
Abstract
Migrasome has recently been reported to regulate cell–cell communication in tumor progression. But the function of migrasome in CRC, especially its prognostic potential and association with long non-coding RNAs and tumor microenvironment has yet to be fully explored. Migrasome-related lncRNAs were identified based on transcriptome data from 650 CRC patients in the TCGA COREAD cohort. A prognostic signature was developed via the least absolute shrinkage and selection operator algorithm and Cox regression. The multimodal predicting model for prognosis of CRC patients was constructed and validated through Kaplan–Meier survival, ROC curve analysis, and nomogram construction. Patients were divided into high- and low-risk groups according to their prognostic signature and functional enrichment, immune cell infiltration, immunotherapy efficacy, and drug sensitivity analyses were performed to characterize their variation. Ten migrasome-related lncRNAs were identified and used to construct a prognostic risk score that effectively stratified patients into high- and low-risk groups, demonstrating significant differences in overall survival (P < 0.001) and survival-related ROC curve analysis. A nomogram based on the multimodal predicting model was constructed with robust calibration curves. The transcriptional variation between high-risk and low-risk patients were mainly associated with signaling receptor activator activity, receptor ligand activity and cytokine-cytokine receptor interaction. High-risk patients exhibited reduced immune cell infiltration and higher potential for immune escape. In addition, drug sensitivity screening revealed that high-risk patients were more likely to resist current targeted drugs including PLX-4720 and JAK-8517 than low-risk patients. This study identifies a novel migrasome-related lncRNA signature as a reliable prognostic tool for CRC, highlighting its potential in patient stratification and personalized therapy.
Similar content being viewed by others


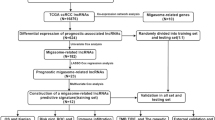
Introduction
Colorectal cancer (CRC) is one of the most prevalent malignancies worldwide, characterized by its high morbidity and mortality rates1,2. Despite the diagnostic and therapeutic strategies having achieved significant advancements in recent years3, the long-time prognosis of CRC patients, especially those with high-risk characteristics, remains unsatisfactory4. It’s important to develop biomarkers which can precisely stratify CRC patients based on their potential malignancy and treatment response5.
Long non-coding RNAs (lncRNAs), a class of transcripts longer than 200 nucleotides while rarely coded protein, have garnered increasing attention for recent cancer research6. LncRNAs played a vital role in various biological processes, including gene regulation, immune modulation, and tumor progression7,8. Previous studies have reported that specific lncRNAs have been implicated in CRC progression by regulating immune cell infiltration9 and promoting tumor metastasis10, underscoring their potential as prognostic biomarkers and therapeutic targets.
Migrasome, a newly discovered type of extracellular vesicle, mediates intercellular communication by transporting bioactive molecules during cell migration11, providing a novel perspective on the mechanisms underlying tumor progression and immune evasion in the tumor microenvironment (TME)12,13. TME has been proved to be essential in shaping CRC progression by regulating immune cell infiltration to affect immunotherapy14,15 and communicating with stromal cells such as cancer-associated fibroblast and macrophages16,17. However, the interplay between migrasome and CRC remained largely unexplored. As key regulators of gene expression, lncRNAs can be stably detected via routine transcriptome sequencing, which allowed preliminary exploration for migrasome functions based on existing data and clinical translation.
In this study, we mainly identified and validated a migrasome-related lncRNA signature for prognosis prediction in CRC, with a particular focus on its implications for tumor immunity and targeted therapy. By elucidating the roles of these lncRNAs in CRC progression, immune regulation, and therapeutic resistance, our findings may pave the way for the development of novel biomarkers for tumor stratification and therapeutic strategies for CRC patients.
Method
Patients and data collection
All the patients enrolled in this study were derive from TCGA COAD and READ cohorts (https://portal.gdc.cancer.gov/), which contained 650 patients with pathologically confirmed colorectal adenocarcinoma. To identify migrasome-related transcriptional variation, the RNA-sequence data and corresponding clinical information of each patient were acquired from TCGA database.
Identification of migrasome-related lncRNAs
The lncRNA expression matrix was screened from the RNA-sequence data based on reference annotation for the human genome from GENCODE. 11 migrasome-related genes were identified from previously published studies (Supplementary Table 1)18,19,20,21,22,23. Then, to identify migrasome-related lncRNAs, correlation analysis was performed for each lncRNA with migrasome-related genes. The lncRNAs with correlation coefficient > 0.6 and P < 0.001 were determined as migrasome-related lncRNAs. Subsequently, we used limma to identify differentially expressed migrasome-related lncRNAs between tumoral and normal samples in TCGA COREAD cohort in R sofware (version 4.0.5). The cutoff was |log2 fold change (FC)|≥ 1.0 and false discovery rate (FDR) < 0.05.
Construction and validation of the migrasome-related lncRNAs prognostic signature
When model training, the TCGA COREAD cohort was divided into training and test sets in a 7:3 ratio, with the training set employed for developing the prognosis prediction model and the testing set utilized to independently assess the prediction performance. To construct a robust signature for predicting prognosis of CRC patients, the differentially expressed migrasome-related lncRNAs were firstly applied into univariate Cox regression analysis and lncRNAs with P < 0.05 were further enrolled into the least absolute shrinkage and selection operator (LASSO) model to reduce overfitting (Supplementary Method). Finally, the independent prognostic indicators were identified for model construction via multivariate Cox regression analysis based on the Akaike information criterion (AIC) value. The prognostic risk score was linearly calculated based on the expression level of lncRNAs and their corresponding coefficients.
The patients were divided into the high-risk and low-risk groups based on the median of risk scores. The Kaplan–Meier analysis was performed to demonstrate the prognostic implications of the developed model based on migrasome-related lncRNAs. Time-dependent ROC curve analysis was also utilized by using “timeROC” R packages to assess the prediction accuracy of the risk score in one, three and five years, respectively.
The predictive nomogram
The clinical characteristics including age, gender, stage was integrated with our risk scores for univariate and multivariate Cox regression analyses to evaluate the independent prognostic ability of each characteristic and construct the multi-parameter predicting model. To facilitate the clinical application of the multi-parameter predicting model we developed, a nomogram was further constructed for prediction of overall survival (OS) of CRC patients. The calibration curves and concordance index (C-index) were performed to assess the robustness of the nomogram.
Functional enrichment analysis
To clarify the genomic variation between patients in the high-risk and low-risk groups, the differentially expression analysis of transcriptome array was performed to screen for differentially expressed genes (DEGs) (|logFC|> 1 and FDR < 0.05). The enrichment analysis for well-known pathways including Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG)24,25,26 based on DEGs were conducted to illustrate the functions and biological pathways linked to the migrasome-related lncRNAs. In addition, the Gene Set enrichment Analysis (GSEA) was performed to further investigate the potential signaling pathways in both groups.
Immune cell infiltration and immunity analysis
As previously reported, migrasome was associated with immune cell infiltration in the tumor microenvironment20,27. Therefore, we also explored the relationship between immune cells compositions and migrasome-related risk scores. The immune cells compositions were determined by CIBERSORT for each patient and the immune-related function of patients in TCGA CRC cohort were quantified via ssGSEA enrichment of well-known pathways (Supplementary Method). The Wilcoxon test was used to analyze the differential expression of immune checkpoint genes between the two groups. In addition, the potential of immune evasion to predict the immune checkpoint blockade (ICB) response was also generated by tumor immune dysfunction and exclusion (TIDE) tool and compared between low-risk and high-risk groups.
Drug sensitivity analysis
Recent studies have proposed various drugs which may exerts potentially antitumor efficacy against CRC including traditional chemotherapy and novel targeted drugs. To assess the predictive ability of our model for drug sensitivity in CRC, the anticancer drug dataset was acquired from Genomics of Drug Sensitivity in Cancer (GDSC, https://www.cancerrxgene. org/) and the relationship of IC50 values for various drugs between two risk groups was calculate using the “oncoPredict” package.
Statistical analysis
Statistical analysis was performed using R sofware (version 4.0.5). P < 0.05 was considered statistically significant.
Results
Identification of migrasome-related differentially expressed lncRNAs
Co-expression analysis revealed 1857 migrasome-related lncRNAs which were highly correlated to the expression level of 10 previously reported migrasome-related genes in 650 CRC samples (Fig. 1A & Supplementary Table 2). Figure 1B presented the differential expression of these migrasome-related lncRNAs between normal and tumor samples. Overall, 463 lncRNAs were up-regulated while 37 lncRNAs were down-regulated in tumors (Fig. 1C & Supplementary Table 3).

Identification of migrasome-related differentially expressed lncRNAs and screening of prognostic lncRNAs (A). Co-expression of lncRNAs that were highly correlated to 10 migrasome-related genes; (B) Volcano plot of 463 up-regulated and 37 down-regulated migrasome-related lncRNAs between normal and tumor samples; (C). Heatmap of differential expressed genes between normal and tumor samples; (D). LASSO coefficient profiles of the ten prognostic LncRNAs; E. Plots of cross-validation partial likelihood deviance of the LASSO model; F. Correlation matrix of the selected 10 prognostic lncRNAs with migrasome-related genes.
To avoid overfitting, 434 migrasome-related lncRNAs which potentially associated with prognosis of CRC were initially screened by the univariable Cox model from the training set. Then we used the LASSO Cox regression model to further select 12 lncRNAs that could predict survival outcomes of CRC more robustly (Fig. 1D, E). Finally, the multivariate Cox regression analysis was performed to avoid covariance of the predicting model and successfully identified 10 prognostic migrasome-related lncRNAs (AC012313.5, AC083967.1, AC019118.1, TNFRSF10A-AS1, ZEB1-AS1, LINC02516, AC114730.3, JARID2-AS1, AC020558.2, and AC016831.4) to build the risk model. Combining the corresponding coefficients, the risk score was computed as follow: AC012313.5 × (-0.99453) + AC083967.1 × (0.72723) + AC019118.1 × (1.21762) + TNFRSF10A-AS1 × (-0.27760) + ZEB1-AS1 × (0.60705) + LINC02516 × (-4.93759) + AC114730.3 × (0.50692) + JARID2-AS1 × (-0.47708) + AC020558.2 × (0.32755) + AC016831.4 × (-2.54820). Figure 1F showed that the selected 10 prognostic lncRNAs were closely related to expression levels of migrasome-related genes.
Development and validation of prognostic signature based on migrasome-related lncRNAs
650 CRC patients were divided into high-risk and low-risk groups according to their respective risk scores. The Principal Component Analysis (PCA) indicated that compared with the whole expression profiles, the selected migrasome-related lncRNAs could better discriminate patients from high-risk and low-risk subsets (Fig. 2A). The Kaplan–Meier analysis showed that patients with higher risk scores had significantly worse overall survival than those with lower risk scores in the training and testing sets (P < 0.001, Fig. 2B, C). The analysis of progression-free survival also confirmed the prognostic efficacy of our risk scores for CRC patients (P < 0.001, Fig. 2D). The risk score distribution map revealed that the risk scores were positively correlated with the mortality rate among CRC patients and the expression profiles of 10 prognostic migrasome-related lncRNAs were various between high-risk and low-risk groups (Fig. 2E). An independent external validation was performed based on the CRC cohort from GEO dataset (GSE72970), which showed that patients in the high-risk group had significantly shorter OS (P < 0.001) compared to the low-risk group (Supplementary Fig. 1A). In addition, we preformed the survival-related ROC curve analysis to assess the sensitivity and specificity of the risk score, which demonstrated that the area under the ROC curves (AUC) of the migrasome-related risk score for 1-, 3- and 5-year OS was 0.700, 0.738, and 0.782, respectively (Fig. 2F). The survival-related ROC curve analysis also achieved a AUC of 0.633 in the GSE72970 cohort (Supplementary Fig. 1B).

Development and validation of the migrasome-related lncRNA prognostic signature (A). PCA analysis of CRC patients based on the whole expression profiles and selected migrasome-related lncRNAs; (B and C). The Kaplan–Meier analysis of overall survival for patients with high risk scores and low risk scores in the training and testing sets; (D). The Kaplan–Meier analysis of progression-free survival for patients with high risk scores and low risk scores; (E). The risk score distribution map and the expression profiles of 10 prognostic migrasome-related lncRNAs among high-risk and low-risk patients; (F). The survival-related ROC curve analysis of patients based on the migrasome-related signature.
Construction of the multimodal model and nomogram for predicting long-term prognosis
Based on the clinical profiles acquired from TCGA COREAD dataset, we first compared the predictive value of the migrasome-related risk score of with classical clinical characteristics. The survival-related ROC curve analysis indicated that the performance of migrasome-related risk score was superior to other clinical factors including age, gender and stage for predicting OS of CRC patients (AUC = 0.782 VS 0.637, 0.492, 0.718), which was further validated by the C-index comparison (Fig. 3A, B).

Construction and validation of the multimodal prognostic model and nomogram (A). The survival-related ROC curve analysis for migrasome-related signature and clinical factors; (B). C-index comparison of the migrasome-related risk score and other clinical factors; (C and D). The Kaplan–Meier analysis of overall survival for patients with high risk scores and low risk scores in the early-onset and later-onset subgroups; (E and F). The Kaplan–Meier analysis of overall survival for patients with high risk scores and low risk scores in the stage I-II and III-IV subgroups; (G). Construction of the nomogram based on the multimodal model; (H). The calibration curves of the nomogram for 1-, 3-, and 5-year OS.
The sub-group survival analysis was also performed to confirm the value of migrasome-related risk score in patients from different clinical background. The result revealed that OS was significantly worse in the low-risk group than the high-risk group in both early-onset (P < 0.001) and later-onset (P < 0.001) CRC patients (Fig. 3C, D). For stage I-II (P < 0.001) or III-IV (P < 0.001) CRC, patients with higher risk scores had significantly worse overall survival than those with lower risk scores (Fig. 3E, F).
To assess the independent predicting value of migrasome-related risk score, we performed the multivariate Cox regression analysis and found that our risk score could effectively predict the survival of CRC patients independent of other clinical characteristics (Table. 1, HR = 1.034, 95% CI 1.018–1.050, P < 0.001). We thereby developed a nomogram based on the multivariate Cox model described above (Fig. 3G). The calibration curves proved the robustness of its consistent predicting value in 1-, 3-, and 5-year OS in CRC patients (Fig. 3H). In addition, the C-index of our nomogram was 0.753 (95% CI, 0.724–0.783) which indicated that the model held a good predictive capacity.
Transcriptional variation comparison and functional enrichment of high-risk and low-risk patients
We performed the differential expression analysis of transcriptome array between high-risk and low-risk groups to explore their genomic variation. 254 up-regulated and 233 down-regulated genes were found in high-risk patients (Fig. 4A, B). For GO enrichment analysis, these DEGs were mainly enriched in signaling receptor activator activity, receptor ligand activity, G protein-coupled receptor binding and endocytic vesicle (Fig. 4C). Enrichment analysis for KEGG pathways indicated that neuroactive ligand-receptor interaction, VEGF signaling pathway and cytokine-cytokine receptor interaction were shown to be important pathways for the tumor progression in patients with high migrasome-related risk scores (Fig. 4D).

Transcriptional variation and functional enrichment analysis of high- and low-risk patients (A). Volcano plot of 254 up-regulated and 233 down-regulated genes between high-risk and low-risk patients; (B). Heatmap of differential expressed genes between high-risk and low-risk patients; C&D. GO and KEGG enrichment analysis of DEGs between high-risk and low-risk patients; E&F. GSEA enrichment analysis of GO pathways for high-risk and low-risk patients; G&H. GSEA enrichment analysis of KEGG pathways for high-risk and low-risk patients.
The GSEA enrichment analysis was further conducted, which showed that nucleosome-related pathways were enriched in low-risk patients, as well as chromatin structural constituents (Fig. 4E, G). For patients with high migrasome-related risk scores, immune-related pathways such as antigen processing and presentation exhibited notable enrichment, which may indicate a close association between migrasome and tumor immunity (Fig. 4F, H).
Immune cell infiltration analysis and immunotherapy efficacy evaluation of the migrasome-related risk score
Considering that migrasome has been previously reported to affect the infiltration and activation of immune cells in the tumor microenvironment, we determine the proportions of 22 types of immune cells and investigated the relationship between immune cell status and migrasome-related risk scores. A significant reduction of infiltrating plasma cells, resting CD4 + memory T cells and activated dendritic cells was observed in the high-risk group (Fig. 5A). As immunotherapy has been increasingly important for clinical application, we further used ssGSEA to quantify the function of immune cells and perform comparison in two groups. We demonstrated that immune scores of HLA and macrophages were significantly higher in the high-risk group (Fig. 5B), which sheds new light for current immunotherapy. The expression levels of 47 immune checkpoint-related genes collected from previously published studies were also compared between patients from the high-risk and low-risk groups, which demonstrated that the risk score had a potential association with 14 key regulator of immune checkpoint including TNF family, CD70 and CD276 (Fig. 5C).

Immune cell infiltration, immunotherapy efficacy, and drug sensitivity analysis (A). Fraction of immune cells in tumor microenvironment for high-risk and low-risk patients; (B). Scores of immune function for high-risk and low-risk patients; (C). The expression levels of immune checkpoint-related genes for high-risk and low-risk patients; (D). TIDE scores for high-risk and low-risk patients; (E-L). The sensitivity for targeted drugs including PLX-4720, JAK-8709, JAK-8517, Irinotecan, ERK-6604, ERK-2440, Epirubicin and Foretinib for high-risk and low-risk patients.
In addition, the sensitivity to immunotherapy and potential of immune escape of CRC patients were evaluated using the TIDE algorithm and compared between patients in the high- and low-risk groups. We found that the TIDE scores were significantly higher in the high-risk group than in the low-risk group (Fig. 5D), which indicated that patients with high migrasome-related risk scores tend to resist immunotherapy. However, whether immunotherapy is better for low-risk CRC patients still needs further exploration.
Drug sensitivity screening for high-risk and low-risk patients
Although adjuvant chemoradiotherapy has been widely used for CRC patients, targeted drugs still faced a significant gap in the treatment of CRC, which hindered precision medicine. It’s essential to develop predicting marker for treatment response of various kinds of targeted drugs, and thus recommend high-sensitivity personalized treatment for each patient. We compared the sensitivity of various targeted drugs of patients in the high-risk and low-risk groups based on the IC50 of each drug. The results showed that patients in the low-risk group were significantly more sensitive to PLX-4720 (B-RafV600E inhibitor), JAK-8517 (JAK/STAT inhibitor), ERK-2440 (ERK pathway inhibitor) and Epirubicin than those in the high-risk group (Fig. 5E–L).
Discussion
In this study, we developed and validated a prognostic signature based on migrasome-related lncRNAs for CRC patients, addressing a critical need for precise prognostic biomarkers and potential therapeutic targets. Our findings highlighted the intricate interplay between migrasome-related lncRNAs, tumor progression, and immune regulation.
The prognostic signature effectively stratified CRC patients into high- and low-risk groups, with significant differences in OS and PFS. This is consistent with prior studies indicating the prognostic value of specific lncRNA signatures in CRC28. For instance, Wang et al. reported a lncRNA-based model predicting the prognosis in colon adenocarcinoma with comparable accuracy29, but their work did not explore the role of migrasome in tumor biology. Our work builds on these findings by focusing on the novel role of migrasome-related lncRNAs and their association with immune features.
The functional enrichment analysis of differentially expressed genes revealed that the variation between high-risk and low-risk patients was mainly enriched in cell communication pathways such as signaling receptor activator activity, endocytic vesicle and cytokine-cytokine receptor interaction. This was in line with previous studies about the importance of migrasome in tumor microenvironment via regulating cell–cell communication30,31. GSEA analysis provided further interpretation that high-risk patients exhibited upregulation of pathways associated with immunity such as antigen processing and presentation, while low-risk patients may achieve better prognosis via nucleosome-related mechanism including chromatin stabilization32,33.
Our analysis of immune cell infiltration revealed reduced levels of plasma cells and activated dendritic cells in high-risk patients. This observation is in line with the findings of Cao et al., who reported that a decrease in dendritic cell activity correlates with inactive response to immunotherapy in tumor microenvironment34. Additionally, the elevated TIDE scores in high-risk patients suggest a greater propensity for immune evasion35, a phenomenon widely observed in advanced CRC stages36. These findings underscore the potential immunosuppressive effects of migrasome-related lncRNAs and their implications for therapy resistance.
Therapeutically, our results demonstrated that low-risk patients are more sensitive to targeted agents such as PLX-4720 and JAK-8517. This aligns with prior studies indicating that patients with active immune microenvironments tend to respond better to targeted therapies37. High-risk patients, in contrast, showed greater resistance to both immunotherapy and certain targeted drugs, potentially due to their immunosuppressive tumor microenvironment38. These insights suggest that migrasome-related lncRNAs could serve as predictive biomarkers for personalized therapy, similar to what has been reported for other lncRNA signatures in recent years39,40.
However, our study has limitations. First, the retrospective design based on TCGA data necessitates prospective validation in independent cohorts. While we identified associations between migrasome-related lncRNAs and immune features, the underlying mechanisms remain speculative. Further experimental studies are required to elucidate how these lncRNAs influence immune cell infiltration and tumor behavior.
Conclusion
This study establishes a novel migrasome-related lncRNA prognostic model for CRC, offering valuable insights into tumor biology and therapeutic responses. By integrating our findings with existing literature, we provide a comprehensive perspective on the potential of migrasome-related lncRNAs as biomarkers and therapeutic targets.
Data availability
The datasets analyzed during the current study are available in the TCGA repository (https://portal.gdc.cancer.gov/).
References
Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer statistics 2023. CA A Cancer J Clin. 73(1), 17–48 (2023).
Xie, Y., Shi, L., He, X. & Luo, Y. Gastrointestinal cancers in China, the USA, and Europe. Gastroenterol. Report 9(2), 91–104 (2021).
Global, regional, and national burden of colorectal cancer and its risk factors, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. The lancet Gastroenterology & hepatology 2022, 7(7):627–647.
Keum, N. & Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 16(12), 713–732 (2019).
Ogunwobi, O. O., Mahmood, F. & Akingboye, A. Biomarkers in colorectal cancer: Current research and future prospects. Int. J. Mol. Sci. 21(15), 5311 (2020).
Mattick, J. S. et al. Long non-coding RNAs: Definitions, functions, challenges and recommendations. Nat. Rev. Mol. Cell Biol. 24(6), 430–447 (2023).
Singh, D., Assaraf, Y. G. & Gacche, R. N. Long non-coding RNA mediated drug resistance in breast cancer. Drug Resis. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 63, 100851 (2022).
Liu, S. J., Dang, H. X., Lim, D. A., Feng, F. Y. & Maher, C. A. Long noncoding RNAs in cancer metastasis. Nat. Rev. Cancer 21(7), 446–460 (2021).
Liu, Z. et al. Machine learning-based integration develops an immune-derived lncRNA signature for improving outcomes in colorectal cancer. Nat. Commun. 13(1), 816 (2022).
Chen, S. & Shen, X. Long noncoding RNAs: Functions and mechanisms in colon cancer. Mol. Cancer 19(1), 167 (2020).
Jiang D, He J, Yu L: The migrasome, an organelle for cell-cell communication. Trends Cell Biol. 2024.
Gu, C. et al. Targeting initial tumour-osteoclast spatiotemporal interaction to prevent bone metastasis. Nat. Nanotechnol. 19(7), 1044–1054 (2024).
Cai, C. & Shen, J. The roles of migrasomes in immunity, barriers, and diseases. Acta Biomater. 189, 88–102 (2024).
Bader, J. E., Voss, K. & Rathmell, J. C. Targeting metabolism to improve the tumor microenvironment for cancer immunotherapy. Mol. Cell 78(6), 1019–1033 (2020).
Elia, I. et al. Tumor cells dictate anti-tumor immune responses by altering pyruvate utilization and succinate signaling in CD8(+) T cells. Cell Metab. 34(8), 1137-1150.e1136 (2022).
Niu, N. et al. Tumor cell-intrinsic epigenetic dysregulation shapes cancer-associated fibroblasts heterogeneity to metabolically support pancreatic cancer. Cancer Cell 42(5), 869-884.e869 (2024).
Ruf, B. et al. Tumor-associated macrophages trigger MAIT cell dysfunction at the HCC invasive margin. Cell 186(17), 3686-3705.e3632 (2023).
Wang, D. & Yu, L. Migrasome biogenesis: when biochemistry meets biophysics on membranes. Trends Biochem. Sci. 49(9), 829–840 (2024).
Jiao, H. et al. Mitocytosis, a migrasome-mediated mitochondrial quality-control process. Cell 184(11), 2896-2910.e2813 (2021).
Chen, W. et al. Migrasome-related ITGA5 for predicting prognosis, immune infiltration and drug sensitivity of hepatocellular carcinoma. Apoptosis Int. J. Program. Cell Death 30(5–6), 1424–1439 (2025).
Tan, Z. et al. Migrasomes, critical players in intercellular communication. Cancer Cell Int. 25(1), 113 (2025).
Huang, Y. et al. Migrasome formation is mediated by assembly of micron-scale tetraspanin macrodomains. Nat. Cell Biol. 21(8), 991–1002 (2019).
Zhao, X. et al. Identification of markers for migrasome detection. Cell Discov. 5, 27 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28(1), 27–30 (2000).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. Pub. Protein Soc. 28(11), 1947–1951 (2019).
Kanehisa, M., Furumichi, M., Sato, Y., Matsuura, Y. & Ishiguro-Watanabe, M. KEGG: Biological systems database as a model of the real world. Nucleic Acids Res. 53(D1), D672-d677 (2025).
Zhang, X. et al. Migrasome-related prognostic signature TSPAN4 correlates with immune infiltrates and metabolic disturbances in hepatocellular carcinoma. J. Gastroenterol. 60(5), 593–606 (2025).
Wu, Y. & Xu, X. Long non-coding RNA signature in colorectal cancer: Research progression and clinical application. Cancer Cell Int. 23(1), 28 (2023).
Wang, K. et al. Disulfidptosis-related long non-coding RNA signature predicts the prognosis, tumor microenvironment, immunotherapy, and antitumor drug options in colon adenocarcinoma. Apoptosis Int. J. Program. Cell Death 29(11–12), 2074–2090 (2024).
Jiang, Y. et al. Migrasomes, a new mode of intercellular communication. Cell Commun. Signal 21(1), 105 (2023).
Zhang, K. et al. CD151-enriched migrasomes mediate hepatocellular carcinoma invasion by conditioning cancer cells and promoting angiogenesis. . Exp. Clin. Cancer Res. CR 43(1), 160 (2024).
Meng, Q. et al. Lactylation stabilizes DCBLD1 activating the pentose phosphate pathway to promote cervical cancer progression. J. Exp. Clin. Cancer Res. CR 43(1), 36 (2024).
Wang, W. et al. Stromal induction of BRD4 phosphorylation results in chromatin remodeling and BET inhibitor resistance in colorectal cancer. Nat. Commun. 12(1), 4441 (2021).
Cao, J. et al. Degradation of PARP1 by MARCHF3 in tumor cells triggers cCAS-STING activation in dendritic cells to regulate antitumor immunity in hepatocellular carcinoma. J. Immunother. Cancer 12(11), e010157 (2024).
Qin, Y. et al. Cuproptosis correlates with immunosuppressive tumor microenvironment based on pan-cancer multiomics and single-cell sequencing analysis. Mol. Cancer 22(1), 59 (2023).
Chu, X. et al. Integrative single-cell analysis of human colorectal cancer reveals patient stratification with distinct immune evasion mechanisms. Nat. Cancer 5(9), 1409–1426 (2024).
Taube, J. M. et al. Implications of the tumor immune microenvironment for staging and therapeutics. Mod. Pathol. Off. J. United States Can. Acad. Pathol. 31(2), 214–234 (2018).
Perez-Penco, M. et al. TGFβ-derived immune modulatory vaccine: targeting the immunosuppressive and fibrotic tumor microenvironment in a murine model of pancreatic cancer. J. Immunother. Cancer 10(12), e005491 (2022).
Vitiello, M., Tuccoli, A. & Poliseno, L. Long non-coding RNAs in cancer: Implications for personalized therapy. Cell. Oncol. 38(1), 17–28 (2015).
Ma, C. et al. Prognosis and personalized treatment prediction in lung adenocarcinoma: An in silico and in vitro strategy adopting cuproptosis related lncRNA towards precision oncology. Front. Pharmacol. 14, 1113808 (2023).
Funding
This work was supported by National Key Clinical Discipline (2012).
Author information
Authors and Affiliations
Contributions
JC and JF collected the data of study cohorts and performed the data analyses under the supervision of JC; JW and QW assisted with the data sorting and analyses; JC and JC jointly interpreted the results; JC, JF and JW wrote the original manuscript; all co-authors critically revised the manuscript and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
The Institutional Review Board of the Sixth Affiliated Hospital of Sun Yat-sen University reviewed and approved the study protocol in accordance with the Declaration of Helsinki.
Consent for publication
Due to the retrospective nature of the study, the Institutional Review Board of the Sixth Affiliated Hospital of Sun Yat-sen University waived the need of obtaining informed consent.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cai, J., Fei, J., Wang, J. et al. A migrasome-related LncRNA signatures for predicting prognosis and immunotherapeutic response in colorectal cancer. Sci Rep 16, 19 (2026). https://doi.org/10.1038/s41598-025-29304-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-29304-8
