
Abstract
Duchenne muscular dystrophy (DMD) is characterized by progressive skeletal muscle degeneration and weakness. DMD is the most common muscular dystrophy; commonly diagnosed in childhood, it has a deadly outcome, typically for respiratory or cardiac failure. In spite of a longer disease course, obtained in recent years thanks to palliative pharmacological treatments, DMD remains one of the foremost public health challenges. The intricate interaction between dysfunctional skeletal muscle fibers and the resident different cell types, including inflammatory and fibroadipogenic progenitors, directly contributes to the progression of the disease, affecting its severity. In this study, we used the remarkable precision of X-ray phase-contrast tomography (XPCT) to conduct an unprecedented three-dimensional (3D) examination of skeletal muscle architecture, comparing healthy and dystrophic mdx mice. The morphological features observed in XPCT images were compared to conventional histological sections, corroborated by morphometric evaluation of bidimensional parameters. Through XPCT, we followed the spatial disposition of degenerating or distorted dystrophic myofibers along a length of approximately 1.3 mm. In addition, the exploitation of micro-XPCT, unveiled significant quantitative differences between healthy and mdx muscles in numerous 3D parameters, such as myofiber length, variability in myofiber caliber, myofiber volume, and the volume of interstitial tissues and cells. We propose XPCT as a novel imaging tool for the ex vivo characterization of the fine architecture of dystrophic muscles: this approach is particularly relevant to highlight the outcomes of a treatment in pre-clinical models and provides the structural bases underlying the functional features of the diseased skeletal muscle.
Similar content being viewed by others


Introduction
Duchenne muscular dystrophy (DMD) is a X-linked, neurodegenerative disorder characterized by progressive muscle wasting. It is still an uncurable disease and, in spite of the ameliorated prognosis characteristic of the most recent years, it leads to premature death of patients, around the age of 40. Whilst DMD is considered a rare disease, with an incidence of 1:3500 born males, the estimated worldwide prevalence is important, with 4.8 per 100,000 people1. Given its degenerative nature, this disease has a very heavy burden for the patients and their families. DMD is caused by mutations in the DMD gene2, which encodes for the dystrophin protein, an important linker between intracellular cytoskeleton and extracellular matrix. The absence of dystrophin alters the assembly of the dystrophin/glycoprotein complex, leading to contraction-induced sarcolemma damage and the consequent degeneration and death of myofibers. In the early stages of the disease, DMD progression is characterized by cycles of myofiber necrosis, inflammation and regeneration, followed by loss of myofibers and replacement with fibrotic and adipose tissues at later stages. Indeed, the exhaustion of the satellite cell pool, i.e. the muscle stem cell reservoir, leads to insufficient muscle regeneration in humans, from the childhood on and ultimately to death for a respiratory crisis or cardiac dysfunction. Despite the enormous research progresses over the years in defining the mechanisms underlying this devasting disease and developing therapeutic approaches with promising results in preclinical studies3, DMD is an uncurable disease, yet.
The most widely used murine model to study DMD is the mdx mouse. In the early 80’s, a spontaneous mutation (mdx) in a C57BL/10ScSn mouse colony produced higher levels of circulating enzymes indicative of muscle damage and induced histological features similar to human muscular dystrophy, such as variation in fiber size, degeneration and necrosis of some fibers, marked presence of inflammatory cells in place of lost fibers and fibers with central nuclei4. Later, it was demonstrated that the mdx mouse has a single base substitution within an exon of the dmd gene, which causes premature termination of the translation of the protein dystrophin5. The lack of dystrophin causes the loss of the dystrophin-associated glycoprotein complex in mdx mice6, as well as in DMD patients7, and consequent sarcolemma leakage. Since the discovery of this mutant colony, mdx mice have been used in many studies all over the world as a tool to understand the signaling pathways underlying DMD and to test in preclinical studies novel therapeutic approaches. To this regard, mdx mice were used in the first study of cell therapy approach8, in the first research on exon skipping with morpholino antisense oligonucleotide9, and even in the new approaches of gene therapy based on CRISPR technology10. Several promising therapies are currently under investigation for DMD, ranging from gene therapies to the treatment of secondary consequences of the lack of dystrophin10. Among the primary therapeutic approaches, antisense oligonucleotides have been employed to induce exon skipping11, while CRISPR-mediated gene editing represents a novel approach for repairing the DMD gene12, and adeno-associated viral vectors are currently used to deliver recombinant micro-dystrophin to replace the mutated dystrophin gene13. Concomitantly, several drug therapies focusing on the secondary effects derived from dystrophin deficiency have been proposed: from the corticosteroids14, the gold standard of care for the treatment of DMD, to the newly FDA-approved histone deacetylase inhibitor givinostat15,16. The current idea for the development of secondary therapies is that the contribution of altered resident or infiltrating cells, including fibroadipogenic progenitors (FAPs) and inflammatory cells, significantly affect the progression of DMD. Understanding the intricate crosstalk between the skeletal muscle fibers and the surrounding environment has opened new possibilities for therapeutic interventions.
In order to evaluate the outcomes of the numerous therapeutic approaches above, there is an unmet need to assess DMD pathology in mdx mice with new techniques to overcome the limitations of the histological analysis in providing a comprehensive morphological characterization of the muscle phenotype. This issue becomes more relevant as the therapeutical approaches get closer to clinical trials: mesoangioblast-based therapies had to be proven efficient in dog before being approved for humans on the basis that scaling up the therapy could represent a problem per se17.
X-ray Phase Contrast Tomography (XPCT) represents a breakthrough in the quality of biomedical imaging, providing detailed insights into morphological features that remain elusive to conventional 2D imaging techniques and other traditional 3D methodologies18,19,20. Indeed, XPCT provides an excellent and highly sensitive tool for the 3D study of ex vivo biological soft tissues without the need to slice, stain or subject biological samples into intensive tissue processing. A major advantage of XPCT is that it enables multi-scale imaging from single cells to whole organs, allowing for the continuous visualization of structural details across different spatial scales. Noticeably, XPCT is a phase-sensitive approach that significantly enhances the visualization of soft tissue structures that exhibit minimal absorption contrast in conventional X-ray imaging. Although section-based 2D imaging, such as immunohistochemistry and electron microscopy, provide high magnification, and high-resolution images, it requires destructive sample preparation, including cutting samples into thin slices21,22. Additionally, these 2D imaging methods can only analyze thin sample sections and may lead to data misinterpretation due to the absence of three-dimensional information, which is only available through serial sectioning. This is particularly relevant for skeletal muscle tissue, which is formed by multinucleated cells, named myofibers, spanning the whole muscle length, thus being gigantic cells. Another major feature of the skeletal muscle tissue is its highly hierarchical organization, characterized by a complex architecture that is essential for the functional output. As the muscles studied gets bigger and bigger it is of pivotal importance to provide techniques that encompass a big amount of muscle to support the results. In previous studies, we have demonstrated the ability of XPCT to image the three-dimensional distribution of disease-relevant anatomical features – e.g. inflammatory cells, vessels, tissue degeneration, calcifications - along with their interaction with the surrounding microenvironment, in anatomical regions of interest in humans and in animal models of neurodegenerative diseases18,19,23,24. This paper presents a groundbreaking application of micro-X-ray phase-contrast tomography (XPCT) to unveil and investigate the 3D skeletal muscle architecture and its alterations in muscular dystrophy, producing 1-micron thick virtual slices covering a depth of about 1.3 mm per scan, without tissue manipulation such as slicing or labeling. For the first time, high-resolution 3D analysis has provided unprecedented skeletal muscle details of the dystrophic muscle, allowing us to execute reliable qualitative and quantitative measurements on intramuscular alterations occurring in DMD murine skeletal muscle.
Results
XPCT identifies the main histological features of healthy and dystrophic skeletal muscle
To deeply investigate the morphological changes occurring in the skeletal muscle of dystrophic mdx mice, we analyzed one of the most affected hindlimb skeletal muscle, i.e. the Gastrocnemius (GA), of five healthy wild-type (CTR) and five mdx mice were analyzed by XPCT. Muscles were isolated at 9 weeks of age, since this corresponds to an acute phase of the disease, embedded in paraffin and analyzed at the ANATOMIX beamline of the synchrotron of Soleil, Saint Aubin (FR). The XPCT images of the GA muscles in transverse and longitudinal sections clearly showed prominent differences in the shape and structural organization of mdx myofibers, when compared to the healthy, wild-type ones (Fig. 1A). Contrary to other imaging techniques, the high resolution of the XPCT images allowed us to discriminate the boundaries of each myofiber, thus highlighting the pathological features present in DMD muscles. Indeed, CTR muscles present myofibers homogeneous in size and intimately in contact with each other in traverse sections. Consistently, the myofibers are well-organized in parallel bundles in longitudinal sections. On the contrary, in transverse sections mdx muscles exhibit hypertrophied fibers co-existing with foci of small new-born myofibers - i.e. with central myonuclei25 - at various stages of maturation; in addition, the presence of degenerating/swollen myofibers and infiltrating mononucleated cells in the interstitial space among the myofibers leads to a high variability of muscle fiber size and a profound heterogeneity of the muscle sections (Fig. 1A). Longitudinal sections of mdx muscles in XPCT highlighted size changes along the longitudinal axis of single fibers, which showed a heterogeneous and irregular caliber, thus corroborating the finding of a disorganized architecture of dystrophic muscle. We also observed the presence of infiltrating mononucleated cells, indicative of muscle inflammation (Fig. 1A).
To assess the observed alterations, a morphometric quantification of the myofiber cross-sectional size was performed in parallel, on H&E-stained cryosections and on a single tomographic slice. For each myofiber we measured two different geometrical parameters: the myofiber cross-sectional area and the minimum Feret’s diameter, as a parameter very insensitive to myofiber orientation26. The myofiber size distribution between CTR and mdx muscles presents a similar profile, for both these parameters, and independently of the section analyzed (i.e. classical histology or XPCT). Indeed, when compared to controls, the mdx muscles display an increase in both the smaller and bigger myofiber classes, mirroring the presence of newborn small myofibers and of bigger hypertrophic ones (Fig. 1B and C). Importantly, the variance coefficient of muscle fiber size calculated for both parameters, which reflects the variability of fiber size, is significantly higher in mdx muscles compared to control ones, formally demonstrating the higher heterogeneity of the muscle fiber size commonly associated with dystrophy (Fig. 1B and C).

X-ray phase-contrast tomography (XPCT) images recapitulate the skeletal muscle features of traditional histological analysis. (A) Representative images of transverse or longitudinal sections stained with hematoxylin and eosin and XPCT images of tomographic slices of GA muscles of wild-type (CTR) and dystrophic mice (mdx) at 9 weeks of age. White arrows indicate centrally nucleated myofibers; red arrows indicate degenerating myofibers. Scale bars: 50 μm for XPCT images and 100 μm for cryosections. Quantification of myofiber size by (B) cross-sectional area or (C) minimum Feret’s diameter, and relative variance coefficients, of H&E cryosections (n = 4 muscles per genotype) and XPCT slices (n = 5 muscles per genotype). Results are shown as mean ± sem. *p < 0.05; #p < 0.01; $p < 0.005 by Student’s t-test.
The data obtained from morphological and morphometric analyses performed on a single tomographic slice of skeletal muscle recapitulates the major features highlighted by a classical histological analysis and are consistent with the data reported in the literature27, thereby confirming the validity of this technique in discriminating against the histopathological features of dystrophic muscles.
XPCT allows detailed 3D analyses of single dystrophic myofibers
An enormous potential of XPCT lies in the possibility of following specific fibers in the three dimensions, exploiting tomographic slices in both transverse and longitudinal orientations. The possibility to observe a fiber along its longitudinal axis for many millimeters is particularly relevant for a tissue such as the skeletal muscle, which is characterized by very long cells, the myofibers, which attain several centimeters in length. In addition, this is crucial to highlight the prevalent pathophysiological features of muscle dystrophy, such as fiber splitting, i.e. the longitudinal halving of a fiber, or focal damage, i.e. localized muscle fiber swamps. Indeed, the use of two-dimensional imaging is often inadequate for detecting these features. XPCT allowed the identification of all degenerating fibers within the analyzed volumes, thus enabling their classification according to the specific type of degeneration exhibited. For instance, we report a case of a degenerating myofiber that disappears (Fig. 2A), as well as a Y-shaped myofiber – which may result from the fusion of two regenerating myofibers or, vice versa, from the splitting of a single myofiber (Fig. 2B).

XPCT allows to follow the fate of a degenerating or fusing fiber. (A) Transverse and longitudinal sequences of XPCT slices showing a degenerating fiber. The sequences visually track the same fiber within the volume. The degenerating fiber (in yellow) is indicated by the dashed squares in the transverse cross-sectional view; the location of the images of the transverse sequence are indicated with a white dashed line in the longitudinal sequence, where the degenerating fiber is highlighted in yellow. Scale bar: 25 μm. (B) XPCT images of a couple of fusing or splitting myofibers in transverse and longitudinal virtual sections of an mdx muscle. The white dashed squares visually help to follow the fibers in the transverse view images. The pale blue and salmon colors help to identify the two splitting/fusing fibers. Scale bar: 25 μm.
Importantly, we were able to fully reconstruct the 3D behavior of apparently splitting myofibers (Fig. 3A-D). This feature is a signature of muscle dystrophy, generally visualized by using serial sectioning and reconstruction techniques, and importantly increases with age and correlates with muscle functional deficit28. Coherently, we have never observed splitting myofibers in CTR muscles. Worth noting, the fact that muscles remain intact during the XPCT analysis makes it unlikely that the presence of splitting fibers is a preparation artifact. We found that about 50% of the analyzed mdx myofibers were splitted: in most cases, the two halves of the splitted fiber fused back together to restore the size of the original fiber, while in very few cases one of the halves degenerated (Fig. 3E).

3D renderings of different types of muscle fiber splitting in intact dystrophic muscles. Representative 3D renderings of myofibers splitting into two or more components. Scale bar = 100 μm. (A) One splitting myofiber in which the two halves are separated. (B) A splitting myofiber where one half is interrupted (light blue), while the opposing half maintains its continuity (blue). (C) A splitted myofiber (blue) whose two halves fuse back together about 400 μm away from the initial event. (D) One myofiber (blue) splitting into three distinct fibers (blue, red, yellow) that subsequently fuse back together. (E) Quantification of splitted myofibers (red), relative to the total analyzed myofibers (blue), in mdx muscles. In yellow: splitted myofibers where the two halves of the splitted fiber fused back together; in light blue: splitted myofiber where one halves is degenerating.
3D morphometric quantification of muscle architecture thanks to XPCT
We performed additional morphometric analyses of about 80 myofibers per sample, selected in three 1.3 mm thick volumes of interest. After semi-manual segmentation of the muscle fibers, several parameters useful to describe the morphological changes typical of the disease condition were quantified. Firstly, mdx myofibers resulted to be significantly shorter than healthy fibers (Fig. 4A). Secondly, mdx muscles present a higher irregularity of the myofiber caliber, along with the single myofiber, when compared to the healthy ones (Fig. 4B). Thirdly, mdx muscles display also a significantly higher average cross-section area (i.e. fiber volume normalized to fiber length) (Fig. 4C). When this parameter is displayed as a fiber distribution, we can appreciate that while healthy muscles present a peak of myofibers around 400–600 µm2, the profile of mdx myofiber distribution is altered, with a significantly higher percentage of hypertrophic fibers bigger than 800 µm2 (Fig. 4D).
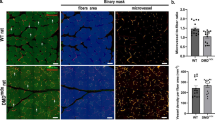
Importantly, similarly to classical histological sections stained with Masson’s trichrome (Fig. 4E), XPCT allowed us to visualize, and thus quantify, the endomysium, i.e. the tissue around single myofibers, consist of in interstitial connective tissue and infiltrating cells, as shown by the 3D rendering of a representative portion of CTR and mdx muscles (Fig. 4F). Since muscle inflammation, which occurs in the connective tissue, is a hallmark of muscle dystrophy, we intended to obtain an objective, reliable parameter to quantify the muscle interstitial tissue. Connective tissue and infiltrating cells appear as very bright objects within the tomographic volume. Thus, their quantification could be performed by segmentation through the setting of an intensity threshold. With this approach, we found that mdx muscles have a significantly higher volume of interstitial tissue than CTR muscles (Fig. 4G).

XPCT-derived quantitative parameters representative of dystrophic muscles. Quantification of (A) myofiber’s length and (B) variability along single myofiber caliber of CTR and mdx muscles. Data are expressed as mean ± sem; n = 5 mice per each genotype; *p < 0.05 by Student’s t test. (C) Quantification of myofiber volume normalized to myofiber length. Data are expressed as mean ± sem; n = 5 mice per each genotype; *p < 0.05 by Student’s t test. (D) Myofiber volume distribution indicating the wider profile of mdx myofiber’s volume, with the presence of hypertrophic myofibers, when compared to CTR one. n = 5 mice per genotype; *p < 0.05 by Student’s t test. (E) Representative picture of CTR and mdx GA muscle cryosections stained with Masson’s trichrome. Scale bar: 100 μm. (F) 3D rendering of myofiber interstitial tissues and cells in CTR and mdx muscles. (G) Quantification of interstitial tissue and cells surrounding myofibers. Data are expressed as mean ± sem; n = 5 mice per each genotype; *p < 0.05 by Student’s t test.
Discussion
There is an urgent demand for sensitive, objective, endpoints and parameters to assess the efficacy of novel therapies for the treatment of DMD and the development of novel techniques to better characterize dystrophic muscles is still a major goal of the worldwide medical research29,30,31,32. In terms of non-invasive imaging techniques used to study skeletal muscle, magnetic resonance imaging (MRI) provides measures in vivo/ex vivo to assess local or generalized disease severity33,34,35. Decades ago, contrast agent-enhanced magnetic resonance has been used to study muscle fiber damage in different animal models of muscle dystrophy36, having the merit of pointing out the importance of non-invasive, tridimensional approaches to study the progression of the pathology and the impact of potential therapeutic interventions. However, MRI provides low resolution images of skeletal muscles from which it is possible to extrapolate only the cross section or the total volume of the entire muscle/leg, differently from XPCT, which allows to visualize and analyze single myofibers. Although MRI has relevant potential applicability in therapeutic screening studies of muscle diseases, it is clearly less suitable for preclinical studies in animal models, where we need to assess fine structural changes, as the resolution is two-orders of magnitude lower than XPCT. Similarly, computed tomography, a well-established diagnostic imaging technique, can serve as a valid tool for the quantitative assessment of skeletal muscle mass in patients37,38, but it does not reach the microscale resolution precluding the detection of pathological alterations at the fiber level. Ultrasonography may be another useful, non-invasive alternative to assess skeletal muscle changes in vivo39, but it suffers the same low resolution as MRI compared to that achievable with XPCT. High-resolution X-ray microtomography, instead, is suitable for a quantitative ex vivo analysis with single-cell resolution of biological samples with different densities40,41. However, visualizing soft tissue remains challenging due to the low intrinsic attenuation contrast between the examined structures. Consequently, staining with heavy elements such as iodine, tungsten, or osmium-based compounds is necessary to enhance contrast42. These contrast staining procedures are often complex, time-consuming and irreversible, thus excluding the investigation of the sample with further experimental techniques. Most importantly, for a meaningful healthy-diseased comparison, it is crucial that the sample remains in the most physiological state possible during the analysis. Imaging analysis of ex vivo skeletal muscles has been performed by optical coherence tomography43,44. This is a non-invasive and in vivo technique - different from XPCT - that enables 3D imaging of biological tissue with a resolution of 1–20 μm at depths up to 1 ~ 2 mm. However, it still lacks the capability to detect individual fibers. Dystrophic myofibers have been studied also by second harmonic generation (SHG) microscopy or multispectral optoacoustic tomography (MSOT), specifically for visualizing collagen deposition45,46 and sarcomeric ultrastructure28. While these approaches are important to implement imaging databases to diagnose and monitor specific morphological alterations in DMD, being non-invasive and label-free, they provide complementary information to XPCT, which allows the morphological identification of the main histological features of muscle samples. To show the potential of XPCT for the investigation of dystrophic muscle, we analyzed the skeletal muscle of mdx mice, since this mouse model has been greatly characterized by histological, biochemical and molecular analyses27,47,48. Our analysis not only revealed the unprecedented level of resolution that is applicable to the study of dystrophy but also demonstrated the feasibility of applying XPCT to the study of soft tissues such as connective tissue and inflammatory cells.
In particular, here we show that XPCT images allow the morphological identification of DMD features of 9-week-old mdx mice, including numerous muscle fibers with central nuclei, indicative of regenerating muscle cells, dispersed hypertrophic fibers, some swollen and degenerating fibers, in line with previous observation of mdx muscles49, and similarly to the human disease50. Qualitative and quantitative analyses performed on XPCT images are comparable to those obtained by the traditional histological analysis performed on muscle sections stained by hematoxylin and eosin. Indeed, morphometric quantification of myofiber size, and the relative variance coefficient, validated the qualitative description of great variation in muscle fiber shapes and diameters, as also described in4,26,27,48,49. Such irregularity of fiber size in dystrophic muscles likely depends in part on the small diameter of newborn myofibers and on the splitted or malformed regenerating ones48,51.
In addition to all the above, XPCT appeared to be essential to highlight the phenomenon of splitted or branched myofibers, present only in mdx muscles, a well-established hallmark of muscle dystrophy52. Branched myofibers appear during muscle regeneration and are more fragile and functionally compromised than the normal ones53,54,55. The presence of splitted fibers, as well as of hypertrophic and atrophied ones, positively correlates with the age of mdx muscles and with the progression of DMD pathology48. Thus, the evaluation and quantification of these parameters is crucial to determining any potential benefits of a therapeutic approach in preclinical studies. The first cinematographic reconstruction from serial sections described a pattern of complex splitting and recombination of mdx fibers, in a confined and small area of dystrophic muscles56. A more accurate reconstruction of 5 to 6 mm long muscle biopsies of two DMD patients, through 1000 to 1200 histological sections, first described splitted dystrophic fibers in human patients57. Alternatively, splitting myofibers have been studied on isolated myofibers in vitro28,58. As a better alternative to following a specific myofiber along the z axis in 2D images, XPCT allows the 3D rendering of altered myofibers, especially the complex and irregular architecture of branching mdx myofibers. Within the 1.3 mm-thick tomographic volumes, thanks to XPCT we could distinguished between subtle alterations, consisting of myofibers that branched once and then remained with two separated halves, or splitted myofibers that lost one of the two halves, or myofibers that branched several times or that formed 3 branches from one single myofiber. Among the speculative causes of branched myofiber, several alternatives have been proposed: defects in fusion of two myoblasts53,59, or of two myofibers60, failure of myoblast migration61, deficiencies in the residual basement membrane which is no longer able to properly confined and control muscle stem cell migration62, presence of cellular debris that physically impedes the formation of linear myofibers62. From the morphological evaluation of splitting myofibers with the surrounding interstitial tissue by XPCT and considering that such phenomenon is often recapitulated in vitro during the differentiation of dystrophic muscle cells into myofibers63, we believe that malformed myofibers depend on intrinsic defects of dystrophic muscle cells, due to aberrant cell fusion or dysfunctional sarcolemma, in line with recent findings64.
Besides the qualitative description of myofiber abnormalities, XPCT analysis allows the morphometric quantification of several volumetric parameters that discriminate between healthy and dystrophic muscles, i.e. myofiber length, the variability in myofiber caliber, myofiber volume/length, and the volume of interstitial tissue. Of note, the myofiber average cross-sectional area was obtained by normalizing the myofiber volume to the fiber length in Fig. 4; in this way, we obtained a myofiber area distribution which nicely correlates with that one measured on XPCT tomographic slices in Fig. 1. The absence of the smallest classes of regenerating fibers in mdx muscles depends on the fact that for the volumetric measurements we needed to select fibers long at least 1.3 mm, not malformed or degenerating.
While XPCT has been used to study the 3D morphological alterations of several tissues in different diseases, including the spinal cords of a mouse model of Amyotrophic Lateral Sclerosis65, brain66 or gut19 of murine models of Alzheimer’s disease, lungs67 of a mouse model of allergic asthma, few studies reported XPCT to study skeletal muscle tissue. XPCT and micro-computed tomography (micro-CT) have been engaged for a merely qualitative description of skeletal muscle morphology of zebrafish68, and for a quantitative description of patients with cerebral palsy69, but very few morphological parameters were measured. In a recent study of Pingel et al.70, micro-CT has been used to visualize, describe and quantify some 3D myofiber features - i.e. fiber splitting, buckling, diameter, shape, and straightness - of three muscle biopsies: one from a healthy control man and two from individuals affected by different degrees of muscle atrophy. Despite the identification of quantifiable morphological features describing changes in 3D muscle architecture and organization, this study lacks statistics, and any conclusions on the use of such parameters as descriptive or representative of muscle atrophy should be corroborated by additional analyses on other muscle biopsies, categorized by disease, age and sex. In the present study, a step forward on establishing XPCT as a valid tool for medical research has been taken, by providing statistics and conclusions on numerous 3D parameters able to discriminate between healthy and dystrophic muscles. XPCT represents a breakthrough approach for studies on the mechanisms underlying the pathology or to assess the effects of innovative therapies in preclinical models as well as in pilot clinical studies.
Although XPCT offers many advantages, several factors currently limit its routine clinical use compared to MRI and CT. High-resolution XPCT often requires specialized X-ray sources, detectors, and advanced reconstruction software. While radiation doses can be optimized, repeated scans may still increase patient exposure, whereas CT protocols are well-standardized to minimize risk. XPCT scans are computationally demanding, and data analysis requires specialized expertise. Additionally, high-resolution imaging typically covers only small tissue volumes, and XPCT remains largely a research tool with limited clinical approval. However, recent developments in laboratory-based XPCT systems and AI-driven data processing are rapidly addressing these challenges. Automated workflows are reducing analysis times, and machine learning is simplifying image reconstruction and interpretation, lowering expertise requirements. The growing availability of compact, user-friendly XPCT systems and collaborative imaging networks is expanding accessibility beyond synchrotron facilities. Together, these advances are paving the way for broader clinical adoption, potentially transforming XPCT from a specialized research tool into a practical diagnostic method for biopsy-based disease assessment and therapeutic monitoring.
In conclusion, the 3D, highly-resolved imaging provided by XPCT represents an optimal tool for the ex vivo characterization of the fine architecture of dystrophic muscles. Therefore, XPCT can considerably contribute to the advancement of preclinical studies on DMD.
Methods
Mice
To study the differences between healthy and dystrophic muscles, C57BL/10ScSn and C57BL/10ScSn-Dmdmdx/J (named mdx) from the Jackson Laboratory were used. In all the experiments, 9-week-old male mice were compared. Five animals per experimental group were used. Animals were housed in groups of 4–5 mice/cage in standard conditions with free access to food and water, at constant temperature (21°-23° C) and relative humidity (50%), with regular 12 h light cycle (light 7AM-7PM). Euthanasia by cervical dislocation was used right before tissue collection. Mice were treated in strict accordance with the guidelines of the Institutional Animal Care and Use Committee, as well as national and European legislation, throughout the experiments. Animal protocols were approved by the Italian Ministry of Health (authorization # 538/2021-PR with the extension 82945.65.EXT.34 and # 485/2024-PR). We confirm the study is reported in accordance with ARRIVE guidelines.
Histological analyses
GA muscles were dissected, embedded in tissue freezing medium (Leica), and frozen in isopentane pre-cooled with liquid nitrogen. Cross- and longitudinal-sections with a thickness of 9 μm in the mid-belly of GA muscles were obtained by using a Leica cryostat. Hematoxylin and eosin staining was performed according to the manufacturer’s instructions (Sigma-Aldrich).
Sample preparation for XPCT
GA muscles were isolated, chemically fixed in formalin over-night, dehydrated with increasing concentrations of ethanol and embedded in paraffin.
Acquisition of XPCT data
Samples were analyzed at the ANATOMIX beamline of the synchrotron of Soleil, Saint Aubin (FR). The XPCT experiment was performed in free-space propagation mode. Samples were mounted and scanned with a filtered white beam with an energy spectrum peaking around 22 keV. The detector was an indirect system consisting of a lutetium aluminum garnet scintillator coupled via a lens system (magnification 10×) to a Hamamatsu Orca Flash 4.0 camera [sensor type, complementary metal-oxide semiconductor (CMOS); sensor array size, 2048 × 2048 pixels; physical pixel size, 6.5 μm; and 16-bit nominal dynamic range]. The measurements were acquired with an effective pixel size of 0.65 μm, at a propagation distance between sample and detector of 27 mm, an exposure time of 30 ms per projection radiograph. The tomography was produced by means of 5000 projections over 360° of rotation, in extended field-of-view (FOV) mode, an acquisition method which allows to almost double the effective horizontal width of the FOV of the detector. The rotation axis is moved close to either left or right side of the FOV and a dataset of projections, having size equal to the detector FOV, is collected over 360°. After properly stitching the sinograms, the reconstruction procedure can be performed as usual. The final FOV is around 2.5 mm × 1.3 mm, resulting in tomographic slices of area of about 2.5 mm × 2.5 mm.
XPCT data processing
Data pre-processing, phase retrieval with Paganin’s method71 and tomographic reconstruction were performed using the standard data processing pipeline at the beamline, based on a Python script as a user front-end and for pre-processing and the PyHST2 software package72 as the backend for the tomographic reconstruction. During phase retrieval step, a δ/β ratio of 120 was applied to optimize image contrast and enhance visualization for subsequent segmentation.
Morphometric analyses
The analysis of XPCT data was performed on three representative volumes of 2.5 × 2.5 × 1.3 mm per muscle. For each sample, about 80 myofibers were considered.
Muscle fibers were segmented using a semi-automatic approach. Specifically, for each fiber, the cross-section was manually outlined every 50 slices (corresponding to ~ 30 μm), after which interpolation was applied to segment the entire fiber within the volume. This approach provided accurate fiber tracing while reducing the amount of manual work required. Fiber size was then quantified through the measure of the myofiber cross-sectional area and the minimum Feret’s diameter, which is defined as the minimum distance between parallel tangents at opposing borders of the muscle fiber. The variance coefficient of the myofiber size was calculated as (standard deviation of the muscle fiber size/mean muscle fiber size) ×100026.
Fiber length was calculated as the summed Euclidean distance between the centroids of the cross-sections taken at equidistant positions (sampled at 30 μm) along the fiber. To quantify the variability of the myofiber caliber of each fiber, minimum Feret’s diameter standard deviation was used. The distribution of the average cross-sectional area was obtained from the volumes of the individual fibers normalized by their length.
To quantify the interstitial tissue and infiltrating cells, we used the image processing package Fiji. Specifically, three regions of interest of about 500 × 500 × 500 µm3 were chosen in each volume/ sample. A segmentation based on intensity thresholding was performed by using Fiji’s iterative implementation of Kittler and Illingworth’s Minimum Error thresholding73 to quantify the volume occupied by infiltrating cells and interstitial tissue. Then the measured volume was normalized to the entire volume of the region of interest and then expressed as a percentage.
Statistics
Statistical significance between the two groups of n = 5 CTR or mdx mice was determined by using two-tailed Student’s t-test. All values were expressed as mean ± standard error of the mean (SEM). All the graphs and statistical analyses were performed by using GraphPad Prism 6 software.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Salari, N. et al. Global prevalence of Duchenne and Becker muscular dystrophy: a systematic review and meta-analysis. J. Orthop. Surg. Res 17, (2022).
Gatto, F., Benemei, S., Piluso, G. & Bello, L. The complex landscape of DMD mutations: moving towards personalized medicine. Front. Genet. 15, (2024).
Johnson, L. M., Pulskamp, T. G. & Berlau, D. J. The latest developments in synthetic approaches to Duchenne muscular dystrophy. Expert Rev. Neurother. https://doi.org/10.1080/14737175.2025.2462281 (2025).
Bulfield, G., Siller, W. G., Wight, P. A. L. & Moore, K. J. X chromosome-linked muscular dystrophy (mdx) in the mouse. Proc. Natl. Acad. Sci. U S A. 81, 1189–1192 (1984).
Sicinski, P. et al. The molecular basis of muscular dystrophy in the Mdx mouse: a point mutation. Science 244, 1578–1580 (1989).
Ohlendieck, K. & Campbell, K. P. Dystrophin-associated proteins are greatly reduced in skeletal muscle from Mdx mice. J. Cell. Biol. 115, 1685–1694 (1991).
Ervasti, J. M., Ohlendieck, K., Kahl, S. D., Gaver, M. G. & Campbell K. P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature 345, 315–319 (1990).
Partridge, T. A., Morgan, J. E., Coulton, G. R., Hoffman, E. P. & Kunkel, L. M. Conversion of Mdx myofibres from dystrophin-negative to -positive by injection of normal myoblasts. Nature 337, 176–179 (1989).
Fletcher, S. et al. Dystrophin expression in the Mdx mouse after localised and systemic administration of a morpholino antisense oligonucleotide. J. Gene Med. 8, 207–216 (2006).
Krishna, L. et al. Molecular and biochemical therapeutic strategies for Duchenne muscular dystrophy. Neurol. Int. 2024. 16, Pages 731–760 (16), 731–760 (2024).
Koo, T. & Wood, M. J. Clinical trials using antisense oligonucleotides in Duchenne muscular dystrophy. Hum. Gene Ther. 24, 479–488 (2013).
Chemello, F., Olson, E. N. & Bassel-Duby, R. CRISPR-Editing therapy for Duchenne muscular dystrophy. Hum. Gene Ther. 34, 379–387 (2023).
Arechavala-Gomeza, V., Anthony, K., Morgan, J. & Muntoni, F. Antisense oligonucleotide-mediated exon skipping for Duchenne muscular dystrophy: progress and challenges. Curr. Gene Ther. 12, 152–160 (2012).
Falzarano, M. S., Scotton, C., Passarelli, C. & Ferlini, A. Duchenne muscular dystrophy: from diagnosis to therapy. Molecules 20, 18168–18184 (2015).
Mercuri, E. et al. Safety and efficacy of givinostat in boys with Duchenne muscular dystrophy (EPIDYS): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Neurol. 23, 393–403 (2024).
Sandonà, M. et al. Histone deacetylases: molecular mechanisms and therapeutic implications for muscular dystrophies. Int. J. Mol. Sci. 2023. 24, 4306 (2023).
Sampaolesi, M. et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 444, 574–579 (2006).
Palermo, F. et al. Multilevel X-ray imaging approach to assess the sequential evolution of multi-organ damage in multiple sclerosis. Commun. Phys. 5(1), 1–12 (2022).
Palermo, F. et al. Investigating gut alterations in alzheimer’s disease: In-depth analysis with micro- and nano-3D X-ray phase contrast tomography. Sci. Adv. 11, (2025).
Cedola, A. et al. X-ray phase contrast tomography reveals early vascular alterations and neuronal loss in a multiple sclerosis model. Sci. Rep. 7(1), 1–11 (2017).
Daou, N. et al. Displaced myonuclei in cancer cachexia suggest altered innervation. Int. J. Mol. Sci. 21, (2020).
Carotenuto, F. et al. How diet intervention via modulation of DNA damage response through MicroRNAs May have an effect on cancer prevention and aging, an in silico study. Int. J. Mol. Sci. 17, (2016).
Palermo, F. et al. X-ray phase contrast tomography serves preclinical investigation of neurodegenerative diseases. Front. Neurosci. 14, 584161 (2020).
Bukreeva, I. et al. Micromorphology of pineal gland calcification in age-related neurodegenerative diseases. Med. Phys. 50, 1601–1613 (2023).
Mazzotti, A. L. & Coletti, D. The need for a consensus on the locution ‘central nuclei’ in striated muscle myopathies. Front. Physiol. 7, 577 (2016).
Briguet, A., Courdier-Fruh, I., Foster, M., Meier, T. & Magyar, J. P. Histological parameters for the quantitative assessment of muscular dystrophy in the mdx-mouse. Neuromuscul. Disord. 14, 675–682 (2004).
Torres, L. F. B. & Duchen, L. W. The mutant MDX: inherited myopathy in the mouse: morphological studies of nerves, muscles and end-plates. Brain 110, 269–299 (1987).
Buttgereit, A., Weber, C., Garbe, C. S. & Friedrich, O. From chaos to split-ups – SHG microscopy reveals a specific remodelling mechanism in ageing dystrophic muscle. J. Pathol. 229, 477–485 (2013).
Vicino, A. & Veltsista, D. & Van Alfen, N. Muscle ultrasound in myopathies. Curr. Opin. Neurol. 37, 549–557 (2024).
Svetlove, A. et al. Non-invasive optical motion tracking allows monitoring of respiratory dynamics in dystrophin-deficient mice. Cells 11, (2022).
Hiyoshi, T. et al. Electrical impedance myography detects dystrophin-related muscle changes in Mdx mice. Skelet. Muscle 13, (2023).
Waters, E. A., Haney, C. R., Vaught, L. A., McNally, E. M. & Demonbreun, A. R. Distribution of MRI-derived T2 values as a biomarker for in vivo rapid screening of phenotype severity in Mdx mice. PLoS ONE. 19, e0310551 (2024).
Zhang, J., Zhang, G., Morrison, B., Mori, S. & Sheikh, K. A. Magnetic resonance imaging of mouse skeletal muscle to measure denervation atrophy. Exp. Neurol. 212, 448 (2008).
Hooijmans, M. T. et al. Compositional and functional MRI of skeletal muscle: A review. J. Magn. Reson. Imaging. 60, 860–877 (2024).
Alic, L., Griffin, J. F., Eresen, A., Kornegay, J. N. & Ji, J. X. Using MRI to quantify skeletal muscle pathology in Duchenne muscular dystrophy: A systematic mapping review. Muscle Nerve. 64, 8–22 (2021).
Straub, V. et al. Contrast agent-enhanced magnetic resonance imaging of skeletal muscle damage in animal models of muscular dystrophy. Magn. Reson. Med. 44, 655–659 (2000).
Nemec, U., Heidinger, B., Sokas, C., Chu, L. & Eisenberg, R. L. Diagnosing sarcopenia on thoracic computed tomography: quantitative assessment of skeletal muscle mass in patients undergoing transcatheter aortic valve replacement. Acad. Radiol. 24, 1154–1161 (2017).
Grindrod, S., Tofts, P. & Edwards, R. Investigation of human skeletal muscle structure and composition by X-ray computerised tomography. Eur. J. Clin. Invest. 13, 465–468 (1983).
Mele, A. et al. Preclinical ultrasonography in rodent models of neuromuscular disorders: the state of the art for diagnostic and therapeutic applications. Int. J. Mol. Sci. 2023. 24, 4976 (2023).
Tesařová, M. et al. A quantitative analysis of 3D-cell distribution in regenerative muscle-skeletal system with synchrotron X-ray computed microtomography. Sci. Rep. 8(1), 1–12 (2018).
de Fonseca, M. et al. C. High-resolution synchrotron-based X-ray microtomography as a tool to unveil the three-dimensional neuronal architecture of the brain. Sci. Rep. 8(1), 1–13 (2018).
Metscher, B. D. MicroCT for developmental biology: A versatile tool for high-contrast 3D imaging at histological resolutions. Dev. Dyn. 238, 632–640 (2009).
Maillet, M., Kammoun, M., Avril, S., Ho Ba Tho, M. C. & Trabelsi, O. Non-destructive characterization of skeletal muscle extracellular matrix morphology by combining optical coherence tomography (OCT) imaging with tissue clearing. Ann. Biomed. Eng. 51, 2323–2336 (2023).
Klyen, B. R. et al. Optical coherence tomography can assess skeletal muscle tissue from mouse models of muscular dystrophy by parametric imaging of the attenuation coefficient. Biomedical Opt. Express. 5(4), 1217–1232 (2014)
Regensburger, A. P. et al. Detection of collagens by multispectral optoacoustic tomography as an imaging biomarker for Duchenne muscular dystrophy. Nat. Med. 25, 1905–1915 (2019).
Brashear, S. E., Wohlgemuth, R. P., Gonzalez, G. & Smith, L. R. Passive stiffness of fibrotic skeletal muscle in Mdx mice relates to collagen architecture. J. Physiol. 599, 943–962 (2021).
Ikeda, T. et al. Degenerative and regenerative features of myofibers differ among skeletal muscles in a murine model of muscular dystrophy. J. Muscle Res. Cell. Motil. 37, 153–164 (2016).
Pastoret, C. & Sebille, A. Mdx mice show progressive weakness and muscle deterioration with age. J. Neurol. Sci. 129, 97–105 (1995).
Tanabe, Y., Esaki, K. & Nomura, T. Skeletal muscle pathology in X chromosome-linked muscular dystrophy (mdx) mouse. Acta Neuropathol. 69, 91–95 (1986).
Bell, C. D. & Conen, P. E. Change in fiber size in Duchenne muscular dystrophy. Neurology 17, 902–913 (1967).
Snow, M. H. & Chortkoff, B. S. Frequency of bifurcated muscle fibers in hypertrophic rat soleus muscle. Muscle Nerve. 10, 312–317 (1987).
Cooper, S. T. & Head, S. I. Membrane injury and repair in the muscular dystrophies. Neuroscientist. 21, 653–668 (2015).
Chan, S. & Head, S. I. The role of branched fibres in the pathogenesis of Duchenne muscular dystrophy. Exp. Physiol. 96, 564–571 (2011).
Pichavant, C. & Pavlath, G. K. Incidence and severity of myofiber branching with regeneration and aging. Skelet. Muscle. 4, 1–11 (2014).
Hernández-Ochoa, E. O., Pratt, S. J. P., Garcia-Pelagio, K. P., Schneider, M. F. & Lovering, R. M. Disruption of action potential and calcium signaling properties in malformed myofibers from dystrophin-deficient mice. Physiol. Rep. 3, e12366 (2015).
Swash, M. & Schwartz, M. S. Implications of longitudinal muscle fibre splitting in neurogenic and myopathic disorders. J. Neurol. Neurosurg. Psychiatry. 40, 1152 (1977).
Schmalbruch, H. Regenerated muscle fibers in Duchenne muscular dystrophy: A serial section study. Neurology 34, 60–65 (1984).
Massopust, R. T. et al. Lifetime analysis of Mdx skeletal muscle reveals a progressive pathology that leads to myofiber loss. Sci. Rep. 10, 1–16 (2020).
Ontell, M., Hughes, D. & Bourke, D. Secondary myogenesis of normal muscle produces abnormal myotubes. Anat. Rec. 204, 199–207 (1982).
Charrin, S. et al. Normal muscle regeneration requires tight control of muscle cell fusion by tetraspanins CD9 and CD81. Nat. Commun. 4, 1–12 (2013).
Griffin, C. A., Kafadar, K. A. & Pavlath, G. K. MOR23 promotes muscle regeneration and regulates cell adhesion and migration. Dev. Cell. 17, 649–661 (2009).
Collins, B. C. et al. Three-dimensional imaging studies in mice identify cellular dynamics of skeletal muscle regeneration. Dev. Cell. 59, 1457–1474.e5 (2024).
Arakawa, M. et al. Negamycin restores dystrophin expression in skeletal and cardiac muscles of Mdx mice. J. Biochem. 134, 751–758 (2003).
Esper, M. E. et al. Intrinsic muscle stem cell dysfunction contributes to impaired regeneration in the Mdx mouse. J. Cachexia Sarcopenia Muscle. 16, e13682 (2025).
Provinciali, G. B. et al. X-ray phase contrast tomography for the investigation of amyotrophic lateral sclerosis. urn:issn:1600–5775 27, 1042–1048 (2020).
Chourrout, M. et al. Brain virtual histology with X-ray phase-contrast tomography part II:3D morphologies of amyloid- β plaques in Alzheimer’s disease models. Biomed. Opt. Express. 13, 1640 (2022).
Dullin, C. et al. Functionalized synchrotron in-line phase-contrast computed tomography: a novel approach for simultaneous quantification of structural alterations and localization of barium-labelled alveolar macrophages within mouse lung samples. J. Synchrotron Radiat. 22, 143–155 (2015).
Vågberg, W., Larsson, D. H., Li, M., Arner, A. & Hertz, H. M. X-ray phase-contrast tomography for high-spatial-resolution zebrafish muscle imaging. Sci. Rep. 5(1), 1–7 (2015).
Borg, L. et al. Muscle fibre morphology and microarchitecture in cerebral palsy patients obtained by 3D synchrotron X-ray computed tomography. Comput. Biol. Med. 107, 265–269 (2019).
Pingel, J., Kjer, H. M., Biering-Sørensen, F., Feidenhans’l, R. & Dyrby, T. B. 3D synchrotron imaging of muscle tissues at different atrophic stages in stroke and spinal cord injury: a proof-of-concept study. Sci. Rep. 12, 1–13 (2022).
Paganin, D., Mayo, S. C., Gureyev, T. E., Miller, P. R. & Wilkins, S. W. Simultaneous phase and amplitude extraction from a single defocused image of a homogeneous object. J. Microsc. 206, 33–40 (2002).
Mirone, A., Brun, E., Gouillart, E., Tafforeau, P. & Kieffer, J. The PyHST2 hybrid distributed code for high speed tomographic reconstruction with iterative reconstruction and a priori knowledge capabilities. Nucl. Instrum. Methods Phys. Res. B. 324, 41–48 (2014).
Kittler, J. & Illingworth, J. Minimum error thresholding. Pattern Recogn. 19, 41–47 (1986).
Acknowledgements
We acknowledge Synchrotron SOLEIL (Saint-Aubin, France) for allocation of beamtime within the scope of proposal 20231706 and we thank Jonathan Perrin and Timm Weitkamp for assistance in using the beamline ANATOMIX. ANATOMIX is an equipment of excellence (EQUIPEX) funded by the investments for the Future program of the French national Research Agency (AnR), project nanoimagesX, grant no. AnR-11-eQPX-0031.
Funding
This work was supported by Fondazione Telethon (GMR23T2003).
Author information
Authors and Affiliations
Contributions
Conceptualization: Viviana Moresi, Alessia CedolaMethodology: Viviana Moresi, Francesca PalermoInvestigation: Francesca Palermo, Viviana Moresi, Arianna Angelini, Micaela Pennetta, Alessandra Renzini, Lorenzo MassimiVisualization: Francesca Palermo, Giorgia CavioliSupervision: Viviana Moresi, Francesca Palermo, Alessia Cedola, Dario ColettiWriting—original draft: Viviana Moresi, Francesca PalermoWriting—review & editing: Dario Coletti, Alessia Cedola, Giuseppe Gigli.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Palermo, F., Moresi, V., Cavioli, G. et al. Quantitative 3D assessment of muscle dystrophy through X-ray phase-contrast tomography. Sci Rep 15, 44996 (2025). https://doi.org/10.1038/s41598-025-28961-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-28961-z
