outlier samples. The criteria for identifying outlier samples were: (1) Spatial distance threshold: The Mahalanobis distance of each sample to the population centroid was calculated, and samples with distances exceeding 3 standard deviations were defined as outliers; (2) Cluster separation: A dendrogram was generated using hierarchical clustering, and samples with a separation greater than 75% of the height were considered outliers. Next, variance analysis was conducted using the adonis2 function from the R package vegan (V 2.6-8, https://CRAN.R-project.org/package=vegan), with a distance matrix to partition sources of variation and fit a linear model, confirming that the separation between groups remained significant after outlier removal. Differential expression analysis of the GSE142153 dataset was then performed, comparing DN and normal groups using the “Limma” package in R (version 3.52.4)17. Differentially expressed genes (DEGs) were identified with a significance threshold of adj P < 0.05 (Benjamini-Hochberg correction, < 0.05 as a criterion for determining differentially expressed genes) and |log2FC| > 0.5 (indicating that the gene expression difference up to a certain fold). The analysis process was as follows: first, genes with expression below the threshold (FPKM/RPKM < 1) in all samples were removed, and genes with expression ≥ 1 in at least 30% of the samples were retained. If multiple probes corresponded to the same gene, the probe with the highest average expression was selected. Next, the Robust Multi-array Average (RMA) algorithm was used for background correction to estimate the true signal and reduce noise by iterative Maximum Likelihood Estimation (MLE). Finally, the batch effect and technical variation were eliminated using the quantile normalization method to ensure consistent expression between samples. In addition, this study used a linear model combined with the empirical Bayes method for differential expression analysis. The model was as follows:
Where: Yij was the expression level of gene j in sample i (after log2 transformation); β0j was the baseline expression of gene j in the control group; β1j represented the expression difference of gene j between the disease group and the control group (log2FC); Diseasei was a binary variable (0 = control group, 1 = disease group); \(\:{\epsilon\:}_{\text{i}\text{j}}\sim\text{N}(0,{{\upsigma\:}}_{\text{j}}^{2})\) was the random error term. The intersection between DEGs and GMRGs defined the pool of DE-GMRGs. Protein-protein interaction (PPI) networks for DE-GMRGs were constructed using Search for Recurring Instances of Neighbouring Genes (STRING, https://string-db.org). Chromosomal localization of the DE-GMRGs was performed using the “RCircos” package in R (version 1.2.2)18 to pinpoint gene locations and provide insights into their functional roles. To explore the functional roles and mechanisms of the genes, Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG)19 enrichment analyses were conducted using the “ClusterProfiler” package in R (version 4.4.4)20, with a significance threshold of adj P < 0.05 (Benjamini-Hochberg correction, < 0.05 indicated significant enrichment). Additionally, Gene Set Enrichment Analysis (GSEA) was performed to identify patterns of gene expression associated with biological processes, functions, or pathways distinguishing normal and DN groups within the training set (|Normalized Enrichment Score (NES)| > 1, Normalized Overlap-based Measure of Pathway (NOMP) < 0.05, q < 0.25). |NES| > 1 reflected the difference in gene set enrichment scores, while NOMP < 0.05 and q < 0.25 were used to assess the significance of the enrichment results and control the false discovery rate.
Identification of key genes
In this study, Least Absolute Shrinkage and Selection Operator (LASSO) and Support Vector Machine-Recursive Feature Elimination (SVM-RFE) algorithms were applied to identify key genes. LASSO regression constrained the sum of the absolute values of the regression coefficients and minimized the residual sum of squares, which resulted in strictly zero coefficients, effectively selecting genes associated with the disease. On the other hand, SVM-RFE utilized the maximum margin principle of support vector machines, combined with multiple iterations of training and feature ranking, to remove the features with the lowest scores, thereby identifying the importance of each gene. At the same time, it selected the gene combination with the lowest error rate, facilitating the identification of key genes. LASSO analysis was performed using the “glmnet” R package21, which implements a regression algorithm with regularization for variable selection. SVM-RFE was executed via the “e1071” R package22. The integration of these two machine learning approaches led to the identification of two pivotal genes. To further refine the selection, the ncvreg package (version 3.14.1) was utilized to screen the key genes using both Smoothly Clipped Absolute Deviation (SCAD) and Minimax Concave Penalty (MCP) algorithms. The diagnostic performance of these genes within the GSE142153 and GSE99325 datasets was evaluated by constructing ROC curves using the “pROC” package (version 1.18.0)23. The expression patterns of these key genes in both datasets were visualized with the “ggplot2” package (version 3.3.6)24, with statistical significance set at P < 0.05.
Clinical correlation analysis and GSEA of two key genes
The correlation between the key genes and relevant clinical indicators, such as glomerular filtration rate (GFR) and serum creatinine, was examined using the Nephroseq v5 online platform (http://v5.nephroseq.org). When performing the correlation analysis, you first entered the key gene names as prompted, ensuring their accuracy. Then, on the homepage, you selected the “Expression"→"Correlation Analysis” module and chose the appropriate kidney disease cohort (such as CKD, DN, etc.) and relevant clinical indicators (eGFR, proteinuria, serum creatinine, etc.) according to your research needs. The analysis used the default Pearson correlation coefficient method (P < 0.05). The platform automatically calculated the correlation between gene expression and clinical indicators, generating correlation coefficients and P-values, which were displayed in tables and scatter plots. Furthermore, GSEA for the two key genes was performed using the “ClusterProfiler” (version 4.4.4) and “org.hs.eg.db” (version 3.15.0) R packages25 (|NES| > 1, NOMP < 0.05, q < 0.25).
Immune infiltration analysis and spearman correlation analysis
To explore the relationship between immune cells and the key genes within the GSE142153 dataset, Single Sample GESA (ssGSEA) was conducted using the “GSVA” package (version 1.46.0)26, revealing connections between immune cell populations and the identified key genes. A heatmap was generated to visually represent the distribution of 26 immune cell types in both DN and normal samples, while a boxplot illustrated the variations in immune cell infiltration levels (Benjamini-Hochberg correction, adj P < 0.05). Additionally, Spearman’s correlation analysis was performed to assess potential correlations between the key genes and various immune cell populations (Benjamini-Hochberg correction, adj P < 0.05).
Construction of PPI network and CeRNA networks
The PPI network was constructed using the GeneMANIA database (http://genemania.org/). To predict potential miRNAs interacting with the key genes, several resources, including miRDB, miRTarbase, and miRWalk, were utilized. Potential lncRNAs interacting with the identified miRNAs were predicted through StarBase (http://starbase.sysu.edu.cn/index.php). The competing endogenous RNA (ceRNA) networks were constructed using Cytoscape. To identify N6-methyladenosine (m6A) methylation sites for the key genes, the Sequence-based RNA Adenosine Methylation Site Predictor (SRAMP, https://www.cuilab.cn/sramp/) was employed.
Potential drug prediction and molecular Docking
Potential drugs targeting the key genes were identified by querying the Comparative Toxicogenomics Database (CTD). The interaction network between key genes and potential drugs was visualized using Cytoscape (https://cytoscape.org/index.html). Additionally, the tertiary structures of receptors (key genes) were downloaded from the Protein Data Bank (PDB), and the structures of small molecule drugs were sourced from the PubChem database. Molecular docking simulations were carried out using AutoDock software.
RNA extraction and quantitative PCR
Five pairs of PBMC samples from both normal and DN cases were collected from clinical settings at The First Affiliated Hospital of Zhengzhou University. All participants provided informed consent for participation. The study protocol was approved by the ethics committee of The First Affiliated Hospital of Zhengzhou University and adhered to the ethical guidelines outlined in the World Medical Association’s Declaration of Helsinki. Total RNA was extracted from the 10 samples using TRIzol reagent (Invitrogen, China) as per the manufacturer’s instructions. cDNA synthesis was performed via reverse transcription using the SureScript First-strand cDNA Synthesis kit (Servicebio, China). Quantitative PCR (qPCR) assays were conducted using the CFX Connect Thermal Cycler (Bio-Rad, USA), and relative mRNA quantification was performed using the 2-ΔΔCT method. Detailed primer sequences are provided in Table S1.
Results
Identification of differentially expressed glutamine metabolism-related genes (DE-GMRGs)
In this study, GSE142153 was utilized as the training set for subsequent analyses. PCA was performed to assess the dataset’s integrity and detect any potential outliers. The analysis identified five abnormal samples (GSM4221586, GSM4221579, GSM4221600, GSM4221581, and GSM4221570). After removing these outliers, the separation between the groups remained significant, and the analysis proceeded with 9 normal samples and 19 DN samples (Supplementary Fig. 1A, B). A total of 2,281 DEGs were identified between the DN and normal groups in the GSE142153 dataset, including 1,035 upregulated genes and 1,247 downregulated genes. These DEGs were visualized using a volcano plot and heatmap (Fig. 1A, B, Table S2). The intersection of DEGs and GMRGs yielded 9 DE-GMRGs: FTCD, SLC7A5, HAL, SIRT4, GLS2, ALDH5A1, PPAT, IDH1, and SLC25A12. The PPI network revealed that these 9 DE-GMRGs interact with 97 DEGs, with SLC7A5 exhibiting more interactions with DEGs than SLC25A12 (Fig. 1C). Chromosome localization analysis showed that all these genes were located on specific chromosomes (Supplementary Fig. 1C).

Differential expression analysis in the GSE142153 dataset. (A,B) Volcano plot (A) and heat map (B) showing the differentially expressed genes (DEGs) between diabetic nephropathy (DN) and normal groups. (C) The interaction between DE-GMRGs and DEGs represented in the protein-protein interaction (PPI) network.
DE-GMRGs were associated with glutamate metabolic process and glutamine family amino acid metabolic process
GO enrichment analysis indicated that, for cell components, DE-GMRGs were predominantly enriched in the mitochondrial matrix. Regarding biological processes, these genes were significantly involved in the glutamate metabolic process and glutamine family amino acid metabolic processes. In terms of molecular function, they were notably enriched in carbon-nitrogen lyase activity (Fig. 2A, Table S3). KEGG enrichment analysis revealed that DE-GMRGs were primarily associated with pathways related to alanine, aspartate, and glutamate metabolism, histidine metabolism, and 2-oxocarboxylic acid metabolism (Fig. 2B, Table S4). Furthermore, GSEA was performed to identify differentially activated pathways between the DN and normal groups within the training set. The results highlighted significant differences in activation pathways, particularly in those linked to mismatch repair, homologous recombination, graft versus host disease, and bladder cancer (Fig. 2C, Table S5).

Functional enrichment analysis of differentially expressed glutamine metabolism-related genes (DE-GMRGs). (A) Gene Ontology (GO) terms enriched in DE-GMRGs. (B) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways enriched in DE-GMRGs. (C) Results of Gene Set Enrichment Analysis (GSEA) for DE-GMRGs.
Selection of key genes via statistical and machine learning algorithms
The LASSO algorithm was implemented to identify signature genes, and an optimal lambda value of 0.007 was determined. Through LASSO regression, four genes were identified as distinctive for DN: GLS2, HAL, SLC7A5, and SLC25A12(Fig. 3A, B). Conversely, the SVM-RFE algorithm identified SLC7A5 and SLC25A12 as characteristic genes (Fig. 3C). By intersecting the feature genes derived from both machine learning algorithms, the key genes SLC7A5 and SLC25A12 were pinpointed (Fig. 3D). Additionally, when λ values were set to 0.043 and 0.107, the SCAD and MCP model error rates were lowest for SLC7A5(SCAD = 5.49; MCP = 3.97) and SLC25A12(SCAD = -1.83; MCP = -1.43) (Fig. 3E-H). Consequently, SLC7A5 and SLC25A12 were considered key genes for further analysis.

Identification of key genes for DN. (A,B) Error plots from 10-fold cross-validation (A) and gene coefficient plot (B) from least absolute shrinkage and selection operator (LASSO) analysis. (C) Results from Support Vector Machine-Recursive Feature Elimination (SVM-RFE). (D) Venn diagram highlighting two key genes. (E,F) Identification of key gene SLC7A5 using Smoothly Clipped Absolute Deviation (SCAD) and Minimax Concave Penalty (MCP) algorithms (SCAD = 5.49; MCP = 3.97). (G,H) Identification of key gene SLC25A12 using SCAD and MCP algorithms (SCAD = -1.83; MCP = -1.43).
Key genes could effectively distinguish between DN and normal samples
The receiver operating characteristic (ROC) curves for SLC7A5 and SLC25A12 revealed area under the curve (AUC) values of 1.000 and 0.988, respectively, in the training set (GSE142153) (Fig. 4A). In the validation set (GSE99325), the AUC values for these two hub genes were 0.819 and 0.875, respectively (Fig. 4B). These results suggest that these key genes could serve as powerful biomarkers for DN diagnosis. Gene expression analysis in both the GSE142153 and GSE99325 datasets revealed that SLC7A5 expression was significantly higher in DN samples compared to normal samples, while SLC25A12 expression was notably lower in DN samples (Fig. 4C).

Diagnostic capabilities of key genes. (A,B) Receiver Operating Characteristic (ROC) curves for key genes in the GSE142153 (A) and GSE99325 (B) datasets. AUC represents the area under the curve. (C) Expression levels of two key genes in the DN and normal groups.
Key genes were significantly correlated with GFR and serum creatinine
The correlation between the expression levels of the key genes and clinical features, such as GFR and serum creatinine, was further examined using Nephroseq v5. Both SLC7A5 and SLC25A12 showed positive correlations with serum creatinine levels (SLC7A5: cor = 0.7118, P = 0.01; SLC25A12: cor = 0.9695, P = 0.03) and negative correlations with GFR (SLC7A5: cor = -0.5639, P = 0.04; SLC25A12: cor = -0.9163, P = 0.03) (Fig. 5A). GSEA revealed that SLC7A5 was significantly enriched in the peroxisome and MAPK signaling pathways, while SLC25A12 was notably enriched in the cytokine receptor interaction pathway (Fig. 5B).

Clinical correlation and functional enrichment analysis. (A) Correlation between two key genes and glomerular filtration rate (GFR) as well as serum creatinine levels. (B) Gene Set Enrichment Analysis (GSEA) of SLC7A5 and SLC25A12.
Key genes were associated with immune cells
Immune infiltration analysis revealed significant differences in 12 types of immune cells between the DN and normal groups (P < 0.05) (Fig. 6A-C). Spearman’s correlation analysis indicated that SLC7A5 exhibited a negative correlation (cor = -0.75) with effector memory CD8 T cells, while SLC25A12 showed a positive correlation (cor = 0.76) with effector memory CD8 T cells. Conversely, SLC7A5 was positively correlated (cor = 0.6) with mast cells, whereas SLC25A12 demonstrated a negative correlation (cor = -0.51) with mast cells (Supplementary Figure S2).

Immune infiltration analysis. (A) Heat map displaying immune cell scores in DN and control samples. (B) Immune cell infiltration discrepancies between DN and control samples. ns, not significant; *P < 0.05; ** P < 0.01; ***P < 0.001; ****P < 0.0001. (C) Correlation between key genes and differential immune cells.
Construction of regulatory network for key genes
PPI network analysis revealed the following interaction types: physical interactions (33.8%), co-expression (25.9%), predicted interactions (2.6%), pathway-based interactions (8.4%), shared protein domains (17.9%), genetic interactions (8.9%), and co-localization interactions (2.1%) (Fig. 7A). The predicted miRNAs and lncRNAs interacting with the two key genes were integrated into a competitive endogenous RNA (ceRNA) network, constructed using Cytoscape, which comprised 23 nodes and 29 edges (Fig. 7B). Additionally, the SRAMP database predicted 2 high-confidence m6A methylation sites in SLC25A12 (bases 165 and 1335) and 29 in SLC7A5 (bases 2013 and 3315) (Fig. 7C,D). These results suggest that methylation alterations in SLC7A5 may have a greater impact on DN development compared to SLC25A12 (Fig. 7B and C, Tables S6 and S7).

Regulatory networks of key genes. (A) Protein-protein interaction (PPI) network of key genes. (B) Competitive endogenous RNA (ceRNA) network of key genes. Red represents mRNA, blue represents microRNA (miRNA), and orange represents long non-coding RNA (lncRNA). (C,D) Prediction of methylation sites in key genes. (C) SLC25A12; (D) SLC7A5.
Drug prediction
To identify potential drugs targeting the key genes, the CTD database was used, establishing interactions between the key genes and identified drugs. The SLC7A5 network contained 234 nodes and 233 edges (Fig. 8A), while the SLC25A12 network had 83 nodes and 82 edges (Fig. 8B). Notably, SLC7A5 exhibited upregulation in DN, while S.3LC25A12 showed downregulation. Drugs capable of inhibiting SLC7A5 expression and promoting SLC25A12 expression were selected. Six drugs met this criterion: D001280 (Atrazine), C006780 (Bisphenol A), C089796 (Hexabromocyclododecane), C009618 (O, O-Diethyl O-3,5,6-trichloro-2-pyridyl phosphate), D013749 (Tetrachlorodibenzodioxin), and D014635 (Valproic Acid). Molecular docking analysis revealed that the docking affinities of SLC25A12 and D014635 (valproic acid) ranged from − 3.4 to -5.1, while the docking affinities of SLC7A5 and D014635 ranged from − 3.8 to -5.1. The pair with the strongest binding affinity was selected for further analysis, showing promising binding potential (Fig. 8C-D).

Drug prediction and molecular docking. (A,B) mRNA-drug interaction network for SLC7A5 (A) and SLC25A12 (B). Red represents key genes and blue represents targeting drugs. (C,D) Molecular docking results for key genes.
Verification of the DE-GMRGs in clinical samples
Finally, the expression of the two key genes was validated using quantitative reverse transcription PCR (qRT-PCR) in clinical samples. The results indicated that the expression of SLC25A12 was significantly higher in normal PBMC samples compared to DN PBMC samples, while SLC7A5 expression was significantly higher in DN PBMC samples (Fig. 9).

Validation of key genes’ expression in DN and control groups through qRT-PCR. ns, not significant; *P < 0.05; **P < 0.01.
Discussion
GMRGs have been identified as biomarkers in various conditions, including gastric adenocarcinoma27, lung adenocarcinoma28, prostate cancer29, breast cancer30, and Alzheimer’s disease31. Animal models have demonstrated that glutamine, a vital amino acid, can mitigate the effects of DN14,15. However, the complex mechanisms of glutamine metabolism in DN remain poorly understood. This study combines comprehensive bioinformatics analysis with experimental validation to uncover biomarkers associated with glutamine metabolism in DN, further investigating their potential molecular and regulatory roles. Consistent with the study by Bao et al., our research also employed machine learning methods such as LASSO and SVM-RFE to identify genes with diagnostic potential. However, while their study focused on Parkinson’s disease, our work explores the potential mechanisms of glutamine metabolism in diabetic nephropathy (DN), identifies novel biomarkers, provides additional insights for DN research, and uncovers potential therapeutic targets32.
In this study, SLC7A5 and SLC25A12 were identified as key indicators of glutamine metabolism in DN. Solute carrier family 7 member 5 (SLC7A5), also known as LAT1, plays a critical role in transmembrane transport, particularly for amino acids and their metabolites33. SLC7A5 primarily functions as an antiporter, mediating the export of glutamine34,35. Amino acid transporters like SLC7A5 are essential for regulating insulin secretion and signaling36. Notably, dysfunctions in the LAT1-4F2hc (SLC7A5-SLC3A2) transporter complex have been implicated in diabetes and immune-related diseases37. Chromosomal localization analysis placed SLC7A5 on chromosome 16, with the rs11865049 locus linked to susceptibility to central serous chorioretinopathy (CSC), suggesting its potential role in genetic predisposition to CSC38. Despite these insights, the role of SLC7A5 in DN had not been previously reported. Interestingly, elevated SLC7A5 expression was observed in both PBMC samples and renal tubulointerstitial tissue samples from patients with DN. This finding suggests that increased SLC7A5 expression may influence the progression of DN. The gene SLC25A12 encodes a mitochondrial carrier protein that facilitates the exchange of aspartate and glutamate within the mitochondrial inner membrane and binds calcium39. Polymorphisms in the SLC25A12 gene have been strongly associated with autism spectrum disorders40,41. Chromosomal localization analysis showed SLC25A12 is located on chromosome 2, with mutations in this gene linked to early infantile epileptic encephalopathy-39 (EIEE39), highlighting its importance in neurological conditions42. Notably, the expression pattern of SLC25A12 was found to be the opposite of SLC7A5. These findings suggest that SLC7A5 and SLC25A12, two key genes related to glutamine metabolism, may serve distinct functions in the pathogenesis of DN. Their contrasting expression patterns and interactions provide novel insights into the underlying mechanisms of DN and offer potential targets for future research and clinical intervention strategies.
GSEA was performed to explore the underlying mechanisms of SLC7A5 and SLC25A12 in DN. The results revealed an association between SLC7A5 and the MAPK signaling pathway, while SLC25A12 was linked to cytokine interactions. Previous studies have shown that the MAPK pathway enhances amino acid uptake in melanoma cells by regulating c-MYC expression and its downstream target, SLC7A5, thereby promoting cell proliferation43. Additionally, natural compounds have been demonstrated to reduce renal inflammation in diabetic patients by inhibiting the MAPK signaling pathway44. Therefore, SLC7A5 may influence DN progression through its modulation of MAPK signaling, potentially interacting with inflammatory responses. Cytokines, produced by immune cells, are active protein molecules that mediate various cellular responses. Hyperglycemia affects both resident and non-resident renal cells, stimulating the production of cytokines and humoral mediators. This cascade triggers functional and phenotypic alterations in renal cells, involving complex interactions among cell growth factors, proteins, and advanced glycation end products, ultimately leading to glomerular and tubular damage and nephropathy45. Moreover, SLC25A12 has been shown to regulate mitochondrial metabolism, influencing cytokine production and the immune response to viral infections46. These findings emphasize the potential immunological significance of SLC7A5 and SLC25A12 in DN. Further immune infiltration analysis revealed a significant increase in mast cell presence in DN, while effector memory CD8 T cells showed a notable decrease. Both SLC7A5 and SLC25A12 exhibited strong associations with mast cells (SLC7A5: cor = 0.6, P = 0.0008; SLC25A12: cor = -0.51, P = 0.0056) and effector memory CD8 T cells (SLC7A5: cor = -0.75, P = 4.58 × 10–6; SLC25A12: cor = 0.76, P = 2.90 × 10–6). Mast cells significantly influence renal fibrosis, as demonstrated in the Yin DN rat model, where inhibiting mast cell infiltration alleviated renal interstitial fibrosis47. This supports the notion that mast cells may exacerbate both the onset and progression of DN, aligning with our findings. In conclusion, the roles of SLC7A5 and SLC25A12 in DN are closely tied to immune cell infiltration and associated pathways. These insights provide a valuable basis for future studies exploring the precise molecular mechanisms of SLC7A5 and SLC25A12 in DN.
ceRNA regulatory mechanisms, including the lncRNA-miRNA-mRNA axis, are crucial in the development and progression of diabetes and its complications48,49,50. Recent studies have highlighted the involvement of the lncRNA-miRNA-mRNA regulatory network in the onset and progression of DN51,52,53. To further investigate the upstream ceRNA regulatory mechanisms associated with SLC7A5 and SLC25A12 in DN, a predictive analysis was conducted to identify relevant miRNAs and lncRNAs from publicly available databases. This effort led to the creation of a ceRNA regulatory network, consisting of the two key genes, six miRNAs, and fifteen lncRNAs. This network provides valuable insight for future studies on the role and regulatory mechanisms of SLC7A5 and SLC25A12 in DN. Further examination of these miRNAs and lncRNAs could offer new avenues for precision treatment in DN.
In the search for potential therapeutic agents for DN, the CTD database was utilized to identify drugs targeting SLC7A5 and SLC25A12. Notably, D014635 (Valproic Acid) has been shown to inhibit cell senescence in DN by blocking the complement C5A receptor54. Additionally, animal model studies have demonstrated that Valproic Acid, a histone deacetylase inhibitor, can alleviate DN55,56,57,58. As such, Valproic Acid emerges as a promising candidate for DN treatment, warranting further clinical investigation.
This study has several limitations. First, the relatively small sample size for qRT-PCR validation may have reduced statistical power, potentially affecting the reliability of the results. Future research should expand the sample size and include kidney tissue samples from patients with DN to enhance the relevance and credibility of the findings. Second, the immune infiltration analysis was not experimentally validated, and further studies are needed to confirm the role of key genes in immune regulation. Finally, while the correlation analysis highlighted potential biological significance, additional research is required to validate these observations.
In conclusion, this study integrated bioinformatics analysis with experimental validation to identify two crucial glutamine-related biomarkers in DN. This effort contributes to a deeper understanding of the molecular mechanisms underlying DN and has the potential to inform diagnostic and therapeutic approaches. However, the findings require further experimental investigation to fully explore their significance. To strengthen the clinical diagnostic and therapeutic potential of these genes, additional clinical samples and data are needed. Future research will continue to explore the multifaceted roles of these genes and their broader implications in DN.
Data availability
The datasets (GSE142153 and GSE99325) analyzed in this study were derived from the Gene Expression Omnibus (GEO) database (https://www.ncbi.nlm.nih.gov/geo/).
Abbreviations
- DN:
-
Diabetic nephropathy
- GEO:
-
Gene Expression Omnibus
- PBMC:
-
peripheral blood mononuclear cell
- GMRGs:
-
glutamine metabolism-related genes
- DEGs:
-
differentially expressed genes
- DE-GMRGs:
-
differentially expressed glutamine metabolism-related genes
- SVM-RFE:
-
Support Vector Machine-Recursive Feature Elimination
- LASSO:
-
Least Absolute Shrinkage and Selection Operator
- ROC:
-
Receiver operating characteristic
- GSEA:
-
Gene Set Enrichment Analysis
- qRT-PCR:
-
real-time fluorescence quantitative PCR
- PCA:
-
Principal Components Analysis
- GO:
-
Gene Ontology
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- ssGSEA:
-
Single Sample Gene Set Enrichment Analysis
- ceRNA:
-
competitive endogenous RNA
References
Almufleh, A. et al. Profound vasoplegia during sacubitril/valsartan treatment after heart transplantation. Can. J. Cardiol. 34(3), 343.e345–343.e347 (2018).
Roy, A. et al. Kidney disease in type 2 diabetes mellitus and benefits of Sodium-Glucose cotransporter 2 inhibitors: A consensus statement. Diabetes Ther. 11(12), 2791–2827 (2020).
Thipsawat, S. Early detection of diabetic nephropathy in patient with type 2 diabetes mellitus: A review of the literature. Diab Vasc Dis. Res. 18(6), 14791641211058856 (2021).
Li, S., Xie, H., Shi, Y. & Liu, H. Prevalence of diabetic nephropathy in the diabetes mellitus population: A protocol for systematic review and meta-analysis. Med. (Baltim). 101(42), e31232 (2022).
Lin, Y. K. et al. The prevalence of diabetic microvascular complications in China and the USA. Curr. Diab Rep. 21(6), 16 (2021).
Qi, C., Mao, X., Zhang, Z. & Wu, H. Classification and differential diagnosis of diabetic nephropathy. J. Diabetes Res. 2017, 8637138 (2017).
Najafian, B., Alpers, C. E. & Fogo, A. B. Pathology of human diabetic nephropathy. Contrib. Nephrol. 170, 36–47 (2011).
Warren, A. M., Knudsen, S. T. & Cooper, M. E. Diabetic nephropathy: an insight into molecular mechanisms and emerging therapies. Expert Opin. Ther. Targets. 23(7), 579–591 (2019).
Li, X. et al. GBP2 promotes M1 macrophage polarization by activating the notch1 signaling pathway in diabetic nephropathy. Front. Immunol. 14, 1127612 (2023).
Zhou, H., Mu, L., Yang, Z. & Shi, Y. Identification of a novel immune landscape signature as effective diagnostic markers related to immune cell infiltration in diabetic nephropathy. Front. Immunol. 14, 1113212 (2023).
Md Dom, Z. I. et al. Circulating proteins protect against renal decline and progression to end-stage renal disease in patients with diabetes. Sci. Transl. Med. 13(600) (2021).
Liu, L. et al. Metabolic homeostasis of amino acids and diabetic kidney disease. Nutrients 15(1) (2022).
Miller, R. A. et al. Targeting hepatic glutaminase activity to ameliorate hyperglycemia. Nat. Med. 24(4), 518–524 (2018).
Nasri, M., Adibhesami, G., Mahdavifard, S., Babaeenezhad, E. & Ahmadvand, H. Exogenous glutamine ameliorates diabetic nephropathy in a rat model of type 2 diabetes mellitus through its antioxidant and anti-inflammatory activities. Arch. Physiol. Biochem. 129(2), 363–372 (2023).
Sadar, S., Kaspate, D. & Vyawahare, N. Protective effect of L-glutamine against diabetes-induced nephropathy in experimental animal: role of KIM-1, NGAL, TGF-β1, and collagen-1. Ren. Fail. 38(9), 1483–1495 (2016).
Chen, K. et al. Genetic variants in glutamine metabolic pathway genes predict cutaneous melanoma-specific survival. Mol. Carcinog. 58(11), 2091–2103 (2019).
Li, Z. et al. Screening of the key genes and signalling pathways for diabetic nephropathy using bioinformatics analysis. Front. Endocrinol. (Lausanne). 13, 864407 (2022).
Zhang, H., Meltzer, P. & Davis, S. RCircos: an R package for circos 2D track plots. BMC Bioinform. 14, 244 (2013).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53(D1), D672–D677 (2025).
Zhang, H., Hu, J., Zhu, J., Li, Q. & Fang, L. Machine learning-based metabolism-related genes signature and immune infiltration landscape in diabetic nephropathy. Front. Endocrinol. (Lausanne). 13, 1026938 (2022).
Friedman, J., Hastie, T. & Tibshirani, R. Regularization paths for generalized linear models via coordinate descent. J. Stat. Softw. 33(1), 1–22 (2010).
Sanz, H., Valim, C., Vegas, E., Oller, J. M. & Reverter, F. SVM-RFE: selection and visualization of the most relevant features through non-linear kernels. BMC Bioinform. 19(1), 432 (2018).
Yan, B., Ren, F., Shang, W. & Gong, X. Transcriptomic Analysis Reveals Genetic Cross-Talk between Periodontitis and Hypothyroidism. Dis. Markers 2022, 5736394 (2022).
Li, T. et al. Data analysis of PD-1 antibody in the treatment of melanoma patients. Data Brief. 30, 105523 (2020).
Qing, J. et al. Differentiation of T helper 17 cells May mediate the abnormal humoral immunity in IgA nephropathy and inflammatory bowel disease based on shared genetic effects. Front. Immunol. 13, 916934 (2022).
Zhao, K., Ma, Z. & Zhang, W. Comprehensive analysis to identify SPP1 as a prognostic biomarker in cervical cancer. Front. Genet. 12, 732822 (2021).
Li, H., Wu, Z., Zhang, Y., Lu, X. & Miao, L. Glutamine metabolism genes prognostic signature for stomach adenocarcinoma and immune infiltration: potential biomarkers for predicting overall survival. Front. Oncol. 13, 1201297 (2023).
Zhang, P. et al. Integrating multiple machine learning methods to construct glutamine metabolism-related signatures in lung adenocarcinoma. Front. Endocrinol. (Lausanne). 14, 1196372 (2023).
Wang, H. et al. A Five glutamine-associated signature predicts prognosis of prostate cancer and links glutamine metabolism with tumor microenvironment. J. Clin. Med. 12(6) (2023).
Pei, S. et al. Integrating single-cell RNA-seq and bulk RNA-seq to construct prognostic signatures to explore the role of glutamine metabolism in breast cancer. Front. Endocrinol. (Lausanne). 14, 1135297 (2023).
Wu, Z. et al. A novel alzheimer’s disease prognostic signature: identification and analysis of glutamine metabolism genes in immunogenicity and immunotherapy efficacy. Sci. Rep. 13(1), 6895 (2023).
Bao, Y., Wang, L., Yu, F., Yang, J. & Huang, D. Parkinson’s disease gene biomarkers screened by the LASSO and SVM algorithms. Brain Sci. 20(2), 175 (2023).
Scalise, M. et al. Chemical approaches for studying the biology and pharmacology of membrane transporters: the histidine/large amino acid transporter SLC7A5 as a benchmark. Molecules 26(21) (2021).
Yanagida, O. et al. Human L-type amino acid transporter 1 (LAT1): characterization of function and expression in tumor cell lines. Biochim. Biophys. Acta. 1514(2), 291–302 (2001).
Fuchs, B. C. & Bode, B. P. Amino acid transporters ASCT2 and LAT1 in cancer: partners in crime? Semin Cancer Biol. 15(4), 254–266 (2005).
Javed, K. & Fairweather, S. J. Amino acid transporters in the regulation of insulin secretion and signalling. Biochem. Soc. Trans. 47(2), 571–590 (2019).
Kahlhofer, J. & Teis, D. The human LAT1-4F2hc (SLC7A5-SLC3A2) transporter complex: physiological and pathophysiological implications. Basic Clin. Pharmacol. Toxicol(2022).
Yuzawa, M. et al. Genome-Wide association study to identify a new susceptibility locus for central serous chorioretinopathy in the Japanese population. Invest. Ophthalmol. Vis. Sci. 59(13), 5542–5547 (2018).
Rueda, C. B. et al. Glutamate excitotoxicity and Ca2+-regulation of respiration: role of the Ca2 + activated mitochondrial transporters (CaMCs). Biochim. Biophys. Acta. 1857(8), 1158–1166 (2016).
Aoki, Y. & Cortese, S. Mitochondrial Aspartate/Glutamate carrier SLC25A12 and autism spectrum disorder: a Meta-Analysis. Mol. Neurobiol. 53(3), 1579–1588 (2016).
Qiu, S., Qiu, Y., Li, Y. & Cong, X. Genetics of autism spectrum disorder: an umbrella review of systematic reviews and meta-analyses. Transl Psychiatry. 12(1), 249 (2022).
Saleh, M., Helmi, M. & Yacop, B. A novel nonsense gene variant responsible for early infantile epileptic encephalopathy type 39: case report. Pak J. Biol. Sci. 23(7), 973–976 (2020).
Pathria, G., Verma, S., Yin, J., Scott, D. A. & Ronai, Z. A. MAPK signaling regulates c-MYC for melanoma cell adaptation to asparagine restriction. EMBO Rep. 3(3), e51436 (2021).
Ma, L. et al. Baicalin alleviates oxidative stress and inflammation in diabetic nephropathy via Nrf2 and MAPK signaling pathway. Drug Des. Devel Ther. 15, 3207–3221 (2021).
Wu, T., Ding, L., Andoh, V., Zhang, J. & Chen, L. The mechanism of Hyperglycemia-Induced renal cell injury in diabetic nephropathy disease: an update. Life (Basel) 13(2) (2023).
Yasukawa, K., Kinoshita, D., Yaku, K., Nakagawa, T. & Koshiba, T. The MicroRNAs miR-302b and miR-372 regulate mitochondrial metabolism via the SLC25A12 transporter, which controls MAVS-mediated antiviral innate immunity. J. Biol. Chem. 295(2), 444–457 (2020).
Yin, D. D., Luo, J. H., Zhao, Z. Y., Liao, Y. J. & Li, Y. Tranilast prevents renal interstitial fibrosis by blocking mast cell infiltration in a rat model of diabetic kidney disease. Mol. Med. Rep. 17(5), 7356–7364 (2018).
Pandey, A. et al. Current Insights into miRNA and lncRNA dysregulation in diabetes: Signal transduction, clinical trials and biomarker discovery. Pharmaceuticals (Basel) 15(10) (2022).
Song, Z., He, C., Wen, J., Yang, J. & Chen, P. Long Non-coding rnas: pivotal epigenetic regulators in diabetic retinopathy. Curr. Genomics. 23(4), 246–261 (2022).
Macvanin, M. T. et al. Diabetic cardiomyopathy: the role of MicroRNAs and long non-coding RNAs. Front. Endocrinol. (Lausanne). 14, 1124613 (2023).
Li, H., Hao, J. & Yu, W. LncRNA CASC15 Inhibition relieves renal fibrosis in diabetic nephropathy through down-regulating SP-A by sponging to miR-424. Open. Med. (Wars). 18(1), 20230710 (2023).
Guo, J. et al. Long non-coding RNA DLX6-AS1 is the key mediator of glomerular podocyte injury and albuminuria in diabetic nephropathy by targeting the miR-346/GSK-3β signaling pathway. Cell. Death Dis. 14(2), 172 (2023).
Xu, B. W., Rao, Y., Wang, L., Chen, S. M. & Zou, S. B. LncRNA MEG3 inhibits renal fibrinoid necrosis of diabetic nephropathy via the MEG3/miR-21/ORAI1 axis. Mol. Biol. Rep. 50(4), 3283–3295 (2023).
Coughlan, M. T., Ziemann, M., Laskowski, A., Woodruff, T. M. & Tan, S. M. Valproic acid attenuates cellular senescence in diabetic kidney disease through the Inhibition of complement C5a receptors. Sci. Rep. 12(1), 20278 (2022).
Khan, S., Jena, G., Tikoo, K. & Kumar, V. Valproate attenuates the proteinuria, podocyte and renal injury by facilitating autophagy and inactivation of NF-κB/iNOS signaling in diabetic rat. Biochimie 110, 1–16 (2015).
Khan, S., Jena, G. & Tikoo, K. Sodium valproate ameliorates diabetes-induced fibrosis and renal damage by the Inhibition of histone deacetylases in diabetic rat. Exp. Mol. Pathol. 98(2), 230–239 (2015).
Sun, X., Sun, Y., Lin, S., Xu, Y. & Zhao, D. Histone deacetylase inhibitor valproic acid attenuates high glucose–induced Endoplasmic reticulum stress and apoptosis in NRK–52E cells. Mol. Med. Rep. 22(5), 4041–4047 (2020).
Sun, X. Y. et al. Valproate attenuates diabetic nephropathy through Inhibition of Endoplasmic reticulum stress–induced apoptosis. Mol. Med. Rep. 13(1), 661–668 (2016).
Author information
Authors and Affiliations
Contributions
Conceptualization, Ruojing Bai; Formal analysis, Yuanyuan Luo and Ruojing Bai; Methodology, Yuanyuan Luo and Ruojing Bai; Software, Yuanyuan Luo; Supervision, Ruojing Bai; Validation, Ruojing Bai; Writing – original draft, Yuanyuan Luo; Writing – review & editing, Yuanyuan Luo and Ruojing Bai.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The studies involving human participants were reviewed and approved by The First Affiliated Hospital of Zhengzhou University and was performed according to the guidance and principles of the World Medical Association’s Declaration of Helsinki. The patients/participants provided their written informed consent to participate in this study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Luo, Y., Bai, R. Identification of key genes related to glutamine metabolism in diabetic nephropathy by machine learning methods. Sci Rep 15, 45703 (2025). https://doi.org/10.1038/s41598-025-28169-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-28169-1
