
Abstract
Benzo[a]pyrene (B[a]P) is a typical environmental persistent organic pollutant and a known nephrotoxicant. However, its toxicological profile under short-term, high-dose exposure conditions remains incompletely characterized. To address this, we established a C57BL/6J mouse model in which a single oral dose of 50 mg/kg B[a]P was administered. The results showed that time-dependent renal injury following Bla]P exposure. Within 3 days serum creatinine (Scr) and blood urea nitrogen (BUN) levels increased significantly (P < 0.05), coinciding with elevated renal malondialdehyde (MDA) content. Concurrently, superoxide dismutase (SOD) and catalase (CAT) activities, as well as total antioxidant capacity (T-AOC) were markedly reduced (P < 0.05). By days 7–14 days, the pathological changes shifted to inflammation and apoptosis, evidenced by upregulated TNF-α, IL-6, and caspase-3 at both gene and protein levels, alongside elevated nitric oxide synthase (NOS) and lactate dehydrogenase (LDH) activities (P < 0.05). While the Rictor/mTORC2 pathway regulates renal pathology, its role in B[a]P-induced injury remains unelucidated. Our study found that B[a]P exposure (7–14 days) significantly upregulated key components of and its downstream effectors (AKT1 and PKC-ζ) at both transcriptional levels (P < 0.05). Mechanistic studies in macrophage-specific Rictor knockout mice (Mac Rictor-/-) showed that, inhibiting Rictor/mTORC2 suppressed B[a]P-induced renal oxidative stress, inflammatory factor release, and apoptosis. This study first revealed that the Rictor/mTORC2 pathway serves as a potential molecular therapeutic target for B[a]P-induced kidney injury.
Similar content being viewed by others

Introduction
The accelerated industrialization process has fueled resource-intensive economic growth, resulting in the global accumulation of polycyclic aromatic hydrocarbons (PAHs), a typical type of persistent organic pollutants (POPs) that pose mounting threats to public health1. Among these, Benzo[a]pyrene (B[a]P) is classified by the UN Environment Programme as a priority pollutant owing to its environmental persistence, potent carcinogenicity, and cross-media mobility2. This pollutant is released mainly from incomplete fossil fuel combustion (the source of > 80% of anthropogenic emissions), industrial activities like aluminum smelting (concentrations up to 100 µg/m³), vehicle exhaust, and high-temperature cooking3,4. Its presence is widespread, with levels in Taiyuan, China, far exceeding background benchmarks1, and dust from classrooms in Jeddah, Saudi Arabia, containing 163.87 ± 68.53 ng/g5. Moreover, smoking significantly increases B[a]P exposure levels while dietary B[a]P formed during food processing demonstrates high bioavailability following gastrointestinal absorption6,7.
In environmental media, B[a]P undergoes complex migration and transformation. Due to low water solubility, high lipophilicity, and strong organic matter affinity, it accumulates in aquatic and terrestrial systems through atmospheric deposition and surface runoff. High detection frequencies in Yangtze River Delta waters and elevated soil levels may expose humans to high doses of B[a]P in daily life8,9, with toxic effects further amplified through the food chain7. The genotoxic effects of B[a]P originate from its bioactivation via the aryl hydrocarbon receptor (AHR). Binds to the AHR, triggering its nuclear translocation and the upregulation of CYP450 enzymes (e.g., CYP1A1/1B1). B[a]P is then metabolized by these enzymes to the ultimate carcinogen, BPDE. This metabolite induces tumorigenesis primarily by forming DNA adducts that facilitate proto-oncogene activation and malignant transformation10. Studies demonstrate that long-term low-dose B[a]P exposure affects animal renal11, hepatic, pulmonary, and neurological system12. Current research prioritizes such models; for example, 90-day gavage administration of 1 mg/kg B[a]P induces renal dysfunction13 or hepatic inflammation14. The public health threat of acute high-dose B[a]P exposure—arising from occupational or accidental incidents—is not adequately addressed by existing studies3,4, which cannot explain its rapid onset of organ damage. This knowledge gap underscores a critical need for a systematic investigation into the distinct toxicological pathways activated under such conditions.
Current evidence implicates oxidative stress, inflammatory responses, and apoptotic dysregulation in B[a]P toxicity mechanisms15,16. Shahid et al.17,18 demonstrated that acute high-dose B[a]P exposure triggers elevated lipid peroxidation, increased xanthine oxidase and LDH activity, impaired glutathione synthesis, and suppressed antioxidant enzymes in murine lung tissue. These alterations synergistically activated the NF-κB/COX-2/TNF-α/IL-6 inflammatory axis while upregulating pro-apoptotic factors (p53/Bax/caspase-3) and downregulating Bcl-2, culminating in DNA damage and pathological changes. Similarly, acute B[a]P intoxication induces multiorgan oxidative stress and DNA damage in murine liver, lung, brain, stomach, and kidney19. Notably, kidneys are particularly vulnerable due to high perfusion and abundant B[a]P-metabolizing enzymes (such as CYP1A1/CYP1B1) and exhibit a first-pass effect in B[a]P biotransformation; they are therefore considered the primary target organ for B[a]P-induced damage20. Adedara et al.21 observed nephrotoxicity in rats after 15-day exposure to 10 mg/kg B[a]P, with significantly increased serum urea/creatinine and compromised antioxidant defenses. While preliminary studies exist on high-dose, short-term B[a]P nephrotoxicity, its renal toxicokinetics and key mechanisms remain unclear, essential for preventing and treating human kidney damage.
The mammalian target of rapamycin (mTOR) precisely regulates critical renal homeostasis through metabolic control, proliferation, and autophagy22. mTOR coordinates protein/energy metabolism via mTORC1/2. The Rictor-dependent mTORC2 phosphorylates AKT1 and PKC-ζ23. Rictor deficiency suppresses AKT/PKC-α signaling and inhibits TGF-β1-driven renal fibrosis by reducing collagen deposition, macrophage M2 polarization, and EndMT24,25,26. Conversely, mTORC2-deficient dendritic cells exacerbate inflammatory injury27. Thus, the Rictor/mTORC2 pathway displays cell-specific duality in renal pathology, its diversity arising from microenvironment-driven signaling remodeling. Analogously, activation of this pathway in cadmium-induced neurotoxicity enhances downstream signaling and apoptosis28, highlighting its potential in pollutant-mediated organ injury. Activation of this pathway also promotes activation of the AKT axis, exacerbating airway inflammation and contributing to pulmonary fibrosis29. Recent studies have further demonstrated that inhibiting Rictor/mTORC2 can alleviate multi-organ dysfunction by reducing levels of reactive oxygen species (ROS), pro-inflammatory factors (p-IκBα/p-NF-κB/IL-6/TNF-α), and apoptosis markers22,30. Although the involvement of the Rictor/mTORC2 pathway in disease pathogenesis is established, systematic studies are lacking on whether B[a]P—a highly toxic environmental pollutant—activates this pathway, mediates B[a]P-induced nephrotoxicity, or whether pharmacological inhibition confers renal protection. Resolving these knowledge gaps will not only elucidate the molecular mechanisms of B[a]P nephrotoxicity, but also provide new drug targets and intervention strategies for the prevention and treatment of pollutant-related kidney damage.
Materials and methods
Animals and chemicals
Adult male C57BL/6J mice (8 weeks old, 24 ± 2 g) were obtained from Shanghai Jihui Laboratory Animal Management Co. Ltd. were acclimatized in an SPF environment for one week. At the same time, C57BL/6J background macrophage-specific Rictor knockout mice (Mac Rictor-/-) were selected, which were provided by Cyagen Biosciences, Inc. All animal protocols were conducted in accordance with ARRIVE guidelines. The authors confirm that animal care and use procedures in this study strictly adhered to the relevant institutional and national guidelines for animal care and use. Specifically, the experimental protocol was oversight and approved by the animal care and use committee of East China Normal University (Approval Number: m20250302).
Benzo[a]pyrene (B[a]P; purity ≥ 98%) was purchased from Sigma-Aldrich. All chemicals and kits were obtained from Beyotime (Shanghai, China) unless stated otherwise. Kit and reagent details are provided in Table S1.
Experimental design
Forty-eight mice were randomly assigned to either a control group (n = 24) or a Benzo[a]pyrene (B[a]P; Sigma-Aldrich, purity ≥ 98%) exposure group (n = 24). Each group was further divided into four subgroups (n = 6) corresponding to different time points: 1, 3, 7, and 14 days. B[a]P was dissolved in corn oil and administered orally at 50 mg/kg to the exposure group, while control animals received an equal volume of corn oil vehicle (5 mg/mL). Daily gavage was performed, and mice from the respective subgroups were euthanized at each designated time point.
The dose of 50 mg/kg B[a]P was selected based on the following considerations. First, it lies within the effective range previously established for inducing acute multi-organ toxicity—including renal injury—in mice19,31. Second, preliminary experiments (Fig. S1) indicated that oral administration of both 50 mg/kg and 100 mg/kg B[a]P resulted in significant and time-dependent renal functional impairment and histopathological alterations across multiple time points (1, 3, 7, and 14 days), with comparable severity between the two doses. Therefore, the 50 mg/kg dose was chosen for subsequent time-course analyses to characterize the toxicological phenotype while minimizing overall animal toxicity. This approach allows systematic evaluation of the renal injury induced by short-term high-dose B[a]P exposure.
As a widely distributed and highly representative persistent organic pollutant among polycyclic aromatic hydrocarbons, B[a]P was selected to align with existing literature and facilitate in-depth mechanistic investigation. Corn oil was used as the solvent due to its excellent lipid solubility, which ensured complete dissolution and consistent delivery of B[a]P. As a biologically inert vehicle, it introduced no additional toxic effects. Comparative analysis revealed no significant differences between the corn oil and saline control groups (Fig. S2), validating corn oil as a suitable vehicle.
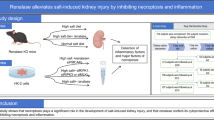
Twelve 8-week-old male Mac Rictor-/- mice were randomly divided into a control group and a B[a]P exposure group (n = 6) and gavaged continuously for 14 days, after which the mice were euthanized. Urine was collected from metabolic cages for biochemical analysis. Mice were anesthetized via intraperitoneal injection of Avertin (1.25%, 15 µL/g). Blood samples were collected from the retro-orbital venous plexus at designated time points, and the mice were then euthanized by cervical dislocation under deep anesthesia. Kidney tissues were rapidly harvested. One portion was immediately snap-frozen in liquid nitrogen and stored at -80 °C for subsequent analysis, while the other portion was fixed in 4% formaldehyde solution. The flowchart of the study procedure was illustrated in Fig. 1.

Overview of the study workflow.
Mouse body weight and organ coefficients
Mouse body weights were recorded daily. Kidneys were bilaterally excised intact at experiment termination for determination of renal organ coefficients.
Biochemical analyses
Serum creatinine (SCr), blood urea nitrogen (BUN), and urinary urea nitrogen concentrations were quantified by using a fully automated biochemical analyzer (BIOBASE BK-200, China) for the assessment of renal function32.
Detection of oxidative stress indicators
Renal cortical tissues were homogenized, and total protein concentration was quantified using the BCA assay. Subsequently, oxidative stress parameters, including malondialdehyde (MDA) content, the activities of superoxide dismutase (SOD) and catalase (CAT), and total antioxidant capacity (T-AOC), were measured in the kidney homogenates using an enzyme-linked immunosorbent assay reader (Thermo Scientific, Multiskan FC, USA). To assess systemic oxidative stress, serum levels of MDA and T-AOC were also evaluated in parallel. All measurements were conducted in strict accordance with the manufacturer’s instructions for the respective commercial kits.
Nitric oxide synthase (NOS) activity assay
NOS activity in kidney tissues was quantified using a commercial assay kit (BioTek Synergy microplate reader, USA; excitation/emission: 495/515 nm).
Enzyme-linked immunosorbent assay (ELISA)
TNF-α content in mouse kidney tissues was quantified using a commercial ELISA kit. Standards and samples were added to pre-coated antibody plates and incubated at 37 °C for 1.5 h in the dark. After washing, biotinylated detection antibody, HRP-streptavidin and TMB color developing solution were added sequentially, and the absorbance at 450 nm was measured following reaction termination. TNF-α concentrations were calculated from the standard curve and normalized to protein content (pg/mg protein).
Lactate dehydrogenase (LDH) activity assay
Quantification of serum LDH activity was performed using a Cytotoxicity Assay Kit. After adding 60 µl of working solution to each well, the samples were thoroughly mixed and incubated in the dark for 30 min at room temperature. Absorbance was measured at 490 nm to calculate cell mortality.
Histopathological observation
Kidney tissues were fixed by 4% paraformaldehyde (PFA), dehydrated by gradient ethanol, transparent by xylene, embedded in paraffin, and sectioned at 5 μm thickness. Hematoxylin and eosin (HE)-stained sections were examined under a light microscope (Leica DM500, Germany), and representative images were acquired.
Immunohistochemistry(IHC)
Sections were dewaxed and hydrated, and antigen repair was performed sequentially using 3% H₂O₂ to inactivate endogenous peroxidase and citrate buffer (pH 6.0). Subsequently, the sections were rabbit-derived anti-caspase-3, HRP-labeled high-sensitivity goat anti-rabbit IgG(H + L) as the secondary antibody, and DAB as the color developer, respectively, and the sections were restained with hematoxylin, dehydrated, and sealed for observation and image acquisition. Antibody Information are shown in Table S2.
Bioinformatics analysis
To characterize the functional implications of the identified differentially expressed genes (DEGs), we conducted a comprehensive bioinformatics workflow. Initially, DEGs were functionally annotated using the GeneCards database (https://www.genecards.org/). Subsequently, Gene Ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed with the DAVID bioinformatics platform (https://david.ncifcrf.gov/). This integrated approach provided systematic insights into the potential biological roles and pathway associations of the DEGs.
Molecular docking
Molecular docking was performed using a semi-flexible approach with AutoDock 1.5.6. The protein structure was prepared and hydrogen atoms were added. The small molecule ligand was optimized for docking, with its rotatable bonds and torsion angles defined using the Torsion Tree module in AutoDock Tools (ADT). A grid box of dimensions 126 × 126 × 126 ų was centered to encompass the entire protein binding site, with a grid spacing of 1.0 Å. Docking simulations were executed using default parameters. The resulting poses were visualized to analyze ligand-receptor binding modes and evaluate complex stability and key molecular interactions.
Real-time fluorescence quantitative PCR (RT-qPCR)
Total RNA was extracted from renal tissues using Trizol reagent and reverse-transcribed into cDNA. Quantitative real-time PCR (qPCR) was performed on a QuantStudio 3 system (Thermo Fisher, USA) using Hieff® qPCR SYBR Green MasterMix to analyze mRNA expression of: SOD1, SOD2, CAT, IL-6, TNF-α, caspase-3, Rictor, mTOR, AKT1, and PKC-ζ. The relative expression was calculated by the 2-ΔΔCt method with GAPDH as the endogenous control. The primer sequences are shown in Table S3.
Statistical analysis
All statistical analyses were performed using GraphPad Prism 9.0 (GraphPad Software, USA). Data are presented as the mean ± standard error of the mean (SEM). Differences between groups were determined by Student’s t-test (for two groups) or two-way ANOVA followed by Šidák’s post-hoc test (for multiple groups and time points). Specifically, the B[a]P-exposed group was compared to the control group at each time point to evaluate treatment effects, and across time points to assess temporal progression. A sample size of n = 6 per group was chosen based on previous studies19,31 and our pilot data, providing ≥ 80% power to detect an effect size of 1.5 at α = 0.05. Differences were considered statistically significant at P < 0.05.
Results
Temporal changes in renal injury induced by B[a]P
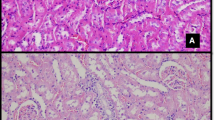
Compared with normal controls, B[a]P-exposed mice showed significantly reduced body weight on days 1 and 14, but not on days 3 or 7 (Fig. 2A). B[a]P exposure caused significant time-dependent kidney damage, evidenced by a decreased renal organ coefficient as early as day 3 post-exposure (Fig. 2B); simultaneously, serum creatinine (SCr), blood urea nitrogen (BUN) and urinary urea nitrogen levels increased significantly (Fig. 2C), with damage severity progressively worsening over exposure time. Histopathological analysis further confirmed the aforementioned damage. H&E-stained sections revealed intact renal architecture in normal controls, featuring regularly aligned tubular epithelia and distinct luminal structures. No discernible pathology on the first day of B[a]P exposure, but by the third day, early glomerular capsule atrophy with periglomerular inflammation and disrupted tubular epithelial alignment. By day 7, advanced glomerular shrinkage, exacerbated epithelial disorganization, and emerging tubular vacuolation. By day 14, renal damage reached maximal severity, characterized by architectural disintegration and luminal obliteration (Fig. 2D). Collectively, our findings demonstrate that short-term high-dose B[a]P exposure elicits rapid body weight reduction during both initial and terminal exposure phases, and induces time-dependent renal impairment and tissue structural lesions from day 3 post-exposure onward.


Temporal progression of B[a]P-induced renal injury. (A) Body weight changes. (B) Kidney morphology and organ coefficient. (C) SCr, BUN, and urinary urea nitrogen. (D) Representative H&E-stained kidney sections (×100 magnification). Key morphological alterations are indicated: black triangles, glomerular capsule lumen atrophy with periglomerular inflammatory infiltration; blue triangles, disorganized arrangement of tubular epithelial cells; red triangles, tubular vacuolization. Data are presented as mean ± SEM (n = 6). Asterisks (*) denote significant differences between the B[a]P group and the corresponding control group: *P < 0.05, **P < 0.01, ***P < 0.001. Hashtags (#) indicate significant differences across time points within the control or B[a]P-treated groups: # P < 0.05, ## P < 0.01, ### P < 0.001.
Renal oxidative stress disruption induced by B[a]P exposure
A systematic comparison between the untreated control and corn oil vehicle control groups confirmed the absence of significant solvent-related effects on all oxidative stress parameters at every time point (Fig. S3). On this basis, the corn oil control was used as the reference baseline in further analyses. Our results show that, Relative to that in control group, exposure to B[a]P significantly increased the content of MDA, a lipid peroxidation end product in the kidney, starting from the third day after exposure, while concurrently reducing SOD and CAT activity and T-AOC (Fig. 3A). This oxidative-antioxidative imbalance persisted through day 14. RT-qPCR further analysis demonstrated significant downregulation of key renal antioxidant genes (SOD1, SOD2, and CAT) mRNA levels by day 3 post-B[a]P exposure (Fig. 3B), with expression patterns corresponding to reduced enzymatic activities. To evaluate the systemic extent of oxidative stress, serum levels of MDA and T-AOC were examined. B[a]P-administered mice exhibited a significant increase in serum MDA and a concurrent decrease in T-AOC, evident from day 3 (Fig. 3C). This systemic redox dysregulation, which mirrors the renal findings, thereby demonstrates the time-dependent and multi-organ nature of B[a]P-induced oxidative stress.

Renal oxidative stress elicited by B[a]P exposure. (A) Renal levels of MDA, T-AOC, activities of SOD and CAT. (B) MDA and T-AOC in Serum. (C) Relative mRNA expression of SOD1, SOD2, and CAT in renal tissue. Data are presented as mean ± SEM (n = 6). Asterisks (*) denote significant differences between the B[a]P group and the corresponding control group: *P < 0.05, **P < 0.01, ***P < 0.001. Hashtags (#) indicate significant differences across time points within the control or B[a]P-treated groups: # P < 0.05, ## P < 0.01, ### P < 0.001.
B[a]P exposure promotes activation of the renal inflammatory response
TNF-α and IL-6 serve as pivotal mediators in renal inflammatory responses. Relative to normal controls, B[a]P exposure significantly elevated TNF-α and IL-6 mRNA expression (Fig. 4A) in renal tissues after 7 and 14 days, as confirmed by RT-qPCR analyses. ELISA further revealed significantly elevated TNF-α protein levels in kidney homogenates (Fig. 4B) and increased NOS activity (Fig. 4C), both of which positively correlated with pro-inflammatory factor upregulation. These results collectively demonstrate that B[a]P exposure drove renal inflammatory injury through time-dependent upregulation of pro-inflammatory mediators, involving coordinated increases in gene transcription, protein expression, and enzymatic activities from day 7 onward.

Renal inflammatory activation elicited by B[a]P exposure. (A) Relative mRNA expression of TNF-α and IL-6 in renal tissues. (B) Renal TNF-α protein quantification by ELISA. (C) NOS activity in mouse renal tissues. Data are presented as mean ± SEM (n = 6). Asterisks (*) denote significant differences between the B[a]P group and the corresponding control group: *P < 0.05, **P < 0.01, ***P < 0.001. Hashtags (#) indicate significant differences across time points within the control or B[a]P-treated groups: # P < 0.05, ## P < 0.01, ### P < 0.001.
B[a]P-induced activation of renal tissue apoptosis
RT-qPCR analysis revealed significantly upregulated caspase-3 mRNA expression in renal tissues relative to that in control group after 7- and 14-day B[a]P exposures (Fig. 5A). This transcriptional upregulation closely matched the spatial localization pattern of caspase-3 protein observed via IHC (Fig. 5C). Although phosphatidylserine ectopia occurs in the early stage of apoptosis, the membrane permeability barrier function is still intact, and LDH is tightly retained; whereas in the late apoptotic or secondary necrotic stage, membrane rupture and extensive LDH release into the cytosol33. In the study, following 7- and 14-day exposures, serum LDH activity was significantly elevated in B[a]P-exposed mice versus normal controls (Fig. 5B), indicating progression to advanced stages of cell death. These findings suggested that B[a]P mediates terminal kidney cell injury by activating the caspase-dependent apoptotic pathway and disrupting membrane integrity.


Renal apoptosis elicited by B[a]P exposure. (A) Relative mRNA expression of caspase-3 in renal tissues. (B) Serum LDH activity. (C) caspase-3 protein localization in renal tissues via IHC (original magnification ×100), Red triangle: Positive cells. Data are presented as mean ± SEM (n = 6). Asterisks (*) denote significant differences between the B[a]P group and the corresponding control group: *P < 0.05, **P < 0.01, ***P < 0.001. Hashtags (#) indicate significant differences across time points within the control or B[a]P-treated groups: # P < 0.05, ## P < 0.01, ### P < 0.001.
B[a]P-Induced renal injury via Rictor/mTORC2 pathway activation
Based on the GeneCards database (https://www.genecards.org/), we identified 420 overlapping differentially expressed genes (DEGs) associated with B[a]P exposure, renal injury, and the mTOR signaling pathway through bioinformatics analysis. Functional enrichment analysis of these genes was performed using the DAVID database (https://david.ncifcrf.gov/). Gene Ontology (GO) analysis suggested potential activation of the mTORC2 signaling pathway in B[a]P-induced renal injury. Specifically, DEGs were significantly enriched in the molecular function categories of “protein kinase binding” and “protein serine/threonine kinase activity,” and in the cellular component categories of “serine/threonine protein kinase complex” and “focal adhesion.” These enriched terms closely align with the known functions and subcellular localization of AKT1 and PKC-ζ, key downstream effectors of mTORC2, indicating their central involvement in this process (Fig. 6A-C).To explore potential molecular initiating events underlying B[a]P-induced mTORC2 activation, we performed molecular docking simulations. The results demonstrated that B[a]P stably binds with high affinity to a putative functional pocket of the Rictor protein, supported by favorable binding energy and interactions with key residues, suggesting a direct mechanism by which B[a]P may stabilize or allosterically activate the mTORC2 complex (Fig. 6D).
AKT1 and PKC-ζ are known regulators of renal cell proliferation, migration, survival, metabolism, cytoskeletal organization, and inflammatory microenvironment remodeling34,35. In order to investigate the mechanistic involvement of the Rictor/mTORC2 pathway in B[a]P-induced renal injury, the activation status of the pathway’s key regulatory nodes and its downstream effector molecules, was examined in the present study. Initial assessment of pathway activation was performed using a wild-type (WT) mouse model. RT-qPCR revealed significantly upregulated mRNA expression of Rictor, mTOR, AKT1, and PKC-ζ in renal tissues following 7- and 14-day B[a]P exposures (Fig. 6E), indicating B[a]P-induced transcriptional activation of core pathway genes. To further define the central role of the Rictor/mTORC2 pathway in B[a]P-induced nephrotoxicity, we validated its function using a macrophage-specific Rictor knockout (Mac Rictor-/-) model. Given that the alterations in pathway-related mRNA expression were more substantial at 14 days than at 7 days post-exposure, the longer duration was therefore chosen for this validation. Notably, in the kidneys of Mac Rictor-/- mice, B[a]P exposure for 14 days failed to induce significant changes in the mRNA levels of key pathway components (Rictor, mTOR, AKT1, and PKC-ζ) (Fig. 6F). This suggests that myeloid Rictor deficiency prevents B[a]P from activating the Rictor/mTORC2 signaling pathway.


Rictor/mTORC2 signaling axis mediates B[a]P-induced renal injury. (A) 420 differentially expressed genes were screened from the databases. (B) KEGG pathway enrichment analysis, generated using the KEGG database (https://www.kegg.jp/)75 with permission from Kanehisa Laboratories (License No. 253975). (C) GO enrichment analysis. (D) Docking View of B[a]P with Rictor. (E) Relative mRNA levels of Rictor, mTOR, AKT1 and PKC-ζ in kidney tissues of WT mice. (F) WT versus Mac Rictor-/- mice comparing the above molecular expression changes. Data are presented as mean ± SEM (n = 6). Asterisks (*) denote significant differences between the B[a]P group and the corresponding control group: *P < 0.05, **P < 0.01, ***P < 0.001. Hashtags (#) indicate significant differences across time points within the control or B[a]P-treated groups: #P < 0.05, ## P < 0.01, ### P < 0.001.
Deficiency of Rictor in macrophages mitigates B[a]P-induced kidney injury through mTORC2 pathway Inhibition
Compared with the control group, wild-type mice exposed to B[a]P for 14 days developed significant renal impairment, as evidenced by a marked reduction in body weight and renal organ coefficients (Fig. S4), together with considerable histopathological damage. In contrast, Mac Rictor-/- mice displayed minimal alterations relative to the control group (Fig. 7B). This indicates that myeloid Rictor deficiency preserved renal structural integrity. More importantly, Rictor deficiency in macrophages significantly reduced B[a]P-induced SCr and BUN levels (Fig. 7A). This indicates that myeloid Rictor deficiency improved renal filtration function. These results emphasize the critical role of Rictor deficiency in macrophages in mitigating B[a]P-induced renal injury.
Biochemical analyses demonstrated significantly reduced renal MDA content in Mac Rictor-/- mice versus B[a]P-exposed WT mice, with concomitant restoration of SOD and CAT activities and T-AOC to normal controls levels (Fig. 7C). RT-qPCR analysis further demonstrated that restoration of SOD1, SOD2, and CAT mRNA levels to those of the control group (Fig. 7D). This indicates that myeloid Rictor deficiency restores renal antioxidant defenses via transcriptional activation. In kidney tissue, TNF-α protein levels in Mac Rictor-/- mice remained comparable to controls. Similarly, IL-6 and TNF-α mRNA levels showed no significant differences from control values (Fig. 7E). This indicates that the deficiency of myeloid Rictor suppresses B[a]P-triggered inflammatory signaling in the kidney, alleviating the renal inflammatory microenvironment. Furthermore, kidney caspase-3 mRNA expression and serum LDH levels showed no significant differences between Mac Rictor-/- mice and controls (Fig. 7F). This indicates that deficiency of Rictor in macrophages modulates B[a]P-induced cell death through transcriptional and non-transcriptional mechanisms.



Deficiency of Rictor in macrophages mitigates B[a]P-induced renal injury via the mTORC2 signaling axis. (A,B) Renal function biomarkers (SCr and BUN) and representative H&E-stained renal tissue sections (×100) in WT versus Mac Rictor-/- mice. Black triangles: glomerular capsule atrophy with inflammatory infiltration; Blue triangles: disorganized tubular epithelium; Red triangles: tubular vacuolization. (C,D) Comparison of renal tissue oxidative stress markers (MDA, SOD, CAT and T-AOC) and relative mRNA levels of SOD1, SOD2 and CAT. (E,F) Comparison of TNF-α protein content (ELISA, pg/mg) and TNF-α and IL-6 mRNA expression. (G,H) Comparison of apoptosis markers serum LDH and relative mRNA levels of caspase-3. Data are expressed as mean ± SEM (n = 6/group), P < 0.05, P < 0.01, P < 0.001, ****P < 0.0001 compared with control.
Discussions
The widespread environmental distribution of B[a]P represents a public health concern; however, the temporal dynamics and underlying mechanisms of its nephrotoxicity following short-term, high-dose exposure—such as in occupational or accidental scenarios—remain incompletely characterized3,4. As the kidney constitutes a major target in B[a]P’s toxic cascade20, this study utilized a single high dose of 50 mg/kg to elicit robust and detectable pathological phenotypes and molecular alterations within a short period, thereby enabling clear identification of key pathogenic pathways. Our results indicate that this regimen effectively induced time-dependent renal pathological injury and markedly activated the Rictor/mTORC2 signaling pathway. Importantly, inhibition of this pathway significantly attenuated associated oxidative stress, inflammatory responses, and apoptotic processes, underscoring its potential as a therapeutic target in B[a]P-induced renal injury.
Body weight dynamics serve as a sensitive indicator of systemic toxicity in renal injury assessments36. In this study, B[a]P exposure induced a non-monotonic pattern of body weight changes in mice. A significant reduction was observed on day 1, suggesting that acute stress and gastrointestinal disturbances following a single high-dose administration may impair food intake and nutrient absorption19. By days 3 and 7, however, body weight recovered to levels comparable to those of the control group, indicating the potential engagement of compensatory mechanisms—such as modulation of feeding behavior, reprogramming of energy metabolism, or hormonal adjustments—to transiently maintain energy homeostasis37. By day 14, body weight again declined significantly, implying that persistent oxidative stress and inflammatory responses led to cumulative tissue damage beyond the body’s compensatory capacity, ultimately resulting in metabolic dysregulation38. This temporal progression outlines the dynamic process of B[a]P toxicity, from acute stress and compensatory adaptation to decompensated failure, providing key phenotypic evidence for its toxic effects.The findings also demonstrate that 3-day B[a]P exposure induces time-dependent renal damage, characterized by significantly elevated Scr, BUN, urinary urea nitrogen, as well as histopathological alterations. This temporal pathological pattern not only corroborates previous studies16,39 but extends beyond chronic toxicity paradigms, advancing early biomarker screening for chemical pollutants.
Oxidative stress-induced kidney injury is directly associated with elevated levels of ROS. MDA, as a lipid peroxidation marker, reflects the degree of oxidative damage, whereas T-AOC, SOD, and CAT collectively characterize the body’s antioxidant defense capacity40. Short-term exposure to B[a]P induces direct ROS generation through metabolic activation, causing immediate oxidative stress that partially activates SOD/CAT compensatory mechanisms. Long-term exposure, however, leads to progressive ROS accumulation, triggering mitochondrial membrane depolarization and DNA damage6,41. This study confirmed that 3-day B[a]P exposure significantly increased renal MDA levels, decreased T-AOC, and suppressed both the activity and gene expression of SOD and CAT, which was consistent with prior reports. These results indicate that B[a]P directly induces lipid peroxidation by inhibiting antioxidant defenses.
Enhanced oxidative stress activates pro-inflammatory signaling pathways including NF-κB and PI3-K/AKT, and upregulates the expression of inflammatory factors such as TNF-α and IL-6, thereby establishing an amplified inflammatory cascade42,43. Thus, inflammatory response is also a core mechanism of nephrotoxicity, exacerbating renal injury through amplified pro-inflammatory cascades and immune cell infiltration44. Previous studies demonstrated that 5- and 14-day doxorubicin (DOX) treatment significantly increases renal expression of pro-inflammatory mediators including IL-6, TNF-α, NLRP3, caspase-1, and IL-1β45,46. This study confirmed significant upregulation of TNF-α and IL-6 expression both at transcriptional and protein levels in renal tissues following 7-day B[a]P exposure, with concomitant elevation of NOS expression, which was consistent with prior reports with DOX-induced nephrotoxicity. These results establish TNF-α and IL-6 as critical mediators of B[a]P-induced renal inflammation.
Furthermore, excessive inflammation coupled with oxidative stress elevates the level of apoptosis47. ROS trigger caspase-3 activation via the p53-dependent mitochondrial apoptotic pathway, disrupting membrane integrity by cleaving cytoskeletal proteins and inducing LDH release33,48. At the same time, excess nitric oxide (NO) produced by NOS can also directly induce apoptotic signaling through nitrosative modifications49. Consequently, apoptosis constitutes a significant pathway for inducing damage at the organismal level. Nigro et al.50 found that high doses of B[a]P administered intraperitoneally for 7 days significantly induced apoptosis in Anguilla anguilla erythrocytes. We further demonstrated that 7-day B[a]P exposure significantly upregulated caspase-3 (gene/protein) expression and elevated LDH release in renal tissues, suggesting B[a]P-mediated renal injury occurs via apoptotic pathways.
Previous studies have established that short-term, high-dose exposure to B[a]P induces multi-organ toxicity, primarily mediated by oxidative stress, inflammatory responses, and apoptosis (Table 1). The temporal sequence observed in this study—early oxidative stress in the kidneys (3 days post-exposure) followed by inflammation and apoptosis (7–14 days)—reflects both shared mechanisms and organ-specific characteristics compared to other target organs. Notably, renal oxidative stress in our model (50 mg/kg, oral, 3 days) manifested earlier than pulmonary toxicity induced by a higher B[a]P dose (125 mg/kg, oral, 7 days) reported by Shahid et al.51. This difference suggests that B[a]P-induced toxicity is likely modulated by multiple determinants, including exposure dosage, tissue susceptibility, and critical temporal windows of response.
The observed “oxidative stress–inflammation–apoptosis” axis is likely mediated by activation of the Rictor/mTORC2 signaling pathway. Reactive oxygen species (ROS) derived from B[a]P metabolism not only act as early mediators of tissue injury but may also directly facilitate mTORC2 assembly and activation via oxidative post-translational modifications56,57. Furthermore, mTORC2 activates its downstream effectors, AKT1 and PKC-ζ. The ensuing activation of AKT1 can subsequently enhance intracellular ROS production by modulating mitochondrial function and suppressing Foxo transcription factors58. Simultaneously, activated PKC-ζ contributes to ROS generation by modulating NADPH oxidase activity59. This process creates a feed-forward cycle wherein ROS-driven mTORC2 activation increases AKT1/PKC-ζ signaling, which further elevates ROS production, thereby creating a self-amplifying loop that perpetuates oxidative damage. Supporting the pathophysiological relevance of this loop, Liu et al.28 reported a similar mechanism in cadmium-induced neurotoxicity, where mTORC2 activation enhanced Akt/Erk1/2 signaling alongside elevated ROS and apoptosis. Wang et al.30 demonstrated that TiO2 nanoparticles induce mitochondrial hyperpolarization, triggering ROS bursts that overactivate mTORC2, disrupt intercellular junctions, and promote cytoskeletal disassembly and apoptosis. In addition, Yin et al.60 showed that NiCl₂-induced renal injury involves autophagic regulation mediated by crosstalk between AMPK and PI3K/AKT/mTOR pathways. In this study, molecular docking further revealed that B[a]P binds directly to Rictor with high affinity, suggesting a structural basis for mTORC2 modulation. GO enrichment analysis showed significant overrepresentation of key genes in categories such as “protein kinase binding,” “serine/threonine kinase activity,” and “serine/threonine protein kinase complex”—functional terms consistent with the known roles of mTORC2 effectors AKT1 and PKC-ζ. This was further validated by RT‑qPCR, which confirmed marked upregulation of AKT1 and PKC‑ζ transcription after B[a]P exposure.Collectively, these findings support a model wherein B[a]P-induced oxidative stress not only initiates renal injury but also engages a Rictor/mTORC2-driven positive feedback loop, sustaining and amplifying pathological progression in the kidney (Fig. 8).

Schematic illustration of the activation mechanism of the Rictor/mTORC2 pathway.
The mTOR pathway, particularly mTORC2, has emerged as a key player in multiple pathological processes—from renal injury to colorectal cancer61, hepatocellular carci*noma62, and Alzheimer’s disease63. In a PTEN-deficient epilepsy model, mTOR pathway inhibition effectively attenuates disease symptoms64. Navroop et al.65 reported that loss of phosphatase and PTEN function synergistically induces hyperactivation of mTORC2, which underlies disease-related phenotypes in human neurons and cortical organoids. Importantly, these abnormalities were reversed by mTORC2 inhibition in PTEN/Rictor mutant models, confirming the pivotal role of mTORC2 signaling in this pathogenic process. Furthermore, The mTORC2 pathway plays a pivotal role in modulating inflammation and oxidative stress. In a yeast polysaccharide-induced non-septic rat model, Sabrie et al.22 showed that the selective mTORC2 inhibitor JR-AB2-011 specifically suppressed AKT1 phosphorylation at Ser473, leading to marked reductions in ROS accumulation and expression of pro-inflammatory mediators, including iNOS, TNF-α, and IL-1β. He et al.66demonstrated that quercetin and its metabolite 7-dihydroquercetin markedly inhibit LPS-induced phosphorylation of AKT1 (Ser473) and mTOR (Ser2448) in macrophages. This suppression resulted in decreased iNOS/NO production and reduced expression of pro-inflammatory cytokines, including IL-6, IL-1β, and TNF-α. Mechanistically, these compounds exert anti-inflammatory effects by stabilizing the Rictor/mTORC2 complex—thereby modulating the balance between pro-apoptotic and survival signals—and by regulating NOS3-mediated pathways to maintain inflammatory microenvironment homeostasis. Studies by Khalid67 and Weske et al.68 indicate that inhibition of the mTORC2 pathway protects the kidney by reducing ROS accumulation, inflammation, and apoptosis. This protective effect is mediated through the concerted modulation of the PI3K/Akt/mTOR and PKC signaling networks. Zhang et al.69 identified a protective role for mTORC2 in atherosclerosis through macrophage-specific knockout models, where it suppressed FoxO1-mediated NLRP3 inflammasome activation and IL-1β release. Rictor deficiency in macrophages may disrupt intercellular communication by altering their polarization state, thereby reducing the release of pathogenic factors70. This disruption could potentially extend to exosome-mediated signaling, as macrophage-derived exosomes are known to carry specific miRNAs, proteins, and lipids that participate in pathological processes69. Rictor deficiency may lead to alterations in the molecular composition of exosomal cargo, thereby converting their functional role from pathogenic to protective71. Collectively, our work establishes macrophage Rictor/mTORC2 signaling as a key regulator of environmental nephrotoxicity. Genetic ablation of Rictor effectively suppressed pathway activation and limited downstream renal injury, highlighting its potential as a multi-faceted therapeutic target.
Notably, the role of mTORC2 in environmental toxicology is highly context-dependent, influenced by both the specific toxic insult and the cellular microenvironment. For instance, in contrast to the results presented here, Zhu et al.72. reported a protective function of the mTORC2–AKT axis in cadmium-induced nephrotoxicity, where it helped maintain autophagy–apoptosis homeostasis. In this study, B[a]P functioned as a genotoxic stressor. Its metabolic activation induced substantial ROS bursts and DNA damage responses, thereby steering mTORC2 signaling toward pro-inflammatory and pro-apoptotic outcomes. Consequently, Rictor knockout conferred marked renal protection under these conditions.Mechanistically, this protection involves two complementary pathways: first, through suppression of the AKT1 cascade, which downregulates pro-apoptotic factors including caspase-3 and impedes G1/S phase transition; second, via inactivation of the mTORC2–PKC axis, which attenuates both the extrinsic apoptosis pathway and the mitochondrial-dependent apoptotic cascade, while also mitigating the DNA damage response73,74. The observed differences indicate that mTORC2 does not exhibit a fixed biological orientation but rather functions as a signaling hub, the output of which is dynamically shaped by toxicological context and cell type68. This understanding carries important implications for the development of mTORC2-targeted intervention strategies, underscoring the necessity of precise, context-aware regulation in toxicological settings.
This study systematically elucidates the temporal pathological progression of B[a]P-induced nephrotoxicity and reveals the potential key role of the Rictor/mTORC2 signaling pathway in this process. Several limitations should be acknowledged. Specifically, renal levels of B[a]P and its metabolites (e.g., BPDE), CYP enzyme activity, and DNA adduct formation were not directly measured, leaving a gap in toxicokinetic evidence. Future work will incorporate comprehensive toxicokinetic analyses to better correlate external exposure with internal dose and pathological outcomes. In addition, systematic comparisons of male and female mouse responses will be conducted to evaluate the generalizability of our findings and explore the influence of sex-specific factors. Notably, this study identifies the Rictor/mTORC2 pathway as a potential therapeutic target. Previous reports indicate that selective mTORC2 inhibitors, such as JR-AB2-011, can alleviate oxidative stress, inflammation, and apoptosis by modulating AKT/mTOR phosphorylation. Thus, targeting this pathway may represent a promising translational strategy for preventing or treating environmentally induced kidney injury.
Conclusions
This study demonstrates that B[a]P exposure induces time-dependent renal injury in mice by activating the Rictor/mTORC2 pathway, which triggers a cascade of oxidative stress, inflammation, and apoptosis, thereby exacerbating pathological damage in the kidneys. In contrast, myeloid-specific knockout of Rictor reverses these pathological alterations. Our findings reveal, for the first time, the Rictor/mTORC2 pathway as a potential molecular target for therapeutic intervention in B[a]P-induced kidney injury. The B[a]P dose (50 mg/kg) employed here represents a high-level, short-term exposure far exceeding typical human intake, and was used specifically to delineate the mechanisms of nephrotoxicity. Informed by these findings, future work will adopt chronic low-dose exposure models that better reflect real-world scenarios. These studies will extend into broader disease contexts, including neurodegeneration, to deeply investigate the role of the Rictor/mTORC2 pathway within toxic microenvironments. Utilizing cell-type-specific silencing and pharmacological inhibition, we will define the pathway’s dynamic regulatory functions and evaluate its translational relevance to human health risk assessment.
Data availability
All data generated or analyzed during this study are included in this article. The data is available. The data that support the findings of this study are available from the corresponding author, Xue-Yun Ma, upon reasonable request (Email: 476643683@qq.com).
References
Li, R. et al. Distribution of the soil PAHs and health risk influenced by coal usage processes in Taiyuan City, Northern China. IJERPH 17 (17), 6319 (2020).
Nwaozuzu, C. C., Abah, S. O. & Patrick-Iwuanyanwu, K. C. Polycyclic aromatic hydrocarbon in community drinking water, Nsisioken, nigeria: source and health risk assessment. Environ. Anal. Health Toxicol. 39 (2), e2024015 (2024).
Bukowska, B., Mokra, K. & Michałowicz, J. Benzo[a]pyrene—Environmental Occurrence, human Exposure, and mechanisms of toxicity. IJMS 23 (11), 6348 (2022).
Song, S. et al. Assessing the contribution of global wildfire biomass burning to bap contamination in the Arctic. Environ. Sci. Ecotechnology. 14, 100232 (2023).
Alghamdi, M. A., Hassan, S. K., Alzahrani, N. A., Al Sharif, M. Y. & Khoder, M. I. Classroom Dust-Bound polycyclic aromatic hydrocarbons in Jeddah primary Schools, Saudi arabia: Level, characteristics and health risk assessment. IJERPH 17 (8), 2779 (2020).
Wannhoff, A., Bölck, B., Kübler, A. C., Bloch, W. & Reuther, T. Oxidative and nitrosative stress and apoptosis in oral mucosa cells after ex vivo exposure to lead and benzo[a]pyrene. Toxicol. In Vitro. 27 (2), 915–921 (2013).
Hummel, J. M. et al. Pharmacokinetics of [14 C]-Benzo[a]pyrene (BaP) in humans: impact of Co-Administration of smoked salmon and bap dietary restriction. Food Chem. Toxicol. 115, 136–147 (2018).
He, Y., Yang, C., He, W. & Xu, F. Nationwide health risk assessment of juvenile exposure to polycyclic aromatic hydrocarbons (PAHs) in the water body of Chinese lakes. Sci. Total Environ. 723, 138099 (2020).
Cao, X. et al. Seasonal variability in multimedia transport and fate of benzo[a]pyrene (BaP) affected by Climatic factors. Environ. Pollut. 292, 118404 (2022).
Zou, H. et al. AHR-mediated DNA damage contributes to BaP-induced cardiac malformations in zebrafish. Sci. Total Environ. 906, 167636 (2024).
Hur, D., Jeon, J. K. & Hong, S. Analysis of immune gene expression modulated by benzo[a]pyrene in head kidney of Olive flounder (Paralichthys olivaceus). Comp. Biochem. Physiol. B: Biochem. Mol. Biol. 165 (1), 49–57 (2013).
Liu, A. et al. BPDE-DNA adduct formation and alterations of mRNA, protein, and DNA methylation of CYP1A1, GSTP1, and GSTM1 induced by benzo[a]pyrene and the intervention of aspirin in mice. Environ. Sci. Pollut Res. 30 (48), 106549–106561 (2023).
Chen, J. et al. DBP and bap co-exposure induces kidney injury via promoting pyroptosis of renal tubular epithelial cells in rats. Chemosphere 314, 137714 (2023).
Chen, W. et al. Long-term co-exposure DBP and bap causes imbalance in liver macrophages polarization via activation of Notch signaling regulated by miR-34a-5p in rats. Chemico-Biol. Interact. 359, 109919 (2022).
Shi, H., Liu, J. & Gao, H. Benzo(α)pyrene induces oxidative stress and inflammation in human vascular endothelial cells through AhR and NF-κB pathways. Microvasc. Res. 137, 104179 (2021).
Ge, J. et al. Secoisolariciresinol diglucoside mitigates benzo[a]pyrene-induced liver and kidney toxicity in mice via miR-101a/MKP-1-mediated p38 and ERK pathway. Food Chem. Toxicol. 159, 112733 (2022).
Shahid, A. et al. Attenuation of genotoxicity, oxidative stress, apoptosis and inflammation by Rutin in benzo(a)pyrene exposed lungs of mice: plausible role of NF-κB, TNF-α and Bcl-2. J. Complement. Integr. Med. 13 (1), 17–29 (2016).
Shahid, A. et al. Modulatory effects of Catechin hydrate against genotoxicity, oxidative stress, inflammation and apoptosis induced by benzo(a)pyrene in mice. Food Chem. Toxicol. 92, 64–74 (2016).
Deng, C. et al. Acute benzo[a]pyrene treatment causes different antioxidant response and DNA damage in liver, lung, brain, stomach and kidney. Heliyon 4 (11), e00898 (2018).
Woo, S. J. Effects of benzo[a]pyrene exposure on black rockfish (Sebastes schlegelii): EROD activity, CYP1A protein, and immunohistochemical and histopathological alterations. Environ. Sci. Pollut Res. 29 (3), 4033–4043 (2022).
Adedara, I. A., Daramola, Y. M., Dagunduro, J. O., Aiyegbusi, M. A. & Farombi, E. O. Renoprotection of Kolaviron against benzo (A) pyrene-induced renal toxicity in rats. Ren. Fail. 37 (3), 497–504 (2015).
Sabrie, Z. et al. Protection by selective mTORC2 Inhibition of Zymosan-induced hypotension and systemic inflammation mediated via IKKα/IκB-α/NF-κB activation. Prostaglandins Other Lipid mediat. 175, 106918 (2024).
Weske, S. et al. Intracellular sphingosine-1‐phosphate induces lipolysis through direct activation of protein Kinase C zeta. FASEB J. 39 (7). https://doi.org/10.1096/fj.202403272R (2025).
Ren, J. et al. Rictor/mammalian target of Rapamycin complex 2 promotes macrophage activation and kidney fibrosis: rictor in macrophage activation and kidney fibrosis. J. Pathol. 242 (4), 488–499 (2017).
Li, J. et al. Rictor/mTORC2 signaling mediates TGFβ1-induced fibroblast activation and kidney fibrosis. Kidney Int. 88 (3), 515–527 (2015).
Feng, D. et al. Rictor/mTORC2 signalling contributes to renal vascular endothelial-to‐mesenchymal transition and renal allograft interstitial fibrosis by regulating BNIP3‐mediated mitophagy. Clin. Translational Med. 14 (5), e1686 (2024).
Dai, H. et al. Rictor deficiency in dendritic cells exacerbates acute kidney injury. Kidney Int. 94 (5), 951–963 (2018).
Liu, C. et al. Neuroprotection of Resveratrol against cadmium-poisoning acts through dual Inhibition of mTORC1/2 signaling. Neuropharmacology 219, 109236 (2022).
Chen, H. et al. M TORC 2 controls Th9 polarization and allergic airway inflammation. Allergy 72 (10), 1510–1520 (2017).
Wang, L. M. et al. TiO2 nanoparticles affect spermatogenesis and adhesion junctions via the ROS-mediated mTOR signalling pathway in eriocheir sinensis testes. Environ. Pollut. 331, 121952 (2023).
Zhang, L. et al. Apoptosis and blood-testis barrier disruption during male reproductive dysfunction induced by PAHs of different molecular weights. Environ. Pollut. 300, 118959 (2022).
Kumari, S., Bahinipati, J., Pradhan, T. & Sahoo, D. Comparison of test performance of biochemical parameters in semiautomatic method and fully automatic analyzer method. J. Family Med. Prim. Care. 9 (8), 3994 (2020).
Jantas-Skotniczna, D., Kajta, M. & Lasoń, W. Memantine attenuates staurosporine-induced activation of caspase-3 and LDH release in mouse primary neuronal cultures. Brain Res. 1069 (1), 145–153 (2006).
Ratnayake, W. S., Apostolatos, A. H., Ostrov, D. A. & Acevedo-Duncan, M. Two novel atypical PKC inhibitors; ACPD and DNDA effectively mitigate cell proliferation and epithelial to mesenchymal transition of metastatic melanoma while inducing apoptosis. Int. J. Oncol. 51 (5), 1370–1382 (2017).
Lin, H. Y. H. et al. Tubular mitochondrial AKT1 is activated during ischemia reperfusion injury and has a critical role in predisposition to chronic kidney disease. Kidney Int. 99 (4), 870–884 (2021).
Tanaka, S. et al. Body and major organ weights of A/J-Chr 11SM consomic mice. Exp. Anim. 58 (4), 357–361 (2009).
Kong, D., Ma, H., Zhu, C., Hao, Y. & Li, C. Unraveling the toxicity response and metabolic compensation mechanism of Tannic acid-Cr(III) complex on Alga Raphidocelis subcapitata. Sci. Total Environ. 930, 172034 (2024).
He, S. et al. Isoorientin attenuates benzo[a]pyrene-induced colonic injury and gut microbiota disorders in mice. Food Res. Int. 126, 108599 (2019).
Owumi, S. E., Adeniyi, G. & Oyelere, A. K. The modulatory effect of taurine on benzo (a) pyrene-induced hepatorenal toxicity. Toxicol. Res. 10 (3), 389–398 (2021).
Zhang, K. et al. Luteolin alleviates Cadmium-Induced kidney injury by inhibiting oxidative DNA damage and repairing autophagic flux Blockade in chickens. Antioxidants 13 (5), 525 (2024).
Qiu, L. et al. Unraveling the protective role of Nrf2 in molluscs: insights into mitochondrial and apoptosis pathways in the defense against Bap-induced oxidative stress. Aquat. Toxicol. 264, 106728 (2023).
Cui, Q., Chen, F. Y., Chen, H. Y., Peng, H. & Wang, K. J. Benzo[a]pyrene (BaP) exposure generates persistent reactive oxygen species (ROS) to inhibit the NF-κB pathway in Medaka (Oryzias melastigma). Environ. Pollut. 251, 502–509 (2019).
Wang, J. et al. Astragaloside IV attenuated 3,4-Benzopyrene-Induced abdominal aortic aneurysm by ameliorating Macrophage-Mediated inflammation. Front. Pharmacol. 9, 496 (2018).
Jin, X. et al. An herbal formulation Shenshuaifu granule alleviates cisplatin-induced nephrotoxicity by suppressing inflammation and apoptosis through Inhibition of the TLR4/MyD88/NF-κB pathway. J. Ethnopharmacol. 306, 116168 (2023).
Wu, Q. et al. Apigenin ameliorates doxorubicin-induced renal injury via Inhibition of oxidative stress and inflammation. Biomed. Pharmacother. 137, 111308 (2021).
Arunachalam, S. et al. α-Bisabolol attenuates doxorubicin induced renal toxicity by modulating NF-κB/MAPK signaling and Caspase-Dependent apoptosis in rats. IJMS 23 (18), 10528 (2022).
Kim, J. et al. Thiobarbiturate-Derived compound MHY1025 alleviates renal fibrosis by modulating oxidative Stress, epithelial Inflammation, and fibroblast activation. Antioxidants 12 (11), 1947 (2023).
Li, X. Q. et al. Inhibiting aberrant p53-PUMA feedback loop activation attenuates ischaemia reperfusion-induced neuroapoptosis and neuroinflammation in rats by downregulating caspase 3 and the NF-κB cytokine pathway. J. Neuroinflammation. 15 (1), 250 (2018).
Young Park, S., Jeong, Y. J., Kim, S. H., Jung, J. Y. & Kim, W. J. Epigallocatechin gallate protects against nitric oxide-induced apoptosis via scavenging ROS and modulating the Bcl-2 family in human dental pulp cells. J. Toxicol. Sci. 38 (3), 371–378 (2013).
Nigro, M., Frenzilli, G., Scarcelli, V., Gorbi, S. & Regoli, F. Induction of DNA strand breakage and apoptosis in the eel Anguilla Anguilla. Mar. Environ. Res. 54 (3–5), 517–520 (2002).
Shahid, A. et al. Methanolic bark extract of Acacia Catechu ameliorates benzo(a)pyrene induced lung toxicity by abrogation of oxidative stress, inflammation, and apoptosis in mice. Environ. Toxicol. 32 (5), 1566–1577 (2017).
Gao, M. et al. Induction of oxidative stress and DNA damage in cervix in acute treatment with benzo[a]pyrene. Mutat. Research/Genetic Toxicol. Environ. Mutagen. 719 (1–2), 52–59 (2011).
Barnwal, P. et al. Benzo(a)pyrene induces lung toxicity and inflammation in mice: prevention by carvacrol. Hum. Exp. Toxicol. 37 (7), 752–761 (2018).
Delgado-Roche, L. et al. Chemoprotective effects of Ulva lactuca (green seaweed) aqueous‐ethanolic extract against subchronic exposure to benzo(a)pyrene by CYP1A1 Inhibition in mice. Phytother. Res. 33 (4), 958–967 (2019).
Khattab, S. A., Hussien, W. F., Raafat, N. & Ahmed Alaa El-Din, E. Modulatory effects of Catechin hydrate on benzo[a]pyrene-induced nephrotoxicity in adult male albino rats. Toxicol. Res. 10 (3), 542–550 (2021).
Sun, N., Hu, S., Zhao, X., Gao, C. & Liu, R. Polystyrene nanoplastics and benzo[a]pyrene co-exposure differentially impacts earthworm intra- and extracellular lysozyme. Int. J. Biol. Macromol. 321, 146255 (2025).
Wang, Z. et al. 6PPD induces apoptosis and autophagy in SH-SY5Y cells via ROS-mediated PI3K/AKT/mTOR pathway: in vitro and in Silico approaches. Toxicology 513, 154091 (2025).
Iskandar, K. et al. A novel MTORC2-AKT-ROS axis triggers mitofission and mitophagy-associated execution of colorectal cancer cells upon drug-induced activation of mutant KRAS. Autophagy 20 (6), 1418–1441 (2024).
Martin-Perez, M. et al. PKC downregulation upon Rapamycin treatment attenuates mitochondrial disease. Nat. Metab. 2 (12), 1472–1481 (2020).
Yin, H. et al. Nickel induces autophagy via PI3K/AKT/mTOR and AMPK pathways in mouse kidney. Ecotoxicol. Environ. Saf. 223, 112583 (2021).
Sane, S. et al. UBXN2A suppresses the Rictor-mTORC2 signaling pathway, an established tumorigenic pathway in human colorectal cancer. Oncogene 42 (21), 1763–1776 (2023).
Zhang, H. et al. Targeting mTORC2/HDAC3 inhibits stemness of liver cancer cells against glutamine starvation. Adv. Sci. 9 (20), 2103887 (2022).
Lee, H. K. et al. mTORC2 (Rictor) in Alzheimer’s disease and reversal of Amyloid-β expression-induced insulin resistance and toxicity in rat primary cortical neurons. JAD 56 (3), 1015–36. (2017).
Cullen, E. R., Safari, M., Mittelstadt, I. & Weston, M. C. Hyperactivity of mTORC1- and mTORC2-dependent signaling mediates epilepsy downstream of somatic PTEN loss. eLife 12, RP91323 (2024).
Dhaliwal, N. K. et al. Synergistic hyperactivation of both mTORC1 and mTORC2 underlies the neural abnormalities of PTEN-deficient human neurons and cortical organoids. Cell. Rep. 43 (5), 114173 (2024).
He, X. et al. Assessment of the anti-inflammatory mechanism of Quercetin 3,7‐dirhamnoside using an integrated Pharmacology strategy. Chem. Biol. Drug Des. 102 (6), 1534–1552 (2023).
Khalid, K. M., Ratnayake, W. S., Apostolatos, C. A. & Acevedo-Duncan, M. Dual Inhibition of atypical PKC signaling and PI3K/Akt signaling dysregulates c-Myc to induce apoptosis in clear cell renal cell carcinoma. Front. Oncol. 13, 1213715 (2024).
Weske, S. et al. Intracellular Sphingosine-1‐Phosphate induces lipolysis through direct activation of protein kinase C zeta. FASEB J. 39 (7), e70528 (2025).
Zhang, X. et al. Loss of macrophage mTORC2 drives atherosclerosis via FoxO1 and IL-1β signaling. Circul. Res. 133 (3), 200–219 (2023).
Wu, M. M. et al. Dioscin ameliorates murine ulcerative colitis by regulating macrophage polarization. Pharmacol. Res. 172, 105796 (2021).
Ni, B. et al. Rictor ameliorates acute Antibody-Mediated rejection following kidney transplantation by suppressing macrophage M1 polarization through p65‐NLRP3 axis. Adv. Sci. 12 (34), e17119 (2025).
Zhu, J. et al. mTORC1 and mTORC2 Co-Protect against Cadmium-Induced renal tubular epithelial cell apoptosis and acute kidney injury by regulating protein kinase B. J. Agric. Food Chem. 72 (36), 19667–19679 (2024).
Zhao, H. M. et al. Erzhi Pill® protected experimental liver injury against apoptosis via the PI3K/Akt/Raptor/Rictor pathway. Front. Pharmacol. 9, 283 (2018).
Fu, Y., Xiang, Y., Zha, J., Chen, G. & Dong, Z. Enhanced STAT3/PIK3R1/mTOR signaling triggers tubular cell inflammation and apoptosis in septic-induced acute kidney injury: implications for therapeutic intervention. Clin. Sci. 138 (6), 351–369 (2024).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: biological systems database as a model of the real world. Nucleic Acids Res. 53 (D1), D672–D677 (2025).
Acknowledgements
The authors wish to thank East China Normal University (ECNU) public platform for the support of this study.
Funding
The current work was supported by the Research and Development Fund of East China Normal University (No. 11300-544000-02731). The authors sincerely thank East China Normal University (ECNU) public platform for its full support.
Author information
Authors and Affiliations
Contributions
Ying Qu: Investigation, Data curation, Writing—Original draft preparation; Jianqiu Han: Investigation,; Data curation ,Writing—Original draft preparation; Yuanrong Zhu: Investigation, Data curation; Ya-Lei Qi: Investigation, Data curation; Tengfei Liu: Investigation, Validation; Yongmei Li: Investigation, Validation; Yanjia Zhang: Investigation, Validation; Honghui Han: Supervision, Writing—Reviewing and Editing. Juan Tan: Conceptualization, Supervision and Writing—Reviewing. Xueyun Ma: Conceptualization, Data curation, Writing—Reviewing and Editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Animal care and use
The authors confirm that all experimental procedures in this study were strictly adherence to the ARRIVE guidelines. Animal care and use procedures in this study strictly adhered to the relevant institutional and national guidelines for animal care and use. Specifically, the experimental protocol was oversight and approved by the animal care and use committee of East China Normal University (Approval Number: m20250302). All efforts were made to minimize animal suffering and ensure compliance with the principles of replacement, reduction, and refinement (3Rs) in animal research. In cases where an equivalent ethics committee oversaw the study, the approval documentation has been retained and is available upon request. A scanned copy of the approval document is also attached.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Han, JQ., Qu, Y., Zhu, Yr. et al. Rictor/mTORC2 signaling pathway mediates Benzo[a]pyrene-induced renal injury. Sci Rep 16, 71 (2026). https://doi.org/10.1038/s41598-025-29025-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-29025-y
