
Abstract
Proximal tubular epithelial cells (PTECs) injury is a driving force toward chronic kidney disease (CKD) progression. However, the characteristics of injured PTECs during CKD pathogenesis and their mechanisms of coordination with specific interstitial cells to promote disease progression require further elucidation. In this study, CKD mouse models were established by administering a 0.2% adenine-containing diet for 2, 4, and 8 weeks to simulate progressive CKD. Renal injury was assessed through histopathology, serum creatinine (Scr), blood urea nitrogen (BUN), and expression of fibrotic markers. Single-cell RNA sequencing (scRNA-seq) was employed to characterize renal cellular heterogeneity during CKD progression. The results showed that adenine feeding induced renal pathological damage, elevated Scr and BUN levels, and upregulated fibrosis-related markers, with the most severe manifestations observed in CKD8w group. In addition, scRNA-seq identified a distinct injured PTEC subset (PT_5) localized to the S1 segment, which progressively expanded during CKD, exhibited elevated injury markers (C3, Havcr1, Vcam1), and activated pathways linked to inflammation and fibrosis. Cell-cell interaction analysis revealed PT_5 coordinates interstitial remodeling via ligand-receptor pairs: CCL2-CCR2/IL34-CSF1R with macrophages, PDGF-PDGFR with fibroblasts, and CXCL10-CXCR3 with T and NK cells. Furthermore, CKD progression may correlate with expansion of Myofibroblasts_Timp1, Macrophages_Mmp12, and CD8Teff_Ccl5 subsets. Our study uncovers PT_5 may be the central orchestrator of CKD progression through multicellular crosstalk, offering novel mechanistic insights and therapeutic targets.
Similar content being viewed by others

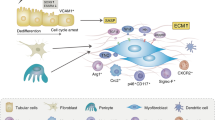

Introduction
It is estimated that approximately 850 million people worldwide are affected by chronic kidney disease (CKD), with the incidence and prevalence of the condition increasing year on year9,11,26. Accompanied by loss of kidney function, CKD can progress to end-stage renal disease requiring dialysis or renal transplantation. Irrespective of the initial cause of the disease, renal tubulointerstitial fibrosis (TIF) is considered to be the ultimate common pathway for the evolution of almost all types of CKD17,30, and increased TIF is the most reliable histological indicator of decreased renal function and poor prognosis14,40. Despite decades of research, the understanding of TIF remains limited. Consequently, a better understanding of the regulatory mechanisms of kidney fibrosis is critical to developing new treatments to better control this chronic disease.
The pathophysiology of TIF is complex and involves multiple cell types, including fibroblasts, tubular epithelial cells, endothelial cells, and infiltrating inflammatory cells36. It is understandable that much attention has been focused on myofibroblasts and their precursors, given that this interstitial cell is thought to be the main producer of extracellular matrix (ECM). However, recent data has reinforced the role of the proximal tubule10, not only as the target of injury but also as an important mediator of TIF progression12,33. In response to various injuries, proximal tubular epithelial cells (PTECs) undergo different changes such as partial epithelial mesenchymal transition (EMT), cell cycle arrest, cellular senescence or metabolic reprogramming15. These alterations cause PTECs to exhibit a secretory phenotype that triggers fibroblast activation49. However, the characteristics of injured PTECs during CKD progression and their interaction with interstitial cells are still not fully understood.
In recent years, there has been a notable advancement in the understanding of CKD pathogenesis at the cellular and molecular levels, which could not have been achieved without the development of single-cell sequencing technology. Single-cell RNA sequencing (scRNA-seq) is a transcriptomic technology that simultaneously measures the expression of multiple genes in tens or even hundreds of thousands of single cells, deepening the understanding of the genes and pathways that characterize kidney cells37,43. This innovative technique allows comparison of transcriptional heterogeneity between healthy and pathological conditions, thus revealing new insights into tissue composition and disease progression28. Here, we established 2-, 4-, and 8-week CKD mice models to simulate different stages of CKD. scRNA-seq was performed to map the transcriptome of renal cells with the aim of identifying injured PTECs and revealing their interactions with renal interstitial cells in CKD progression.
Materials and methods
Animals
The male C57BL/6 mice (6 ~ 8 weeks, 20 ~ 25 g) used in this study were obtained from Guangdong Medical Laboratory Animal Center (Foshan, China). The mice were raised in a standard environment with a 12-h light/dark cycle and 50 ± 5% humidity at 25 ± 2 °C. After 1 week of adapted feeding, a total of 24 mice were randomly divided into 4 groups: the control group (n = 6); the CKD2w group (n = 6); the CKD4w group (n = 6); and the CKD8w group (n = 6). CKD was induced by feeding a diet containing 0.2% adenine for 2, 4, and 8 weeks, respectively. All mice had free access to food and water. At the experimental endpoint, mice were anesthetized with isoflurane gas for terminal blood collection, followed by euthanasia via cervical dislocation prior to regaining consciousness. Kidney samples were rapidly harvested and scRNA-seq was performed on the kidneys of three mice from each group.
Histopathology
Kidney tissue was fixed overnight in 4% paraformaldehyde, dehydrated with an alcohol gradient and embedded in paraffin. Wax blocks containing kidney tissue were cut into 4 μm sections and stained with periodic acid-Schiff (PAS), Masson’s trichrome, and Sirius red. Representative images were taken with an Axio Imager M2 microscope and ZEN 2.6 software (Carl Zeiss, Jena, Germany). Tubular injury was scored according to renal tubular epithelial cell atrophy, detachment, and tubular dilatation. The scoring rules were as follows: no renal tubular injury = 0; less than 10% = 1; 10%−25% = 2; 26%−50% = 3; 51%−75% = 4; greater than 75% = 5. Area of fibrosis by Masson and Sirius staining was calculated by Image J software (Bethesda, Maryland, NIH, USA).
Renal function indexes
The evaluation of serum creatinine (Scr) and blood urea nitrogen (BUN) were measured by Creatinine Serum Detection Kit and Blood Urea Nitrogen Detection Kit (StressMarq Biosciences, British Columbia, Canada), respectively.
Western blotting
Frozen kidney cortexes were pulverized in liquid nitrogen and homogenized in RIPA lysis buffer (Cell Signaling Technology, Beverly, MA, United States) containing a protease and phosphatase inhibitors cocktail. Equal amounts of proteins were separated on 7% or 10% SDS-PAGE gels and then transferred to nitrocellulose membranes (Millipore, Billerica, MA, United States). The membranes were blocked with 5% skim milk for 1 h at room temperature and the blots were cut prior to hybridization with primary antibodies against fibronectin (FN, 1:1,000), type I collagen (Col-I, 1:1,000; Proteintech, Wuhan, China), type IV collagen (Col-IV, 1:1,000; Abcam, Cambridge, United Kingdom), Vimentin (1:1,000;), α-Tubulin (1:1000; Cell Signaling Technology, Beverly, MA, United States), and α-smooth muscle actin (α-SMA, 1:1,000; Sigma-Aldrich, St Louis, MO, United States) overnight at 4 °C. The next day the blots were incubated with horseradish peroxidase-conjugated secondary antibodies (1:2,000; Life Technologies, Carlsbad, CA, United States) secondary antibodies for 1 h at room temperature and then were visualized and quantified by ChemiDoc MP imaging system and Image Lab software version 5.1 (Bio-Rad Laboratories, Hercules, CA, United States).
Single-cell Preparation
The kidney samples of mice in different feeding groups were collected for the scRNA-seq assay. Fresh tissues were preserved on ice in sCelLive® Tissue Preservation Solution (Singleron Biotechnologies, Nanjing, China). Specimens were washed three times with Hanks Balanced Salt Solution, minced into small pieces, and then digested with 3 mL of sCelLive® Tissue Dissociation Solution for 15 min at 37 °C using the Singleron PythoN™ Tissue Dissociation System. The cell suspension was collected and filtered through a 40 μm sterile filter. GEXSCOPE® Erythrocyte Lysate (RCLB, Singleron) was then added and the mixture [cells: RCLB = 1:2 (v/v)] was incubated at room temperature for 5–8 min to remove erythrocytes. The supernatant was then removed by centrifugation at 300 × g for 5 min at 4 °C to obtain a soft suspension with PBS. The scripts of key steps can be found at the GitHub repository: https://github.com/SingleronBio/celelenscloud. The data generated in this study have been uploaded to the China National Genebank (CNGB) with accession number CNP0007462: https://db.cngb.org/data_resources/?query=CNP0007462.
Construction of scRNA-seq library
Single-cell suspensions (2 × 105 cells/mL) with PBS (HyClone, Utah, United States) were loaded onto microwell chip using the Singleron Matrix® Single Cell Processing System. Barcoding Beads were subsequently collected from the microwell chip, followed by reverse transcription of the mRNA captured by the Barcoding Beads and to obtain cDNA, and PCR amplification. The amplified cDNA was then fragmented and ligated with sequencing adapters. The scRNA-seq libraries were constructed according to the protocol of the GEXSCOPE® Single Cell RNA Library Kits (Singleron Biotechnologies, Nanjing, China). Individual libraries were diluted to 4 nM, pooled, and sequenced on Illumina novaseq 6000 with 150 bp paired end reads.
Quality control, dimension-reduction and clustering
Scanpy v1.8.2 was used for quality control, dimensionality reduction and clustering under Python 3.7. For each sample dataset, we filtered expression matrix by the following criteria: (1) cells with gene count less than 200 or with top 2% gene count were excluded; (2) cells with top 2% UMI count were excluded; (3) cells with mitochondrial content 50% were excluded; (4) genes expressed in less than 5 cells were excluded. After filtering, 109,010 cells were retained for the downstream analyses, with on average 1193 genes and 3246 UMIs per cell. The raw count matrix was normalized by total counts per cell and logarithmically transformed into normalized data matrix. Top 2000 variable genes were selected by setting flavor = ‘seurat’. Principle Component Analysis (PCA) was performed on the scaled variable gene matrix, and top 22 principal components were used for clustering and dimensional reduction. Cells were separated into 33 clusters by using Louvain algorithm and setting resolution parameter at 1.2. Cell clusters were visualized by using Uniform Manifold Approximation and Projection (UMAP).
Batch effect removal
Harmony: Batch effect between samples was removed by Harmony v1.0 using the top 22 principal components from PCA.
scRNA-seq data analysis
Raw reads were processed to generate gene expression profiles using CeleScope (version 1.5.2, Singleron Biotechnologies) with default parameters. Scanpy (version 1.8.2) was used for quality control, dimensionality reduction and clustering under Python 3.744. Briefly, Barcodes and UMIs were extracted from R1 reads and corrected. Adapter sequences and poly A tails were trimmed from R2 reads and the trimmed R2 reads were aligned against the GRCm38 (mm10) transcriptome using STAR (version 2.6.1b). Uniquely mapped reads were then assigned to exons with FeatureCounts (version 2.0.1). Successfully Assigned Reads with the same cell barcode, UMI and gene were grouped together to generate the gene expression matrix for further analysis.
Differentially expressed genes (DEGs) analysis
To identify differentially expressed genes (DEGs), we used the scanpy.tl.rank_genes_groups function based on Wilcoxon rank sum test with default parameters, and selected the genes expressed in more than 10% of the cells in either of the compared groups of cells and with an average log (Fold Change) value greater than 1 as DEGs. Adjusted p value was calculated by benjamini-hochberg correction and the value 0.05 was used as the criterion to evaluate the statistical significance.
Pathway enrichment analysis
Gene Ontology (GO) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG)21,22,23 pathway enrichment analysis were conducted with the “clusterProfiler” package in R software (version 4.0.0) to identify distinct biological functions and pathways48. Gene set variation analysis (GSVA) was performed to determine further the underlying biological functions of significant genes13.
Cell-cell interaction analysis: cellphonedb
Cell-cell interaction (CCI) between PT_5 and renal interstitial cells were predicted based on known ligand–receptor pairs by Cellphone DB (version 2.1.0)8. Permutation number for calculating the null distribution of average ligand-receptor pair expression in randomized cell identities was set to 1,000. Individual ligand or receptor expression was thresholded by a cutoff based on the average log gene expression distribution for all genes across each cell type. Predicted interaction pairs with p value < 0.05 and of average log expression > 0.1 were considered as significant and visualized by heatmap plot and dot plot in CellphoneDB.
Pseudotime trajectory analysis: monocle2
Cell differentiation trajectory of monocyte subtypes was reconstructed with the Monocle2 (version 2.22.0)38. For constructing the trajectory, top 2,000 highly variable genes were selected by Seurat (version 4.1.0) FindVairableFeatures, and dimension-reduction was performed by DDRTree. The trajectory was visualized by plot_cell_trajectory function in Monocle2.
Statistical analysis
The data are presented as mean ± standard error of mean (SEM). One-way ANOVA with Dunnett post hoc analysis was conducted to compare significant differences among groups using GraphPad Prism 9 (La Jolla, CA, United States). A p value of less than 0.05 was considered significantly different.
Results
Renal pathological and functional changes in adenine-induced CKD mice
Compared with the control group, CKD mice showed tubular atrophy, tubular lumen dilatation, and interstitial collagen deposition, which were most obvious in the CKD8w group (Fig. 1A). The results of quantitative analysis were consistent with the pathological findings that tubular injury score and percentage of fibrotic areas increased progressively with the progression of CKD (Fig. 1B-D). The Scr level (1.20 ± 0.08 mg/dL vs. 0.66 ± 0.03 mg/dL, p < 0.01) and BUN level (91.57 ± 2.26 mg/dL vs. 21.73 ± 0.43 mg/dL, p < 0.001) were significantly elevated in the CKD8w group as compared to the control group (Fig. 1E, F). The presence of FN, Col-IV, Col-I, Vimentin, and α-SMA in the renal tissue is indicative of renal fibrosis. Western blot analysis revealed a significant increase in the expression of these five indicators in the kidneys of CKD mice in the CKD8w group (p < 0.05) (Fig. 1G-L). These data indicated that the adenine model exhibited the hallmark characteristics of CKD.

Renal histopathology and functional changes in adenine-induced CKD mice. A Representative images of PAS, Masson, and Sirius staining. All images are shown at identical magnification, ×200, scale bar = 50 μm. B-D Quantitative analysis of tubular injury and fibrosis. E Serum creatinine levels. F Blood urea nitrogen levels. G Representative Western blot images of FN, Col-IV, Col-I, Vimentin, and α-SMA expression in the kidneys of mice in all groups. The original blots are presented in Supplementary Fig. 1. H-L Densitometric analysis of FN, Col-IV, Col-I, Vimentin, and α-SMA normalized to α-Tubulin content. Data are expressed as mean ± SEM, n = 3. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with indicated group, ns = no significance.
Overview of mouse kidney single‑cell transcriptome landscape
Following sample preparation and quality control, a total of 109,010 cells (21,048 cells from the control group, 19,979 cells from the CKD2w group, 27,031 cells from the CKD4w group, and 40,952 cells from the CKD8w group) were isolated and used for further scRNA-seq analysis. Unsupervised clustering analysis was employed to distinguish and characterize eleven cell types, including epithelial cells, endothelial cells, fibroblasts, mural cells, mesangial cells, proliferating cells, B cells, T and NK cells, neutrophils, mononuclear phagocytes (MPs), and erythrocytes (Fig. 2A). The top 10 and top 3 DEGs in all cell types were displayed in Fig. 2B and C, respectively, which demonstrated distinct transcriptome features of the eleven cell types. The top 3 marker genes used for cell type annotation were summarized in Fig. 2D. Subsequently, the changes in the proportions of each cell subset in the kidneys of different groups of mice were analyzed. The results demonstrated a reduction in the proportion of B cells and epithelial cells. In contrast, the proportion of fibroblasts, neutrophils, and MPs increased in the CKD8w group (Fig. 2E and F). Specific to the epithelial cells, unsupervised cluster analysis of 55,128 epithelial cells identified nine distinct subsets including collecting duct intercalated cells (ICs), collecting duct principal cells (PCs), renal loop of Henle epithelial cells (LOH), proximal tubule (PT) cells, distal convoluted tubule (DCT) cells, epithelial cells, kidney pelvis urothelial cells, podocytes, and proliferating epithelial cells (Fig. 2G). Analysis of the top 10 and top 3 DEGs shown significant differences in transcription levels among epithelial cell subsets (Fig. 2H and I). The marker genes utilized for the annotation of epithelial cell subsets were shown in Fig. 2J. The proportion of PT cells in epithelial cells fluctuated as CKD progressed (Fig. 2K), but the proportion of PT cells in total kidney cells decreased continuously (Fig. 2L). The above data displayed the basic characteristics of total cell subsets and epithelial cell subsets in normal mice and CKD mice at different stages.

Overview of mouse kidney single‑cell transcriptome landscape. A Uniform manifold approximation and projection (UMAP) of renal cells colored by groups (left), samples (middle), and distinct cell types (right). B Heatmap of the top 10 differentially expressed genes (DEGs) among all cell types according to log fold changes. C Violin plot of the top 3 DEGs among all cell types according to log fold changes. D Bubble dot plot of the top 3 marker genes used for cell type annotation in the scRNA-seq. E Proportion of each cell type in each kidney sample. F Differences in cell proportion among groups. G UMAP of epithelial cells colored by four groups (left) and epithelial cell subsets (right). H Heatmap of the top 10 DEGs among epithelial cell subsets according to log fold changes. I Violin plot of the top 3 DEGs among epithelial cell subsets according to log fold changes. J Bubble dot plot of the top 3 marker genes used for epithelial cell type annotation. K Proportion of epithelial cell subsets for twelve samples. L Differences in epithelial cell proportion among groups. Data are expressed as mean ± SEM, n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 compared with indicated group, ns = no significance. Abbreviations: DCT, distal convoluted tubule cells; ECs, endothelial cells; ICs, collecting duct intercalated cells; LOH, renal loop of Henle epithelial cells; PCs, collecting duct principal cells; PelvisUrothelial, kidney pelvis urothelial cells; PT, proximal tubule cells; MPs, mononuclear phagocytes.
Identification of injured PTECs associated with CKD progression
Subclustering of 17,358 PTECs yielded seven distinct subsets named PT_1, PT_2, PT_3, PT_4, PT_5, PT_6, and PT_7 (Fig. 3A). The top 10 and top 3 DEGs in PT subsets were summarized in Fig. 3B and C, respectively. Quantitatively, the proportion of PT_5 subset in either PT cells or total kidney cells was all gradually increased with CKD progression (Fig. 3D and E). The top 10 DEGs of PT_5 subset were Spp1, C3, Cp, Cryab, Atf3, Cd74, Sparc, Igfbp7, Nfkbia, and H2-K1 (Fig. 3B). The marker genes of tubular injury, C3, Havcr1, and Vcam1, were highly expressed in the PT_5 subset, but hardly expressed in other PT subsets (Fig. 3F). In addition to expressing universal markers of proximal tubules such as Slc34a1 and Lrp2, the PT_5 subset specifically expressed the S1 segment marker genes Slc5a2 and Slc5a12 (Fig. 3G). Subsequently, we performed functional enrichment analysis to explore the unique functional characteristics of PT_5 subset. GO pathway analysis revealed that response to endoplasmic reticulum stress, cytosolic ribosome, and structural constituent of ribosome pathways were up-regulated in PT_5 subset (Fig. 3H). Transcription factor analysis of subsets of PTs showed that the activity of Trp53, Sox4, Relb, Batf, and Irf1 were up-regulated in PT_5 subset (Fig. 3I). The enrichment of downstream target genes for Relb and Irf1 demonstrated that they mainly regulated inflammation- and fibrosis-related pathways, respectively (Fig. 3J). Furthermore, intergroup differential gene analysis of PT_5 revealed that Spp1, Fgg, and Lyz2 were the top three up-regulated gene expressions in the CKD8w group compared with the control group (Fig. 3K). Intergroup differential analysis of transcription factors in PT_5 demonstrated that Stat3, Cebpd, Mbd2, Xbp1, and Spi1 were markedly up-regulated in the CKD8w group (Fig. 3L). Similarly, the enrichment of Cebpd and Spi1 downstream target genes showed that they mainly regulated inflammation-related pathways (Fig. 3M). From the above analysis, we identified a subset of injured PTECs associated with CKD progression, named PT_5, which was mainly located in the S1 segment of proximal tubule and had pro-inflammatory and pro-fibrotic functions.

Identification, characterization, and functional analysis of injured PTECs associated with CKD progression. A UMAP of PTECs colored by groups (left) and subsets (right). B Heatmap of the top 10 DEGs among PTECs types according to log fold changes. C Violin plot of the top 3 DEGs among PTECs types according to log fold changes. D Proportion of PTECs subsets for twelve samples. E Differences in PTECs subsets proportion among groups. F Heatmap of specific expression of the injury genes C3, Havcr1, and Vcam1 in the PTECs subsets. G Bubble dot plot of proximal tubule marker genes expressed in PT_5 subset. H GO enrichment analysis of up-regulated pathways in PT_5 subset. I Heatmap of the transcription factors activity of each PTECs subset. J KEGG enrichment of downstream target genes of transcription factor Relb and Irf1. K Heatmap of the top 20 up-regulated genes and the top 20 down-regulated genes sorted by avg_logFC for control vs. CKD8w intergroup differential genes. L Heatmap of the transcription factors activity of PT_5 in each group. M KEGG enrichment of downstream target genes of transcription factor Cebpd and Spi1. Data are expressed as mean ± SEM, n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 compared with indicated group, ns = no significance.
Interaction between PT_5 and renal interstitial cells
As shown in Fig. 4A, the interaction between PT_5 and renal interstitial cells increased gradually with CKD progression. Specifically, PT_5 increased the effects on fibroblasts, MPs, and T and NK cells most significantly, reaching a maximum at 8 weeks of CKD (Fig. 4B). We then analyzed the potential crosstalk pathway between PT_5, serving as a ligand, and renal interstitial cells through ligand-receptor interactions. The results revealed a gradual augmentation in the gene interaction pair interleukin 34 (IL34) and colony stimulating factor receptor-1 (CSF1R) for PT_5 and MPs alongside the progression of CKD (Fig. 4C and E). Notably, the chemokine ligand 2 (CCL2) and C-C chemokine receptor type 2 (CCR2) interaction was significantly elevated exclusively in the CKD8w group, with no discernible changes observed in the other three groups (Fig. 4D). The interaction between PT_5 and fibroblasts was mediated by the binding of platelet-derived growth factor (PDGF) and its receptors (PDGFR), including PDGFA_PDGFRA, PDGFB_PDGFRB, PDGFC_PDGFRA, and PDGFD_PDGFRB, and the effect was obvious in the CKD8w group (Fig. 4C and E). Regarding the interaction between PT_5 and T and NK cells, the interaction pair CXCL10_CXCR3 exhibited a sudden decline in the CKD8w group (Fig. 4D). The aforementioned results highlighted the multifaceted role of PT_5 in modulating MPs, fibroblasts, and T and NK cells by secreting growth factors, chemokines, and cytokines.

Interaction between PT_5 and renal interstitial cells. A Interaction networks among PT_5, neutrophils, MPs, fibroblasts, and T and NK cells in each group. B Heatmaps of the amount of interaction gene pairs per two cell types (up) and networks of the intensity of PT_5 action as a ligand on other cell types (down) in each group. C Bubble plot of growth factor ligand-receptor pairs between PT_5 and renal interstitial cells. D Bubble plot of chemokines ligand-receptor pairs between PT_5 and renal interstitial cells. E Bubble plot of cytokine ligand-receptor pairs between PT_5 and renal interstitial cells. In C, D, and E, PT_5 acted as ligand and MPs, T and NK cells, fibroblasts, and neutrophils acted as receptors.
Characterization of T and NK cells in CKD progression
Subcluster analysis of 5,593 T and NK cells identified twelve subsets including proliferatingT_Mcm5, group 2 innate lymphoid cells (ILC2), GDTCells_Trdc, NKT_Xcl1, NK_Ncr1, Treg_Foxp3, NaiveT_Ccr7, CD8Teff_Ccl5, CD8Teff_Cxcl10, CD8Teff_Ifng, CD8Tex_Xcl1, and HelperT_Tnfsf8 (Fig. 5A). The top 10 and top 3 DEGs among T and NK cell subsets were visually displayed in Fig. 5B and C, respectively. The marker genes used for annotation of T and NK cell subsets were shown in Fig. 5D. Notably, in the CKD8w group, there was a reduction in the proportion of NaiveT_Ccr7, NK_Ncr1, and NKT_Xcl1, while an increase in the abundance of GDTCells_Trdc, HelperT_Tnfsf8, and Treg_Foxp3 (Fig. 5E and F). To further explore the heterogeneity of T and NK cells, we assessed the gene set scores for each subset using Ucell score. Nk_Ncr1 subset exhibited a significant T and NK cells cytotoxicity score and CD8Tex_Xcl1 subset exhibited a significant T and NK cells exhaustion score (Fig. 5G). Pseudotime analysis illuminated that the vast majority of CD8 T cells in the CKD8w group were at the initial stage of differentiation, in contrast to the other three groups. CD8Teff_Ccl5 subset was located at the origin of differentiation and was the most abundant CD8 T cell in the CKD8w group (Fig. 5H). Then, intergroup differential gene analysis of CD8Teff_Ccl5 revealed that Apoe, Lyz2 and Spp1 were the top three up-regulated gene expressions in the CKD8w group compared with the control group (Fig. 5I). GO enrichment results showed that the up-regulated genes were mainly enriched in ribonucleoprotein complex biogenesis, ribosome, and structural constituent of ribosome pathways (Fig. 5J). These data suggested that the CD8Teff_Ccl5 subset might be involved in CKD progression.

Characterization of T and NK cells in CKD progression. A UMAP of T and NK cells colored by groups (left) and subsets (right). B Heatmap of the top 10 DEGs among T and NK cell types according to log fold changes. C Violin plot of the top 3 DEGs among T and NK cell types according to log fold changes. D Bubble dot plot of top 3 marker genes used for cell type annotation in T and NK cells. E Proportion of T and NK cell subsets for twelve samples. F Differences in T and NK cell proportion among groups. G Heatmap of UCell score in distinct T and NK cell subsets. H Pseudotime trajectory analysis colored by CD8 T cell subsets at varying pseudotime stages (left) and by individual CD8 T cell subsets (right). I Heatmap of the top 20 up-regulated genes and the top 20 down-regulated genes sorted by avg_logFC for control vs. CKD8w intergroup differential genes in the CD8Teff_Ccl5 subset. J GO enrichment analysis of the up-regulation pathways of CD8Teff_Ccl5 subset. Data are expressed as mean ± SEM, n = 3. *p < 0.05, **p < 0.01, and ***p < 0.001 compared with indicated group, ns = no significance.
Characterization of fibroblasts in CKD progression
Subcluster analysis of 982 fibroblasts resulted six subsets, including Myofibroblasts_Phlda1, Myofibroblasts_Ifitm1, Myofibroblasts_Hspa1a, Myofibroblasts_Prkg1, Myofibroblasts_Timp1, and Fibroblasts_Dcn (Fig. 6A). Of note, there were limited number of fibroblasts in the control (18 cells), CKD2w (16 cells), and CKD4w (21 cells) groups, and the vast majority of fibroblasts distributed in the CKD8w group. The top 10 DEGs among fibroblasts subsets were visually displayed in Fig. 6B, and the marker genes utilized for annotation of fibroblasts were shown in Fig. 6C. It was evident that Myofibroblasts_Timp1 cells accounted for the largest proportion in the CKD8w group (Fig. 6D). The GSVA heatmap analysis demonstrated that Myofibroblasts_Timp1 subset markedly enhanced myofibroblasts, pro-angiogenic, inflammatory, pro-epidermal growth factor, and adipose-derived pathways (Fig. 6E). In addition, GO enrichment analysis showed that endoplasmic reticulum to Golgi vesicle-mediated transport, collagen-containing extracellular matrix, and extracellular matrix structural constituent-related pathways were enriched in Myofibroblasts_Timp1 subset (Fig. 6F). Single-cell pseudotime analysis revealed that Myofibroblasts_Timp1 subset represented the earliest differentiation stage of fibroblasts, and Fibroblasts_Dcn subset were in the late stages of fibroblasts differentiation (Fig. 6G). The top 8 genes exhibiting differential expression with pseudotime progression were visually depicted in Fig. 6H. Expression of genes, including Spp1 and Timp1 were highest in early status, while expression of genes such as C3, Clec3b, Cxcl12, Dcn, and Gsn peaked in late ones. These results indicated that Myofibroblasts_Timp1 subset contributed to renal fibrosis in CKD.

Characterization of fibroblasts in CKD progression. A UMAP of fibroblasts colored by groups (left) and subsets (right). B Heatmap of the top 10 DEGs among fibroblast types according to log fold changes. C Bubble dot plot of top 3 marker genes used for cell type annotation in fibroblasts. D Proportion of fibroblasts subsets for twelve samples. E Heatmap of gene set variation analysis (GSVA) in distinct fibroblast subsets. F GO enrichment analysis of up-regulation pathways of Myofibroblasts_Timp1 subset. G Pseudotime trajectory analysis colored by fibroblast subsets at varying pseudotime stages (up) and by individual fibroblast subsets (down). H The top 8 differential expression genes with pseudotime progression.
Characterization of MPs and macrophages in CKD progression
Through unsupervised clustering analysis, we identified four distinct cell subsets among 14,964 MPs, including macrophages, monocytes, conventional dendritic cells (cDCs), and conventional type 2 dendritic cells (cDC2) (Fig. 7A). The proportion of macrophages in the CKD8w group was significantly higher than that in the control group, while the proportion of other MPs subsets did not differ significantly among groups (Fig. 7B and C). We then focused on macrophages for further analysis. Subclustering of 11,176 macrophages obtained nine distinct subsets, named Macrophages_Tgfbr1, Macrophages_Mcm2, Macrophages_Cxcl7, Macrophages_Cxcl8, Macrophages_Cxcl10, Macrophages_Mmp12, Macrophages_Mmp13, Macrophages_S100a6, and Macrophages_Tmem119 (Fig. 7D). The most significant DEGs among macrophage subsets were visually illustrated in Fig. 7E and F. The proportion of macrophage_Cxcl7 subset and macrophage_Mmp12 subset were increased and the proportion of Macrophage_Tgfbr1 subset and Macrophage_Mmp13 subset were decreased in the CKD8w group (Fig. 7G and H). Next, we calculated the M1-macrophage and M2-macrophage polarization scores for each distinct subset (M1-macrophage: IL1B, IL6, TNF; M2-macrophage: MRC1, CD163). Macrophages_S100a6 and Macrophages_Tmem119 exhibited characteristics similar to M1-macrophages, while Macrophages_Cxcl7 and Macrophages_Cxcl8 displayed similarities to M2-macrophages (Fig. 7I). The Ucell score revealed the up-regulation of pathways related to lipid metabolism, ferroptosis, ECM remodeling, immune regulation, and pro-angiogenic pathways in Macrophages_Mmp12 subset (Fig. 7J). Then, Pseudotime analyses shown that Macrophage_Mmp12 subset represented a later stage of differentiation of macrophages (Fig. 7K). Additionally, we performed a GO enrichment analysis of up-regulation genes of Macrophages_Mmp12 and found that pathways related to ATP metabolic process, mitochondrial inner membrane, and proton transmembrane transporter activity were enriched (Fig. 7L). Transcription factor analysis shown that the activity of Pou2f3, Six1, Klf9, Tbx19, and Nr1h3 were up-regulated in Macrophages_Mmp12 subset (Fig. 7M). The enrichment of downstream target genes for Klf9 and Six1 showed that pathways related to lipid metabolism were mainly enriched (Fig. 7N). Overall, these results demonstrated that macrophages were highly plastic and heterogeneous during the progression of CKD. Notably, the Macrophages_Mmp12 subset emerged as a prominent player, exhibiting pronounced pro-inflammatory and pro-fibrotic traits that might contribute to the disease pathology.

Characterization of MPs and macrophages in CKD progression. A UMAP of MPs colored by groups (left) and subsets (right). B Proportion of MPs subsets for twelve samples. C Differences in MPs proportion among groups. D UMAP of macrophages colored by groups (left) and subsets (right). E Heatmap of the top 10 DEGs among macrophage subsets according to log fold changes. F Violin plot of the top 3 DEGs among macrophage subsets according to log fold changes. G Proportion of macrophage subsets for twelve samples. H Differences in macrophage proportion among groups. I Relative expression level of macrophage markers in distinct macrophage subsets. J Heatmap of UCell score in distinct macrophage subsets. K Pseudotime trajectory analysis colored by macrophage subsets at varying pseudotime stages (left) and by individual macrophage subsets (right). L GO enrichment analysis of up-regulation pathways of Macrophages_Mmp12 subset. M Heatmap of the transcription factors activity of each macrophage subset. N GO enrichment of downstream target genes of transcription factor Klf9 and Six1. Data are expressed as mean ± SEM, n = 3. *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 compared with indicated group, ns = no significance.
Discussion
In this study, we identified an injured PTECs cell type, named PT_5, which was associated with CKD progression. PT_5 cells expressed the markers of tubule injury, mainly localized in S1 segment of proximal tubule, and had pro-inflammatory and pro-fibrotic functions. Moreover, PT_5 cells had regulatory effects on T and NK cells, fibroblasts, and MPs by secreting growth factors, chemokines, and cytokines. Further analysis revealed that the CD8Teff_Ccl5 subset, Myofibroblasts_Timp1 subset, and Macrophages_Mmp12 subset contributed to CKD progression. These results provided new insights into the coordination of PTECs with renal interstitial cells in the process of CKD.
CKD is characterized by a sustained reduction in kidney function and structural damage over time. Recent studies have shown that the PTECs are primary targets of kidney injury and progression of kidney disease. The PTECs act as primary sensors in response to stress conditions (e.g., obstructive, ischemic, hypoxic, oxidative, or metabolic stress), which result in cell death, and thus serve as effector in the progression of CKD10,33. The damage of PTECs is considered a central event in the pathogenesis of CKD and deregulated repair processes of PTECs result in EMT and TIF, which in turn aggravates tubular injury and kidney fibrosis. scRNA-seq has identified a subset of PTECs with damage characteristics that could predict future decline in renal function. These cells expressed Havcr1, Krit8, Spp1, Spp2, and Vcam1 to varying degrees and are known by various names in the literature, including PT_VCAM1, scattered tubular cells, adaptive/maladapted PTECSs, failed-repair PTECs, and injured PTECs25,32,46,51. In the present study, PT_5 was defined by the expression of Spp1, C3, and Cp. C3, Havcr1, and Vcam1 were highly expressed in PT_5, which was involved in the inflammatory response and fibrotic process. The up-regulation of pathways related to response to endoplasmic reticulum stress (ERS), cytosolic ribosome, and structural constituent of ribosome suggested that protein synthesis was at a higher level in PT_5. Notably, sustained or intense ERS may trigger programmed cell death or apoptosis4,19, and targeting ERS may be a valuable strategy for treating CKD. In addition, in the CKD8w group, there was a significant expression of Spp1, a pleiotropic glycoprotein expressed in a variety of cell types and involved in multiple functions such as inflammation, cell adhesion, and migration1,20. Therefore, Spp1 maybe a noteworthy target in the progression of CKD promoted by PT_5.
The kidney is generally regarded as having a highly distinctive microenvironment, where a multitude of cells engage in a highly diverse interactions47. In physiological conditions, the interactions between these different cell types are well ordered to fine-tune kidney function. However, in pathogenic conditions, injured kidney cells significantly alter their cell fate, which in turn leads to dysregulated intercellular communication and ultimately exacerbates kidney injury18. Thus, interactions between these different cell types are critical for signaling exchange in the renal microenvironment42. In our study, PT_5 crosstalked with MPs through gene interaction pairs CCL2_CCR2 and IL34_CSF1R, suggesting that PT_5 was involved in the recruitment and activation of these immune cells, which are critical for inflammatory infiltration and injury of renal tissue. Furthermore, our data indicated that PT_5 cells could interact with fibroblasts through multiple pathways involving the PDGF family members, suggested a direct involvement of PT_5 cells in the regulation of fibroblast activity, which is central to fibrogenesis in the kidney. IL34 is a key cytokine that stimulates macrophage and monocyte inflow by binding to its receptor CSF1R2. Previous studies have confirmed the critical role of PT-derived IL34 and macrophage inflow in acute kidney injury and unilateral ureteral obstruction (UUO) nephropathy models2,41. CCL2, also known as monocyte chemokine protein-1, belongs to a broad family of chemokines. It is a potent macrophage chemokine inducer acting through its receptor CCR2, and has been shown to direct macrophage infiltration and injury in a variety of renal and non-renal diseases7. CCL2 mediates and promotes renal fibrosis by recruiting monocytes and promoting macrophage activation and transdifferentiation24,50. Interestingly, the interaction between CCL2 and CCR2 was significantly intensified in the CKD8w group, highlighting a complex mechanism that warrants further exploration. Furthermore, PDGF, specifically the PDGFB ligand, emerges as another critical mediator in the epithelial-fibroblast crosstalk. PDGFB is highly up-regulated in injured renal tubules, contrasting its absence in healthy renal epithelial cells27,35, underscoring its role in disease progression.
For T and NK cells, their importance in CKD remains in question. In some studies, T-cell-deficient mice damaged by UUO had reduced TIF, but fibrosis was unchanged in other studies5,31. C-C chemokine ligand 5 (CCL5), which is mainly secreted by activated CD8 + T lymphocytes, plays an active role in leukocyte recruitment and is a potent leukocyte activator6. This is similar to our results, the CD8Teff_Ccl5 subset of T and NK cells might be involved in CKD progression. For fibroblasts and myofibroblasts, it is undeniable that myofibroblast activation and subsequent ECM accumulation are the main events in renal fibrosis. Activated myofibroblasts are the main contributors to renal fibrosis as they are able to produce the most matrix15. Cytokines produced by injured PTECs such as transforming growth factor-β, Wnt, and PDGFB can stimulate myofibroblast formation, proliferation and ECM production3,34. Our study showed that Myofibroblasts_Timp1 was the most abundant fibroblast subset in the CKD8w group, and closely related to myofibroblast activation, inflammation, and extracellular matrix remodelling. Several studies have demonstrated that tissue inhibitor of metalloproteinases-1 (TIMP-1) played a role in kidney injury by regulating the synthesis and degradation of the extracellular matrix, promoting tubulointerstitial fibrosis through the inhibition of proteolytic matrix metalloproteinases, and exacerbating inflammation and renal scarring39,45. Macrophages act as the main immune cells for inflammatory infiltration during the CKD process, which ultimately leads to the development of renal fibrosis. In our study, Macrophage_Mmp12 subset represented a later stage of differentiation of macrophages, and related to ferroptosis, ECM remodelling, and immune regulation. Macrophages are a potent source of matrix metalloproteinases (MMPs). Although some MMP can slow TIF progression through degradation of ECM components, MMP2, MMP9, and MMP12 are thought to be associated with TIF progression16. It has been reported that abnormal expression of MMP12 could contribute to coronary artery and cerebral vascular disease, metastatic cancer, and pulmonary disease29. However, the mechanism of action of MMP12 in the progression of CKD remains unclear and requires further exploration.
Conclusions
In conclusion, our study has uncovered an injured PTEC subset, PT_5, displaying a coordinative pattern involving T and NK cells, fibroblasts, and MPs in CKD progression.
Data availability
All data generated or analyzed in the current study are available from the corresponding author on reasonable request. The scripts of key steps can be found at the GitHub repository: https://github.com/SingleronBio/celelenscloud. The data generated in this study have been uploaded to the China National Genebank (CNGB) with accession number CNP0007462: https://db.cngb.org/data_resources/?query=CNP0007462.
Abbreviations
- BUN:
-
Blood urea nitrogen
- CCL5:
-
C-C chemokine ligand 5
- CCR2:
-
C-C chemokine receptor type 2
- CCI:
-
Cell-cell interaction
- CCL2:
-
C-C chemokine ligand 2
- CKD:
-
Chronic kidney disease
- CSF1R:
-
Colony stimulating factor receptor-1
- DEGs:
-
Differentially expressed genes
- EMT:
-
Epithelial mesenchymal transition
- ECM:
-
Extracellular matrix
- GO:
-
Gene Ontology
- GSVA:
-
Gene set variation analysis
- IL34:
-
Interleukin 34
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MMPs:
-
Matrix metalloproteinases
- MPs:
-
Mononuclear phagocytes
- PAS:
-
Periodic acid-Schiff
- PDGF:
-
Platelet-derived growth factor
- PDGFR:
-
Platelet-derived growth factor receptor
- ERS:
-
Endoplasmic reticulum stress
- PTECs:
-
Proximal tubular epithelial cells
- Scr:
-
Serum creatinine
- scRNA-seq:
-
single-cell RNA sequencing
- TIMP-1:
-
Tissue inhibitor of metalloproteinases-1
- TIF:
-
Tubulointerstitial fibrosis
References
Abedini, A. et al. Single-cell transcriptomics and chromatin accessibility profiling elucidate the kidney-protective mechanism of mineralocorticoid receptor antagonists. J. CLIN. INVEST. 134 https://doi.org/10.1172/JCI157165 (2024).
Baek, J. H. et al. IL-34 mediates acute kidney injury and worsens subsequent chronic kidney disease. J. CLIN. INVEST. 125, 3198–3214. https://doi.org/10.1172/JCI81166 (2015).
Borges, F. T. et al. TGF-beta1-containing exosomes from injured epithelial cells activate fibroblasts to initiate tissue regenerative responses and fibrosis. J. AM. SOC. NEPHROL. 24, 385–392. https://doi.org/10.1681/ASN.2012101031 (2013).
Boyce, M. & Yuan, J. Cellular response to Endoplasmic reticulum stress: a matter of life or death. CELL. DEATH DIFFER. 13, 363–373. https://doi.org/10.1038/sj.cdd.4401817 (2006).
Braga, T. T. et al. MyD88 signaling pathway is involved in renal fibrosis by favoring a TH2 immune response and activating alternative M2 macrophages. MOL. MED. 18, 1231–1239. https://doi.org/10.2119/molmed.2012.00131 (2012).
Chen, J. et al. Investigation of the correlation between the Polymorphism/Expression level of RANTES and its receptor CCR5 gene promoter and type 2 diabetes mellitus. DIABET. METAB. SYND OB. 16, 213–223. https://doi.org/10.2147/DMSO.S398264 (2023).
Cianci, R. et al. Stem cells in kidney ischemia: from inflammation and fibrosis to renal tissue regeneration. INT. J. MOL. SCI. 24 https://doi.org/10.3390/ijms24054631 (2023).
Efremova, M., Vento-Tormo, M., Teichmann, S. A. & Vento-Tormo, R. CellPhoneDB: inferring cell-cell communication from combined expression of multi-subunit ligand-receptor complexes. NAT. PROTOC. 15, 1484–1506. https://doi.org/10.1038/s41596-020-0292-x (2020).
Francis, A. et al. Chronic kidney disease and the global public health agenda: an international consensus. NAT. REV. NEPHROL. https://doi.org/10.1038/s41581-024-00820-6 (2024).
Gewin, L. S. Renal fibrosis: primacy of the proximal tubule. MATRIX BIOL. 68–69, 248–262. https://doi.org/10.1016/j.matbio.2018.02.006 (2018).
Global & national burden of chronic kidney disease. Global, regional, and national burden of chronic kidney disease, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. LANCET 395, 709–733. https://doi.org/10.1016/S0140-6736(20)30045-3 (2020). 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017.
Guo, C. et al. Crosstalk between proximal tubular epithelial cells and other interstitial cells in tubulointerstitial fibrosis after renal injury. FRONT. ENDOCRINOL. 14, 1256375. https://doi.org/10.3389/fendo.2023.1256375 (2023).
Hanzelmann, S., Castelo, R. & Guinney, J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7. https://doi.org/10.1186/1471-2105-14-7 (2013).
Hewitson, T. D. Renal tubulointerstitial fibrosis: common but never simple. AM. J. PHYSIOL-RENAL. 296, F1239–F1244. https://doi.org/10.1152/ajprenal.90521.2008 (2009).
Huang, R., Fu, P. & Ma, L. Kidney fibrosis: from mechanisms to therapeutic medicines. SIGNAL. TRANSDUCT. TAR. 8, 129. https://doi.org/10.1038/s41392-023-01379-7 (2023).
Huen, S. C. & Cantley, L. G. Macrophages in renal injury and repair. ANNU. REV. PHYSIOL. 79, 449–469. https://doi.org/10.1146/annurev-physiol-022516-034219 (2017).
Humphreys, B. D. Mechanisms of renal fibrosis. ANNU. REV. PHYSIOL. 80, 309–326. https://doi.org/10.1146/annurev-physiol-022516-034227 (2018).
Inagi, R. & Yoshioka, K. Stray tubules: the varied and dynamic fate of tubular cells. KIDNEY INT. 104, 19–21. https://doi.org/10.1016/j.kint.2023.02.005 (2023).
Joazeiro, C. Mechanisms and functions of ribosome-associated protein quality control. NAT. REV. MOL. CELL. BIO. 20, 368–383. https://doi.org/10.1038/s41580-019-0118-2 (2019).
Kaleta, B. The role of osteopontin in kidney diseases. INFLAMM. RES. 68, 93–102. https://doi.org/10.1007/s00011-018-1200-5 (2019).
Kanehisa, M. Toward Understanding the origin and evolution of cellular organisms. PROTEIN SCI. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. NUCLEIC ACIDS RES. 28, 27–30. https://doi.org/10.1093/nar/28.1.27 (2000).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. NUCLEIC ACIDS RES. 51, D587–D592. https://doi.org/10.1093/nar/gkac963 (2023).
Kashyap, S. et al. Blockade of CCR2 reduces macrophage influx and development of chronic renal damage in murine renovascular hypertension. AM. J. PHYSIOL-RENAL. 310, F372–F384. https://doi.org/10.1152/ajprenal.00131.2015 (2016).
Kirita, Y., Wu, H., Uchimura, K., Wilson, P. C. & Humphreys, B. D. Cell profiling of mouse acute kidney injury reveals conserved cellular responses to injury. P NATL. ACAD. SCI. USA. 117, 15874–15883. https://doi.org/10.1073/pnas.2005477117 (2020).
Kitty, J. J. et al. A single number for advocacy and communication-worldwide more than 850 million individuals have kidney diseases. KIDNEY INT. 96. https://doi.org/10.1016/j.kint.2019.07.012 (2019).
Kok, H. M., Falke, L. L., Goldschmeding, R. & Nguyen, T. Q. Targeting CTGF, EGF and PDGF pathways to prevent progression of kidney disease. NAT. REV. NEPHROL. 10, 700–711. https://doi.org/10.1038/nrneph.2014.184 (2014).
Lake, B. B. et al. An atlas of healthy and injured cell States and niches in the human kidney. NATURE 619, 585–594. https://doi.org/10.1038/s41586-023-05769-3 (2023).
Lee, K. B., Dunn, Z. S., Lopez, T., Mustafa, Z. & Ge, X. Generation of highly selective monoclonal antibodies inhibiting a recalcitrant protease using decoy designs. BIOTECHNOL. BIOENG. 117, 3664–3676. https://doi.org/10.1002/bit.27519 (2020).
Li, L., Fu, H. & Liu, Y. The fibrogenic niche in kidney fibrosis: components and mechanisms. NAT. REV. NEPHROL. 18, 545–557. https://doi.org/10.1038/s41581-022-00590-z (2022).
Lili, L. et al. CD4 + T Lymphocytes, especially Th2 cells, contribute to the progress of renal fibrosis. Am. J. Nephrol. 36 https://doi.org/10.1159/000343283 (2012).
Lindgren, D. et al. Isolation and characterization of progenitor-like cells from human renal proximal tubules. AM. J. PATHOL. 178, 828–837. https://doi.org/10.1016/j.ajpath.2010.10.026 (2011).
Liu, B. C., Tang, T. T., Lv, L. L. & Lan, H. Y. Renal tubule injury: a driving force toward chronic kidney disease. KIDNEY INT. 93, 568–579. https://doi.org/10.1016/j.kint.2017.09.033 (2018).
Maarouf, O. H. et al. Paracrine Wnt1 drives interstitial fibrosis without inflammation by tubulointerstitial Cross-Talk. J. AM. SOC. NEPHROL. 27, 781–790. https://doi.org/10.1681/ASN.2014121188 (2016).
Masato, K. et al. Role of atrophic changes in proximal tubular cells in the peritubular deposition of type IV collagen in a rat renal ablation model. Nephrol. Dial Transpl. 20 https://doi.org/10.1093/ndt/gfh872 (2005).
Michael, Z. & Eric, G. N. Mechanisms of tubulointerstitial fibrosis. J. Am. Soc. NEPHROL. 21 https://doi.org/10.1681/ASN.2010080793 (2010).
Muto, Y. et al. Single cell transcriptional and chromatin accessibility profiling redefine cellular heterogeneity in the adult human kidney. NAT. COMMUN. 12, 2190. https://doi.org/10.1038/s41467-021-22368-w (2021).
Qiu, X. et al. Single-cell mRNA quantification and differential analysis with census. NAT. METHODS. 14, 309–315. https://doi.org/10.1038/nmeth.4150 (2017).
Ries, C. Cytokine functions of TIMP-1. CELL. MOL. LIFE SCI. 71, 659–672. https://doi.org/10.1007/s00018-013-1457-3 (2014).
Ruilope, L. M. et al. Design and baseline characteristics of the finerenone in reducing cardiovascular mortality and morbidity in diabetic kidney disease trial. AM. J. NEPHROL. 50, 345–356. https://doi.org/10.1159/000503712 (2019).
Sanchez-Nino, M. D., Sanz, A. B. & Ortiz, A. Chronicity following ischaemia-reperfusion injury depends on tubular-macrophage crosstalk involving two tubular cell-derived CSF-1R activators: CSF-1 and IL-34. NEPHROL. DIAL TRANSPL. 31, 1409–1416. https://doi.org/10.1093/ndt/gfw026 (2016).
Tan, R. J., Zhou, D. & Liu, Y. Signaling crosstalk between tubular epithelial cells and interstitial fibroblasts after kidney injury. KIDNEY DIS-BASEL. 2, 136–144. https://doi.org/10.1159/000446336 (2016).
Wilson, P. C. & Humphreys, B. D. Kidney and organoid single-cell transcriptomics: the end of the beginning. PEDIATR. NEPHROL. 35, 191–197. https://doi.org/10.1007/s00467-018-4177-y (2020).
Wolf, F. A., Angerer, P. & Theis, F. J. SCANPY: large-scale single-cell gene expression data analysis. GENOME BIOL. 19, 15. https://doi.org/10.1186/s13059-017-1382-0 (2018).
Won, A. J. et al. Discovery of urinary metabolomic biomarkers for early detection of acute kidney injury. Mol. Biosyst. 12, 133–144. https://doi.org/10.1039/c5mb00492f (2016).
Wu, H. et al. Mapping the single-cell transcriptomic response of murine diabetic kidney disease to therapies. CELL. METAB. 34, 1064–1078. https://doi.org/10.1016/j.cmet.2022.05.010 (2022).
Yao, W. et al. Single cell RNA sequencing identifies a unique inflammatory macrophage subset as a druggable target for alleviating acute kidney injury. ADV. SCI. 9, e2103675. https://doi.org/10.1002/advs.202103675 (2022).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. ClusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16, 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Zhou, D. & Liu, Y. Renal fibrosis in 2015: Understanding the mechanisms of kidney fibrosis. Nat. rev. nephrol. 12, 68–70. https://doi.org/10.1038/nrneph.2015.215 (2016).
Zhou, H., Mu, L., Yang, Z. & Shi, Y. Identification of a novel immune landscape signature as effective diagnostic markers related to immune cell infiltration in diabetic nephropathy. FRONT. IMMUNOL. 14, 1113212. https://doi.org/10.3389/fimmu.2023.1113212 (2023).
Zhou, M. et al. Decoding the intercellular Cross-Talking between immune cells and renal innate cells in diabetic kidney disease by bioinformatics. J. INFLAMM. RES. 16, 3049–3062. https://doi.org/10.2147/JIR.S409017 (2023).
Funding
This work was supported by the National Natural Science Foundation of China (grant number 81973602) and Sanming Project of Medicine in Shenzhen (grant numbers SZZYSM202311004).
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation and animal experiments were performed by Shanshan Wu, Liwen Gao, and Xi Huang. Data analysis was performed by Youcai Xu, Xiaoqin Ye, and Yu Peng. The first draft of the manuscript was written by Youcai Xu, Xiaoqin Ye, and Yu Peng. Lingfei Lu, Jiandong Lu, Xinhui Liu reviewed and edited the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study was approved by the Ethics Committee of Shenzhen Glorybay Biotech Co., Ltd (approved ID: RW-IACUC-22-0029). Animal experiments were carried out in accordance with National Research Council’s Guide for the Care and Use of Laboratory Animals. The reporting of animal experimental protocols in the manuscript follows the recommendations in the ARRIVE guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, Y., Ye, X., Peng, Y. et al. Single-cell RNA sequencing revealed the association between proximal tubular epithelial cells and renal interstitial cells in chronic kidney disease progression. Sci Rep 15, 45256 (2025). https://doi.org/10.1038/s41598-025-28988-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-28988-2
