
Abstract
Pathogenic variants in the STRC gene are among the most common causes of autosomal recessive non-syndromic hearing loss, particularly in cases with mild-to-moderate sensorineural hearing loss (SNHL). Despite its prevalence, the clinical phenotype and natural history of STRC-related SNHL remain undercharacterized due to diagnostic challenges posed by a highly homologous pseudogene, pSTRC. This study included 23 families enrolled in the Yonsei University Hearing Loss cohort. Genetic testing was performed using either targeted deafness gene panels or whole-exome sequencing, followed by multiplex ligation-dependent probe amplification and confirmatory Sanger sequencing. A total of 23 patients with STRC-related SNHL were identified, including 12 with homozygous STRC/CATSPER2 gene deletions and 11 with other combinations of pathogenic variants. Most patients exhibited mild-to-moderate SNHL with flat or gently sloping audiometric configurations, predominantly affecting mid-to-high frequencies. No significant differences in mean PTA thresholds were observed between the two genotypic groups. Longitudinal analysis over a follow-up period of up to 4 years demonstrated stable hearing thresholds in 75% of ears, with no significant progression detected using linear mixed model analysis. Linear regression showed no age-dependent threshold shift in either ear across all genotypic subgroups. In conclusion, STRC-related hearing loss is typically mild-to-moderate, stable over time, and audiometrically similar regardless of genotypic subclassification. Given its subtle phenotype and diagnostic complexity, STRC mutations may be underrecognized without targeted screening. Incorporating STRC-specific MLPA assay into routine genetic diagnostics in patients with mild-to-moderate hearing loss may improve early detection and guide timely precision intervention.
Similar content being viewed by others

Introduction
Hearing loss is the most common inherited sensory impairment in humans. More than 50% of prelingual sensorineural hearing loss (SNHL) is attributed to genetic causes, and approximately 80% of these cases are classified as non-syndromic SNHL (NSHL)1. Non-syndromic prelingual SNHL is genetically heterogeneous, but autosomal recessive inheritance accounts for the majority of cases, representing approximately 80%2,3. To date, the most frequent genetic cause of AR-NSHL is a mutation in the GJB2 gene located at the DFNB1 locus4. The second most commonly affected locus is DFNB16, caused by pathogenic variants in the STRC gene. STRC variants are increasingly recognized as a major cause of non-syndromic hearing loss and have been reported at relatively high prevalence across various ethnic populations5,6. In mixed-ethnicity cohorts with hearing loss, STRC deletions or mutations have been reported at frequencies ranging from 5.4% to 16.1%. In the United States and Italy, prevalence rates have been reported at 6–11.2% and 6%, respectively. In Asian populations, the estimated prevalence is 1.7–2.4% in Japan and 10.8% in Korea. Notably, in Korean patients with mild-to-moderate SNHL, STRC mutations were identified as the most common genetic cause, while in a comparable Japanese cohort, they were found to be the second most frequent cause following GJB2 single nucleotide variants7,8,9,10,11,12,13,14.
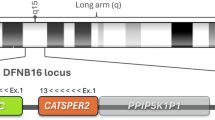
The STRC gene encodes stereocilin, a protein localized to the stereocilia bundles of outer hair cells. STRC is located on chromosome 15q15.3, within a complex segmental duplication region that also includes three other genes: PPIP5K1 (MIM: 610979), CATSPER2 (MIM: 607249), and CKMT1A (MIM: 613415)15. Genetic testing of STRC is particularly challenging due to the presence of a highly homologous pseudogene, pSTRC, in close proximity. STRC and pSTRC share 99.6% sequence identity in coding regions and 98.9% when intronic sequences are included, making them nearly indistinguishable by conventional sequencing methods. Moreover, pSTRC is a nonfunctional pseudogene due to a nonsense mutation in exon 206,16. These structural complexities necessitate specialized approaches for accurate STRC analysis. Although most STRC-related pathogenic variants are copy number variations (CNVs) involving the entire gene or large portions of it, point mutations, small insertions/deletions, and gene conversions have also been reported. Therefore, variants detected by next-generation sequencing (NGS) must be validated using gene-specific long-range PCR followed by Sanger sequencing to avoid misinterpretation caused by pseudogene contamination. Additionally, for deletion screening, a quantitative competitive fluorescence PCR (QCF-PCR) assay—exploiting length differences between STRC and pSTRC—has been implemented in diagnostic workflows17,18.
Among the various NGS approaches, massively parallel sequencing of targeted deafness genes has become the most frequently used and effective method for patients without biallelic GJB2 mutations. Currently, large-scale NGS is widely applied in clinical practice, enabling accurate diagnosis through dedicated bioinformatics pipelines. In this study, we aimed to identify patients with STRC mutations using NGS-based methods and to characterize their clinical and genetic features, including auditory genotype–phenotype correlations.
Results
We identified and clinically evaluated 14 children and 9 adults with pathogenic variants in the STRC gene (Table 1). Genetic diagnoses were confirmed by a board of otolaryngologists and clinical geneticists according to the hearing loss-specified American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) guidelines in the Deafness Variation Database19. STRC gene deletion ,defined as a contiguous loss of exons 19–25 detected by P461-A1 multiplex ligation-dependent probe amplification (MLPA) kit, or one exon deletion in STRC was confirmed pathogenic according to the ACMG/AMP guideline. In all cases with STRC gene deletion, a CATSPER2 gene deletion, defined as a contiguous loss of exons 1,2,4,7, and 12 detected by P461-A1 MLPA kit, was also identified (Table 1). Pure-tone audiometry (PTA) could not be performed in patients younger than 4 years; instead, auditory brainstem response (ABR) and/or auditory steady-state response (ASSR) tests were used. For patients aged 4 to 5 years, behavioral audiometry with headphones was conducted. Due to differences in testing modalities, patients who underwent ASSR instead of PTA—primarily because of their young age—were excluded from the audiometric comparison analysis. To ensure consistency in measurement, only patients who underwent behavioral audiometry were included.
Among the 19 patients who underwent PTA, most exhibited flat or gently sloping audiometric configurations, with elevated thresholds particularly in the mid-to-high frequencies. When comparing mean thresholds across frequencies, the threshold at 500 Hz was significantly different from those at higher frequencies. The mean hearing thresholds at each frequency were as follows: right ear – 32.9, 43.4, 44.7, 41.8, and 44.2 dB HL; left ear – 32.9, 43.4, 44.5, 45.0, and 45.3 dB HL (at 500, 1,000, 2,000, 4,000, and 8,000 Hz, respectively) (Fig. 1).

Mean hearing thresholds by frequency in patients with STRC-related hearing loss (n = 19). Only patients who were able to undergo behavioral audiometry were included.
We compared the distribution of average pure-tone hearing thresholds between two genetically defined groups: patients with homozygous STRC/CATSPER2 gene deletion and those with other pathogenic variants (e.g., heterozygous exon deletions, nonsense mutations, or compound heterozygous mutations). Audiometric comparisons were based on the mean initial PTA thresholds at 0.5, 1, 2, and 4 kHz for the better ear (Fig. 2). For the group with homozygous STRC/CATSPER2 gene deletions, individual audiograms were presented alongside family pedigrees (Supplemental Fig. 1).

Comparison of mean hearing thresholds among STRC genotypic groups. Comparison of mean hearing thresholds (better ear) between genotypic groups: patients with homozygous STRC/CATSPER2 deletions (homo del, n = 10) and those with other pathogenic variants (other, n = 9), including exon deletions, nonsense or missense mutations, and compound heterozygous combination of a STRC gene deletion with another variant (p = 0.549 by Mann–Whitney U test).
Linear mixed model analysis was used to compare serial hearing levels over time. Nineteen ears were assessed at the initial test, 10 ears at the 2-year follow-up, and 6 ears at the 4-year follow-up, analyzing thresholds from the better ear. No statistically significant differences in hearing thresholds were observed over time at any frequency (Fig. 3). In 75% of ears (9 out of 12), the difference in average hearing thresholds between the first and last tests was within 5 dB or not statistically significant. The relationship between age and mean PTA thresholds was analyzed using linear regression, and no significant age-related progression was observed in the better ear across all genotypic subgroups (Fig. 4).

Longitudinal changes in hearing thresholds in patients with STRC mutations. Better ear thresholds at 500–8000 Hz measured at baseline (●, n = 19), 2 years (■, n = 10), and 4 years (▲, n = 6); no significant time effect (linear mixed model, F(2, 4.79) = 1.75, p = 0.268).

Age-related change in mean pure tone audiometry (PTA) thresholds by genotype. (A-C) Mean PTA thresholds (average of 0.5, 1, 2, and 4 kHz) of the better ear are plotted as a function of age. Solid lines represent best-fit linear regression models, and dashed lines indicate 95% confidence intervals. No statistically significant age-related progression was observed in any group. (A) Better ear thresholds in all patients (n = 19). Regression: Y = − 0.03·X + 39.80 (95% CI: − 0.37 to 0.30, r² = 0.002, p = 0.84). (B) Better ear thresholds in patients with homozygous STRC/CATSPER2 gene deletions (n = 10). Regression: Y = 0.21·X + 35.83 (95% CI: − 0.38 to 0.80, r² = 0.076, p = 0.44). (C) Better ear thresholds in patients with other STRC variants (n = 9). Regression: Y = − 0.29·X + 45.32 (95% CI: − 0.77 to 0.18, r² = 0.237, p = 0.18).
Discussion
In this study, we demonstrated that patients with STRC-related hearing loss predominantly exhibited mild-to-moderate SNHL, characterized by flat or gently sloping audiometric configurations affecting the mid-to-high frequencies. Notably, no significant differences in hearing thresholds were observed between patients with homozygous STRC gene deletions and those with other pathogenic variants, such as heterozygous exon deletions, nonsense mutations, or compound heterozygous mutations. Furthermore, longitudinal audiometric evaluations revealed no statistically significant progression of hearing loss over time.
One of the major challenges in analyzing STRC variants lies in its high sequence similarity with the adjacent pseudogene, STRCP1, which complicates read mapping and variant interpretation when using short-read sequencing technologies. In this study, gene-specific long-range PCR was not employed to confirm the origin of detected variants. For cases involving novel single nucleotide variants, we applied multiple layers of bioinformatic and manual validation. This included visual inspection of mapped reads using Integrative Genomics Viewer (IGV) to assess mapping quality and rule out ambiguous alignments, as well as sequence similarity analysis using UCSC BLAT. In these analyses, the reads demonstrated alignment patterns more consistent with STRC than STRCP1. While these approaches provide reasonable confidence in variant localization, we acknowledge that long-range PCR remains the gold standard for definitive confirmation, especially in cases involving novel or uncertain variants. We recommend that future studies incorporate orthogonal validation methods to further reduce the risk of pseudogene-related misinterpretation, particularly in clinical diagnostic settings.
The STRC gene encodes stereocilin, an extracellular structural protein localized in the stereocilia of outer hair cells (OHCs). Stereocilin plays a crucial role in anchoring the OHC bundle to the tectorial membrane20,21,22. The absence of upper horizontal connectors or abnormalities in tip-link structures between stereocilia may impair cochlear amplification, particularly affecting high-frequency hearing22,23. Consistent with previous findings, our study confirmed a statistically significant threshold difference between 500 Hz and higher frequencies.
Previous reports investigating STRC-related SNHL have identified patients across a wide age range, consistently revealing a high-frequency hearing loss pattern. Most individuals demonstrated mild-to-moderate hearing loss without progression to severe impairment8,9,13,14,24,25. In agreement with these studies, our findings show that thresholds at frequencies ≥ 1,000 Hz were indicative of moderate hearing loss. Prior studies have also demonstrated that this hearing profile typically persists into the fifth or sixth decade of life, with no evidence of substantial progression9,14,25,26. In our cohort, 75% of ears showed stable thresholds (change < 5 dB), aligning with previous reports of similar stability rates25. Although some studies have reported progressive hearing loss, the changes were generally statistically insignificant or occurred at a slow rate (e.g., 0.6 dB/year)27,28.
We further categorized patients based on genotypic patterns and compared their audiologic outcomes. Our data support previous findings that no significant differences in mean hearing thresholds exist between genotypes. Simi et al. reported no statistical difference in baseline pure-tone averages between patients with homozygous STRC deletions and those with compound heterozygous variants28. Similarly, Markova et al. compared average hearing thresholds among three genotype-based subgroups—homozygous deletions, compound heterozygotes with two distinct deletions, and compound heterozygotes with a deletion and a point mutation in trans—and found comparable thresholds across all groups, although a few individuals in the homozygous deletion and deletion/point mutation groups exhibited relatively better hearing25.
This study has several limitations. Audiologic assessments were not uniformly applied across all participants, and hearing loss progression may not have been detected in some patients with shorter follow-up periods. To enhance statistical power, we performed linear mixed model analysis.
Given that STRC-related SNHL typically presents with mild-to-moderate severity and may remain undetected without active screening, the importance of accurate molecular diagnosis is heightened. Compared to severe-to-profound hearing loss, mild-to-moderate SNHL is more likely to be overlooked or underestimated. While severe hearing loss is often identified early, enabling timely intervention that can mitigate significant medical, educational, and socioeconomic burdens, mild-to-moderate SNHL may go undetected and, if unmanaged, result in greater academic challenges than in patients with properly treated hearing loss29,30. The STRC gene is known to be technically challenging to analyze due to the presence of a highly homologous pseudogene and frequent genomic duplications, which limit the sensitivity of standard sequencing methods13,18. Therefore, supplementary genetic testing methods, such as MLPA or CNV analysis, are recommended to ensure accurate detection of pathogenic variants.
In conclusion, this study characterized the clinical and audiologic features of STRC-related SNHL, demonstrating that most patients exhibited mild-to-moderate hearing loss with a stable, non-progressive course. Notably, hearing thresholds were comparable between patients with homozygous STRC gene deletions and those with other genotypes. Given the subtle nature of STRC-related SNHL, such cases may be easily overlooked without active screening. Incorporating STRC analysis into broader genetic testing strategies may facilitate earlier diagnosis and intervention, potentially improving long-term developmental and educational outcomes.
Methods
Patient enrollment
This study enrolled patients from the Yonsei University Hearing Loss (YUHL) cohort, a registry for individuals with genetic hearing loss. Patients included in the cohort either had a family history of hearing loss or voluntarily underwent genetic testing. From this larger cohort, 23 probands were retrospectively selected between 2017 and 2024 based on the identification of biallelic pathogenic variants in the STRC gene. The cohort comprised 6 males and 17 females, with a mean age of 13.4 ± 9.7 years. The study was approved by the Severance Hospital Institutional Review Board (approval no. 4–2015-0659), and written informed consent was obtained from all participants. All procedures performed in this study were in accordance with the ethical standards of the institutional review board and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. Eight patients did not have any relatives with hearing loss within two generations (Table 1).
Evaluation of hearing function
Audiologic evaluations were conducted using age-appropriate methods. For children under 4 years of age, ABR and/or ASSR tests were performed. Children aged 4–5 years underwent behavioral audiometry with headphones (play audiometry), while conventional (PTA was used for older patients. Medical and family histories, clinical information, and age of onset of hearing loss were documented. Patients under serial follow-up received repeated audiologic assessments at each visit. Specifically, three children underwent ABR and ASSR, and four children were evaluated using behavioral pure-tone audiometry. Audiologic evaluations were conducted during the same study period, from 2017 to 2024. One patient did not undergo audiologic testing at the time of enrollment; therefore, hearing assessments were available for 22 of the 23 patients.
Genetic analysis
Genetic testing was performed on affected individuals and their family members using NGS, as previously described31,32,33,34,35,36. A two-track approach was employed, utilizing either whole-exome/genome sequencing (WES/WGS) or a targeted deafness gene panel, depending on insurance coverage and patient preference. For panel-based testing, a custom-designed panel targeting 207 deafness-associated genes was used, as previously reported37,38. WES was performed using the Agilent SureSelect V5 enrichment capture kit (Agilent Technologies, Santa Clara, CA, USA) following the manufacturer’s protocol. Sequencing was carried out on the Illumina MiSeq platform (San Diego, CA, USA) using the MiSeq Reagent Kit v2 (300 cycles). Segregation analysis was conducted using Sanger sequencing. Variant detection was performed using the “Basic Variant Caller” function in CLC software, applying thresholds of a minimum of five reads, 20× coverage, and a variant allele frequency ≥ 20%. Variants with minor allele frequencies > 0.5% (for recessive genes) or > 0.05% (for dominant genes) in the dbSNP and gnomAD databases were excluded.
CNVs were detected using MLPA for panel-tested patients and ExomeDepth (v1.1.16) for WES data. CNV calls from WES were confirmed by MLPA using MLPA Probemix P461 and reagent kit (MRC-Holland, Amsterdam, Netherlands). CNV of “STRC gene deletion” was defined as a contiguous loss of exons 19–28 (covered exons by P461 kit) on at least one allele, which we interpret as complete loss of the gene because exons 1–18 cannot be reliably assessed due to pseudogene homology. Similarly, CNV of “CATSPER2 gene deletion” was defined as a contiguous loss of exons 1, 2, 3, and 7 (covered exons by P461 kit). Final genetic diagnoses were made by a multidisciplinary team of otolaryngologists and clinical geneticists in accordance with hearing loss–specific interpretation guidelines from the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP), as implemented in the Deafness Variation Database. Patients with one pathogenic/likely pathogenic variant in trans with a VUS were also included when segregation and phenotype supported a molecular diagnosis of STRC-related hearing loss.
Statistical analysis
A p-value < 0.05 was considered statistically significant. All statistical analyses were performed using IBM SPSS for Windows, version 20.0 (IBM Corp., Armonk, NY, USA). Data are presented as mean ± standard deviation. The Mann–Whitney U test or linear mixed model analysis were used to compare average thresholds between groups and to evaluate longitudinal changes in hearing levels, respectively. In addition, linear regression analysis was performed using GraphPad Prism version 8.0 (GraphPad Software, San Diego, CA, USA) to assess the relationship between age and hearing thresholds.
Data availability
The datasets generated and analysed during the current study are available in the BioProject repository, accession number PRJNA1290888.
References
Petersen, M. & Willems, P. Non-syndromic, autosomal‐recessive deafness. Clin. Genet. 69, 371–392 (2006).
Faundes, V. & Pardo, R. A. Genetics of congenital deafness. Med. Clin. 139, 446–451 (2012).
Hilgert, N., Smith, R. J. & Van Camp, G. Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics? Mutat. Research/Reviews Mutat. Res. 681, 189–196 (2009).
Tranebjærg, L. Genetics of congenital hearing impairment: a clinical approach. Int. J. Audiol. 47, 535–545 (2008).
Han, S., Zhang, D., Guo, Y., Fu, Z. & Guan, G. Prevalence and characteristics of STRC gene mutations (DFNB16): a systematic review and meta-analysis. Front. Genet. 12, 707845 (2021).
Verpy, E. et al. Mutations in a new gene encoding a protein of the hair bundle cause non-syndromic deafness at the DFNB16 locus. Nat. Genet. 29, 345–349 (2001).
Amr, S. S. et al. Allele-specific droplet digital PCR combined with a next-generation sequencing-based algorithm for diagnostic copy number analysis in genes with high homology: proof of concept using stereocilin. Clin. Chem. 64, 705–714 (2018).
Ito, T. et al. Rapid screening of copy number variations in STR C by droplet digital PCR in patients with mild-to-moderate hearing loss. Hum. Genome Variation. 6, 41 (2019).
KimBJ, O. SignificantMendelian genetic contribution to pediatric mild-to-moderate hearingloss andits comprehensive diagnostic approach. GenetMed 22, 1119–1128 (2020).
Mandelker, D. et al. Comprehensive diagnostic testing for stereocilin: an approach for analyzing medically important genes with high homology. J. Mol. Diagn. 16, 639–647 (2014).
Morgan, A. et al. Lights and shadows in the genetics of syndromic and non-syndromic hearing loss in the Italian population. Genes 11, 1237 (2020).
Shearer, A. E. et al. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 6, 1–10 (2014).
Vona, B. et al. DFNB16 is a frequent cause of congenital hearing impairment: implementation of STRC mutation analysis in routine diagnostics. Clin. Genet. 87, 49–55 (2015).
Yokota, Y. et al. Frequency and clinical features of hearing loss caused by STRC deletions. Sci. Rep. 9, 4408 (2019).
Knijnenburg, J. et al. A homozygous deletion of a normal variation locus in a patient with hearing loss from non-consanguineous parents. J. Med. Genet. 46, 412–417. https://doi.org/10.1136/jmg.2008.063685 (2009).
Francey, L. J. et al. Genome-wide SNP genotyping identifies the stereocilin (STRC) gene as a major contributor to pediatric bilateral sensorineural hearing impairment. Am. J. Med. Genet. Part. A. 158, 298–308 (2012).
Back, D. et al. Phenotypic characterization of DFNB16-associated hearing loss. Otology Neurotology. 40, e48–e55 (2019).
Marková, S. P. et al. STRC gene mutations, mainly large deletions, are a very important cause of early-onset hereditary hearing loss in the Czech population. Genetic Test. Mol. Biomarkers. 22, 127–134 (2018).
Oza, A. M. et al. Expert specification of the ACMG/AMP variant interpretation guidelines for genetic hearing loss. Hum. Mutat. 39, 1593–1613. https://doi.org/10.1002/humu.23630 (2018).
McGrath, J., Roy, P. & Perrin, B. J. in Seminars in Cell & Developmental Biology, 88–95 (Elsevier).
Verpy, E. et al. Stereocilin connects outer hair cell stereocilia to one another and to the tectorial membrane. J. Comp. Neurol. 519, 194–210 (2011).
Verpy, E. et al. Stereocilin-deficient mice reveal the origin of cochlear waveform distortions. Nature 456, 255–258 (2008).
Ashmore, J. A fast motile response in guinea-pig outer hair cells: the cellular basis of the cochlear amplifier. J. Physiol. 388, 323–347 (1987).
Čada, Z. et al. Moderate sensorineural hearing loss is typical for DFNB16 caused by various types of mutations affecting the STRC gene. Eur. Arch. Otorhinolaryngol. 276, 3353–3358 (2019).
Markova, T. et al. Clinical features of hearing loss caused by STRC gene deletions/mutations in Russian population. Int. J. Pediatr. Otorhinolaryngol. 138, 110247 (2020).
Nishio, S. & Usami, S. -i. Frequency of the STRC-CATSPER2 deletion in STRC-associated hearing loss patients. Sci. Rep. 12, 634 (2022).
Domínguez-Ruiz, M. et al. Novel pathogenic variants in the gene encoding stereocilin (STRC) causing non-syndromic moderate hearing loss in Spanish and Argentinean subjects. Biomedicines 11, 2943 (2023).
Simi, A. et al. Audiologic phenotype and progression in pediatric STRC-related autosomal recessive hearing loss. Laryngoscope 131, E2897–E2903 (2021).
Marschark, M., Shaver, D. M., Nagle, K. M. & Newman, L. A. Predicting the academic achievement of deaf and hard-of-hearing students from individual, household, communication, and educational factors. Except. Child. 81, 350–369 (2015).
Schlieper, A., Kisilevsky, H., Mattingly, S. & Yorke, L. Mild conductive hearing loss and Language development: a one year follow-up study. J. Dev. Behav. Pediatr. 6, 65–68 (1985).
Jung, J. et al. MYH1 deficiency disrupts outer hair cell electromotility, resulting in hearing loss. Exp. Mol. Med. 1–13 (2024).
Han, J. H. et al. Comprehensive prediction model, including genetic testing, for the outcomes of cochlear implantation. Ear Hear. 44, 223–231 (2023).
Rim, J. H. et al. Differential genetic diagnoses of adult post-lingual hearing loss according to the audiogram pattern and novel candidate gene evaluation. Hum. Genet. 1–13 (2022).
Bae, S. H. et al. The audiological phenotype of patients with a variant in MYH9 and MYH14 genes. Sci. Rep. 15, 22324 (2025).
Joo, S. Y. et al. Biallelic variants of SEMA3F are associated with nonsyndromic hearing loss. Mol. Cells. 48, 100190 (2025).
Kim, J. A. et al. Systematic genetic assessment of hearing loss using whole-genome sequencing identifies pathogenic variants. Exp. Mol. Med. 1–13 (2025).
Nam, J. et al. Natural history of auditory function in patients with Alport syndrome: A case series study. J. Clin. Med. 13, 6639 (2024).
Jung, J. et al. Clinical characteristics and audiological profiles of patients with pathogenic variants of WFS1. J. Clin. Med. 13, 4851 (2024).
Shearer, A. E. et al. Comprehensive genetic testing for hereditary hearing loss using massively parallel sequencing. Proc. Natl. Acad. Sci. 107, 21104–21109 (2010).
Funding
This research was supported by National Research Foundation of Korea grants funded by the Korean government (MSIT) (RS-2024-00346485 and RS-2025-18362970 to J.J.); the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: RS-2024-00404555 to J.J., and RS-2024-00439403 to J.J.).
Author information
Authors and Affiliations
Contributions
J.J. conceived and designed the study; T.U.C. and S.Y.J. performed the experiments; S.H.K., J.Y.C., D.W., and H.Y.G. performed data analyses and contributed to data interpretation; and T.U.C., S.Y.J., and J.J. wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cheon, T.U., Joo, S.Y., Kim, S.H. et al. Auditory genotype-phenotype correlation of patients with variants in STRC. Sci Rep 15, 44763 (2025). https://doi.org/10.1038/s41598-025-28499-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-28499-0
