
Abstract
Hepatocellular carcinoma (HCC) is a highly heterogeneous malignant tumor characterized by a high recurrence rate and poor prognosis. In recent years, the study of miRNAs as potential prognostic markers and therapeutic targets, as well as their regulation of the glucose metabolism pathway in HCC, has attracted widespread attention. This study aims to construct a risk model for predicting the prognosis of HCC by analyzing differentially expressed glycolysis-related miRNAs and further explore their relationship with the immune microenvironment and drug sensitivity. In this study, the original mRNA and miRNA expression data of HCC were downloaded from the TCGA and GEO databases, respectively, with a total of 374 TCGA samples and 97 GSE30297 samples collected. A prognostic risk score model for HCC was constructed using LASSO regression analysis, and survival differences between different risk groups were analyzed using Kaplan-Meier curves. Metascape and GSEA analyses were performed for functional enrichment to explore the potential molecular mechanisms of the model miRNAs. Additionally, the CIBERSORT algorithm was used to analyze the immune microenvironment, and the “pRRophetic” package was employed to predict the sensitivity of HCC patients to commonly used chemotherapy drugs. Real-time quantitative PCR (RT-qPCR) was used to detect the expression levels of these glycolysis-metabolism-related miRNAs with prognostic value in tumor tissues and adjacent normal tissues of HCC patients. Through differential expression analysis, a total of 4,421 differentially expressed mRNAs and 106 differentially expressed miRNAs were screened, and 59 glycolysis-metabolism-related differential miRNAs were identified. Cox univariate regression and LASSO regression analysis were used to select 10 prognosis-related miRNAs, and a risk score model based on these miRNAs was constructed. The validation results of the model in both the training and test sets showed that the overall survival (OS) of the high-risk group was significantly lower than that of the low-risk group (P < 0.05). The nomogram model further validated the independent predictive value of the risk score for the prognosis of HCC patients. Immune microenvironment analysis revealed that the content of immune cells, such as M0 macrophages and regulatory T cells (Tregs), was higher in the high-risk group, while the content of immune cells, such as resting NK cells, was lower. Drug sensitivity analysis showed that the risk score was significantly correlated with the sensitivity to various chemotherapeutic drugs (e.g., Methotrexate, Paclitaxel). The results of RT-qPCR showed that the expression levels of hsa-mir-454 and hsa-mir-149 were up-regulated in HCC, and the expression level of hsa-mir-621 was down-regulated in HCC. However, the expression level of hsa-mir-653 was not significant in HCC, and the difference was not statistically significant. In patients with recurrent and primary HCC, the results showed significant expression differences between hsa-mir-454 and hsa-mir-621. Specifically, hsa-mir-454 was upregulated in recurrent tumor samples, while hsa-mir-621 was downregulated. Notably, hsa-mir-149 and hsa-mir-653 showed no statistically significant differences between the two groups. This study established a reliable prognostic risk scoring model for HCC by screening differentially expressed glycolysis-related miRNAs, which effectively distinguishes between high-risk and low-risk patients and predicts patient survival. Additionally, the model is closely associated with the immune microenvironment and drug sensitivity, offering strong support for personalized treatment and clinical decision-making in HCC.
Similar content being viewed by others

Introduction
Hepatocellular carcinoma (HCC), the sixth most prevalent malignancy worldwide and the third leading cause of cancer-related mortality, continues to exhibits increasing incidence and death rates globally1. Current treatment modalities primarily involve surgical resection, radiotherapy, and locoregional therapies. Nonetheless, HCC displays aggressive pathological features, including rapid cellular proliferation, high metastatic potential, and frequent neurovascular invasion, culminating in postoperative recurrence and distant metastasis in over 70% of cases. Consequently, the prognosis remains poor, with a 5-year survival rate below 20%, representing a substantial global health burden2,3. At present, the search for potential and reliable molecular targets is particularly important for the prognosis, recurrence risk and therapeutic efficacy evaluation of HCC. Ye W et al.4 discovered that association between higher expression of Vav1 in HCC and unfavourable clinicopathological features and prognosis. Mu R et al.5 reported that the expression of PRDX6 may be closely related to the prognosis of HCC. However, the underlying molecular mechanisms driving HCC development remain incompletely understood, underscoring the clinical importance of identifying novel biomarkers for prognostic evaluation and recurrence risk stratification.
MicroRNAs (miRNAs), a class of endogenous, single-stranded non-coding RNAs (~ 22 nucleotides), modulate gene expression at the post-transcriptional level by binding to complementary sequences within the 3′-untranslated regions (3′-UTRs) of target mRNAs. This interaction induces mRNA degradation or translational repression, thereby regulating critical biological processes such as cell proliferation, cycle progression, differentiation, and apoptosis6. Increasing evidence demonstrates that miRNAs exert context-dependent oncogenic or tumor-suppressive functions through their interaction with diverse molecular targets7. Aberrant miRNA expression is mechanistically associated with tumor initiation, progression, and therapeutic resistance, positioning them as promising candidates for diagnostic and therapeutic applications.
Cellular energy metabolism is predominantly sustained through glucose catabolism via two principal ATP-generating pathways: mitochondrial oxidative phosphorylation and cytoplasmic glycolysis8. Under normoxic conditions, glucose is transported into cells via glucose transporters (GLUTs) and sequentially metabolized through glycolysis, the tricarboxylic acid (TCA) cycle, and oxidative phosphorylation, yielding carbon dioxide, water, and up to 36 ATP molecules per glucose molecule9. In contrast, tumor cells frequently undergo metabolic reprogramming, characterized by persistent glycolysis despite adequate oxygen availability—a phenomenon termed the Warburg effect or aerobic glycolysis10. This adaptation supplies biosynthetic precursors to sustain uncontrolled proliferation and contributes to an acidic tumor microenvironment that facilitates immune evasion and metastatic progression11. As such, targeting aerobic glycolysis represents a viable strategy for disrupting cancer-specific metabolic dependencies.
Recent studies implicate miRNA-mediated regulation of key glycolytic enzymes (e.g., HK2, LDHA) and signaling cascades (e.g., PI3K/AKT/mTOR) in governing tumor growth, invasion, and metastatic dissemination across multiple malignancies, including breast cancer, oral squamous cell carcinoma, prostate cancer, and pancreatic ductal adenocarcinoma12,13,14,15. In light of this, a glycolysis-related miRNA-based prognostic model was developed and validated using bioinformatics approaches to evaluate survival outcomes in HCC. The model further investigates its associations with tumor recurrence, immune microenvironment remodeling, and responsiveness to immunotherapy, thereby offering a robust, evidence-driven framework for prognostic assessment and therapeutic decision-making in HCC. Despite emerging insights, the prognostic potential of glycolysis-related miRNA signatures in HCC remains underexplored. To address this, the proposed model integrates survival prediction for both overall survival (OS) and recurrence-free survival (RFS), while also correlating with immune infiltration patterns, recurrence risk, and immunotherapeutic sensitivity. These findings contribute to a translational foundation for refined risk stratification and individualized treatment strategies in HCC.
Materials and methods
Data download and clinical data of patients with HCC
The TCGA database (https://portal.gdc.cancer.gov/) is currently the largest repository of cancer-related genetic data, encompassing gene expression data, copy number variations, SNPs, and more. The original mRNA expression data for HCC were downloaded, obtaining a total of 374 samples, which included 371 primary tumor samples and 3 recurrent tumor samples. Additionally, the original miRNA expression data for HCC were retrieved, comprising 375 samples, with 372 from primary tumors and 3 from recurrent tumors.
The GEO database (https://www.ncbi.nlm.nih.gov/geo/info/datasets.html), or Gene Expression Omnibus, is a gene expression database maintained by the National Center for Biotechnology Information (NCBI). The Series Matrix File for GSE30297 was downloaded from the NCBI GEO public repository, using the annotation file GPL8786. This dataset includes 97 samples, consisting of 36 primary tumor samples and 61 recurrent tumor samples.
From January 2025 to September 2025, we collected fresh tumor and adjacent tissue samples from 16 HCC patients who underwent radical surgery from the First Affiliated Hospital of Kunming Medical University. Among them, 10 cases were primary and 6 cases were recurrent. Complete clinical history information was available for all patients, and the diagnosis of HCC was confirmed microscopically by two pathologists. Then, RT-qPCR was used to determine the levels of four miRNAs expression.
Ethical considerations
The Ethics Committee of the First Affiliated Hospital of Kunming Medical University examined and approved this study, which involved human data, in accordance with the Declaration of Helsinki [(2023) Ethics L No. 205]. Every participant provided written informed permission. All participants’ personal information was kept private, participation in the study was entirely voluntary, and all data sets were coded and maintained over the entire data gathering and analysis process.
Model construction and prognosis
To construct a prognosis-related model, miRNAs associated with prognosis were selected and analyzed using Lasso regression. The expression values of each selected miRNA were incorporated into a risk score formula for each sample, with the regression coefficients derived from Lasso regression analysis. Based on the median risk score, the samples were divided into low-risk and high-risk groups. Survival differences between the two groups were assessed using Kaplan-Meier analysis, with comparison via the log-rank test. The prognostic predictive value of the risk score was further evaluated through stratified analysis and Lasso regression. The model’s prediction accuracy was assessed by generating ROC curves. The cross-validation multiple is 10,lambda.min = 0.03382 (the penalty coefficient corresponding to the minimum error).
Construction of nomogram model
The nomogram, based on multivariate regression analysis, was constructed by incorporating both miRNA expression data and clinical factors. In the nomogram, line segments with scales are drawn on a shared plane to represent the relationships between variables. Each factor’s contribution to the outcome is determined by its regression coefficient. The values for each influencing factor are assigned scores, and the total score is calculated by summing these values to derive a predicted outcome.
Metascape enrichment analysis
The Metascape database (www.metascape.org) was utilized for functional annotation of the model miRNAs to explore their functional relevance comprehensively. Gene Ontology (GO) pathway analysis was conducted on specific miRNAs, with a minimum overlap of 3 and a p-value threshold of ≤ 0.01 considered statistically significant.
GSEA
Based on the sample risk scores, patients were stratified into high-risk and low-risk groups. Subsequently, differences in signaling pathways between the two groups were assessed using Gene Set Enrichment Analysis (GSEA). The background gene set used in the analysis was version 7.0 of the MsigDB annotation gene set, serving as the reference for subtype pathway annotations. Differential expression analysis of the pathways was performed, with significantly enriched gene sets (adjusted p-value < 0.05) ranked according to their consistency score. GSEA is commonly employed to explore the integration of disease classification and its biological significance.
Immune microenvironment analysis
The CIBERSORT method, based on support vector regression, was employed to evaluate immune cell types within the tumor microenvironment. This method performs deconvolution analysis on the expression matrix of immune cell subtypes, distinguishing 22 human immune cell phenotypes, including T cells, B cells, plasma cells, and myeloid cell subsets. In this study, the CIBERSORT algorithm was applied to analyze patient data, inferring the relative proportions of 22 immune cell types and performing correlation analysis between gene expression and immune cell content.
Drug sensitivity analysis
To predict the chemotherapy sensitivity of each tumor sample, the largest pharmacogenomics database, GDSC (Cancer Drug Sensitivity Genomics Database, https://www.cancerrxgene.org/), was utilized. The R package “pRRophetic” was applied to predict the IC50 values for each chemotherapy drug treatment. These values were estimated using a regression method, with the prediction accuracy validated by 10-fold cross-validation using the GDSC training set. All parameters, including “combat” for batch effect correction, were set to default values, and the average of repeated gene expressions was taken for analysis.
RT-qPCR analysis
Total RNA was isolated from fresh tumor tissue and adjacent tissue samples of HCC by using RNAiso Plus (TAKARA). For miRNAs, the reverse transcription of obtained RNA to cDNA via The PrimeScript™ RT reagent Kit (TAKARA). Then, The HieffTM qPCR SYBR Green Master Mix (YEASEN) was used to detect The expression of miRNAs. Each sample was needed three replicates wells. The 2−ΔΔCt method was used to calculated the relative expression. Total synthesized primers were purchased Beijing TsingkeBiotech and listed in Table S1.
Statistical analysis
Statistical analyses were performed using R language (version 4.3.0) and Quantitative data were presented as mean ± standard deviation. Data of non-normal distribution were analysed with Wilcoxon test between two groups, with p < 0.05 was considered statistically significant.
Results
Differential analysis
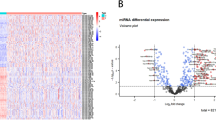
RNA-seq expression data for HCC were downloaded from the TCGA database, comprising 374 samples, including 371 primary tumor samples and 3 recurrent tumor samples. Additionally, original miRNA expression data for HCC were retrieved, consisting of 375 samples, with 372 from primary tumors and 3 from recurrent tumors. Differential gene expression analysis between the two groups was conducted using the limma package, applying a threshold of p-value < 0.05. From the RNA-seq dataset, a total of 4421 differential genes were identified, including 3746 upregulated genes and 675 downregulated genes (Fig. 1A-B). Similarly, 106 differential miRNAs were identified from the miRNA-seq dataset, with 96 upregulated miRNAs and 10 downregulated miRNAs (Fig. 1C-D). Subsequently, 200 glycolysis-related genes were obtained from the HALLMARK_GLYCOLYSIS gene set of the MSigDB molecular signature database (https://www.gsea-msigdb.org/gsea/msigdb/), along with 5102 metabolism-related genes from the literature16. Using a Venn diagram, 20 differential glycolysis genes and 528 differential metabolism genes were identified (Fig. 1E-F). To explore the relationships between differential miRNAs and differential glycolysis and metabolism genes, correlation coefficients were calculated using the cor.test() function, with filtering conditions set at a correlation coefficient ≥ 0.3 and p-value ≤ 0.05. The results were visualized in a scatter plot, ultimately identifying 59 differentially expressed glycolysis-metabolism-related miRNAs. Furthermore, the regulatory network between these differential miRNAs, glycolysis genes, and metabolism genes was visualized using Cytoscape to illustrate their interactions (Fig. 2).

Differential analysis. (A) Volcano diagram of mRNA differentially expressed genes, with blue representing downregulated differentially expressed genes and pink representing upregulated differentially expressed genes. (B) A heatmap of mRNA differentially expressed genes, with blue representing downregulated differentially expressed genes and pink representing upregulated differentially expressed genes. (C) Volcano diagram of miRNA differentially expressed genes, with blue representing downregulated differentially expressed genes and pink representing upregulated differentially expressed genes. (D) A heatmap of miRNA differentially expressed genes, with blue representing downregulated differentially expressed genes and pink representing upregulated differentially expressed genes. (E) Venn diagram of the intersection between mRNA differentially expressed genes and glycolytic genes. (F) Wayne diagram of the intersection between mRNA differentially expressed genes and metabolic genes.

PPI network. Glycolysis metabolism related genes and miRNA networks, with green representing glycolysis metabolism genes and red representing miRNAs.
Construction of the prognostic model
Clinical data from HCC samples were collected from the TCGA database, and prognostic miRNAs in HCC were identified by performing Cox univariate regression on differentially expressed glycolysis-metabolism-related miRNAs. Ten prognostic-related miRNAs were identified (p-value < 0.05) (Fig. 3A). Based on these prognostic miRNAs, characteristic miRNAs in HCC were selected using the Lasso regression feature selection algorithm. The processed samples with survival data from the TCGA-LIHC dataset were randomly divided into a training set and a test set in a 4:1 ratio. After performing Lasso regression analysis (Fig. 3B-D), the optimal risk score value for each sample was determined for subsequent analysis: RiskScore = hsa-mir-621 × (−0.121994968) + hsa-mir-454 × (−0.047356538) + hsa-mir-653 × 0.0801983486745445 + hsa-mir-149 × 0.265842951894282. Based on the risk score, samples were categorized into high-risk and low-risk groups. Kaplan-Meier survival analysis revealed that the OS of the high-risk group was significantly lower than that of the low-risk group in both the training and test sets (Fig. 3E-F). Furthermore, ROC curve analysis of both the training and test sets confirmed the good validation efficiency of the model (Fig. 3G-H).

Construction of prognostic model. (A) Single factor forest plot of prognostic miRNA. (B) The LASSO coefficient distribution of prognostic genes and the gene combination at the minimum lambda value. (C) Ten cross validation of tuning parameter selection in LASSO model to determine the minimum lambda value. (D) The coefficient of Lasso gene. (E-F) The survival curve of the TCGA model training set and testing set. (G-H) The ROC curve of the training and testing sets for the TCGA model.
Verification of MiRNA differential expression by external data sets
Transcriptome data of HCC(GSE30297) was analyzed to assess the expression differences of model miRNAs between primary and recurrent tumor samples. The results revealed significant expression differences for hsa-mir-454 and hsa-mir-621, with hsa-mir-454 exhibiting higher expression levels in recurrent tumor samples (Fig. 4).

Differential expression of model miRNA. Differential expression of model differential miRNA in two groups of patient samples, blue represents primary samples, pink represents relapse samples.
Clinical correlation analysis and univariate and multivariate analysis
Clinical correlation analysis, along with univariate and multivariate analyses, categorized risk score values according to clinical index values, presenting the results of each clinical grouping through box plots (Fig. 5A-H). The rank sum test showed significant differences in the distribution of risk scores between groups based on Fustat, tumor stage, and tumor T stage (p-value < 0.05), demonstrating the applicability of the risk score derived from the modeling analysis for classifying HCCsamples. Additionally, univariate and multivariate Cox regression analyses identified the risk score as an independent prognostic factor for patients with HCC (Fig. 6A-B).

Clinical correlation. (A-H) Correlation between risk scores and clinical indicators.

Construction of univariate and multivariate and nomogram models for risk assessment. (A-B) Forest plots of risk scores in univariate and multivariate Cox regression. (C) Model correlation nomogram of risk score and clinical characteristics. (D) Prediction analysis of OS in one, two and three years. (E-F) ROC curves and DCA curves.
Construction of the nomogram model
The results of regression analysis for clinical characteristics were presented in the form of a nomogram, incorporating the risk scores of the samples. This analysis demonstrated varying contributions of different clinical indicators and the risk score to the overall scoring process across all samples (Fig. 6C). Additionally, predictive analyses for OS at 1, 2, and 3 years were performed (Fig. 6D), accompanied by ROC and DCA curves. The findings indicated that the predicted OS closely matched the observed OS, confirming the Nomogram model’s strong predictive efficiency (Fig. 6E-F).
Functional enrichment analysis
Metascape analysis revealed that the model miRNAs were enriched in the RISC complex pathway (Fig. 7A). Further investigation into the signaling pathways associated with high- and low-risk models aimed to elucidate the molecular mechanisms by which risk scores influence tumor progression. GSEA results identified several involved pathways, including the Cytosolic DNA-sensing pathway, DNA replication, and Notch signaling pathway (Fig. 7B-C). A total of 1503 mRNAs, corresponding to 1574 mRNA-miRNA pairs, were predicted for the 4 model miRNAs using the miRDB database, with visualization through Cytoscape (Fig. 8).

Functional enrichment analysis. (A) GO-KEGG enrichment analysis using Metascape database. (B-C) KEGG signaling pathways involved in risk scores, as well as pathway regulation and genes involved.

miRNA network. Interaction network of key miRNA and mRNA, red represents miRNA, green represents mRNA.
Immune microenvironment analysis
The tumor microenvironment primarily consists of tumor-associated fibroblasts, immune cells, extracellular matrix, growth factors, inflammatory factors, specific physical and chemical characteristics, and the cancer cells themselves. It plays a pivotal role in influencing tumor diagnosis, survival outcomes, and clinical treatment responses. To further investigate the molecular mechanisms by which risk scores affect HCC progression, the relationship between risk scores and tumor immune infiltration was analyzed. Immune cell content and immune cell correlations were presented for high- and low-risk groups in various formats (Fig. 9A-B). Compared to the low-risk group, the high-risk group exhibited significantly lower levels of Monocytes and resting NK cells, while levels of Macrophages M0 and regulatory T cells (Tregs) were significantly higher (Fig. 9C). Further exploration of the correlation between risk scores and immune cells revealed a significant positive correlation between risk scores and resting Dendritic cells, Tregs, and Macrophages M0, as well as a significant negative correlation with resting Mast cells, NK cells, and memory resting CD4 + T cells (Fig. 9D).

Immune infiltration analysis. (A) Relative percentage of immune cell subsets. (B) Correlation between immune cells, blue indicates negative correlation and red indicates positive correlation. (C) Difference in immune cell content between low-risk samples and high-risk samples. (D) Correlation between risk score and immune cells.
Correlation analysis between immune cell markers and risk score
Immune cell markers were obtained from the CellMarker2.0 database, and correlation analysis was performed using the cor.test() function. The results indicated a significant positive correlation between the risk score and immune cell markers such as T cells, Monocytes, and Dendritic cells (Fig S1). Additionally, immune regulatory genes from the TISIDB database, including immunosuppressive factors, immunostimulatory factors, chemokines, and chemokine receptors, were analyzed for differences between high- and low-risk groups (Fig. 10A-E). In terms of Chemokines, genes such as CCL2, CCL20, and CCL21 were significantly more expressed in the high-risk group. In Immunoinhibitors, the expression of genes like CSF1R, CTLA4, and HAVCR2 was also significantly higher in the high-risk group. Similarly, Immunostimulators such as CD86, CXCR4, and ENTPD1, as well as MHC-related genes like HLA-DMA, HLA-DMB, and HLA-DOA, exhibited higher expression in the high-risk group. Receptor genes, including CCR1, CCR10, and CCR2, were also expressed at significantly higher levels in the high-risk group.

Differences between risk scores and immune factors. (A-E) Differential expression of risk scores and chemokine, Immunoinhibitor, Immunostimulator, MHC and receptor.
Drug sensitivity
In early HCC, the combination of surgery and chemotherapy has proven effective. This study, utilizing drug sensitivity data from the GDSC database, applied the R package “pRRophetic” to predict chemotherapy sensitivity for each tumor sample and to further assess the relationship between risk scores and sensitivity to common HCC chemotherapy drugs. The results demonstrated a significant correlation between the risk score and the sensitivity of patients with HCC to methotrexate, paclitaxel, docetaxel, vinblastine, gemcitabine, and erlotinib (Fig S2).
Validation of the expression levels of four MiRNAs in HCC
RT-qPCR was used to detect fresh tumor tissue and adjacent tissue samples from 16 HCC patients, and the results showed that the expression levels of hsa-mir-454 and hsa-mir-149 were up-regulated in HCC, and the expression level of hsa-mir-621 was down-regulated in HCC. However, the expression level of hsa-mir-653 was not significant in HCC, and the difference was not statistically significant (Fig. 11A). In patients with recurrent and primary HCC, the results showed significant expression differences between hsa-mir-454 and hsa-mir-621. Specifically, hsa-mir-454 was upregulated in recurrent tumor samples, while hsa-mir-621 was downregulated. Notably, hsa-mir-149 and hsa-mir-653 showed no statistically significant differences between the two groups (Fig. 11B).

The expression levels of four miRNAs were detected by RT-qPCR. (A) The expression levels of hsa-mir-454 and hsa-mir-149 were up-regulated in HCC, and the expression level of hsa-mir-621 were down-regulated in HCC. The expression level of hsa-mir-653 was not statistically significant in HCC. (B) hsa-mir-454 was upregulated in the recurrent tumor samples, while hsa-mir-621 was downregulated in the recurrent tumor samples, and hsa-mir-149 and hsa-mir-653 showed no statistical difference between the two groups.* p < 0.05; *** p < 0.001; **** p < 0.0001.
Disscussion
HCC remains one of the most prevalent malignancies globally, with particularly high incidence rates in Asia17. Traditional prognostic stratification largely relies on clinicopathological features such as tumor size, multifocality, and vascular invasion. However, these parameters often lack the accuracy necessary to predict long-term survival outcomes and recurrence risks18. As a result, identifying novel biomarkers and developing more precise prognostic models have become central objectives in HCC research. Wang J et al.19reported FIGNL1 could modulate the ECM-receptor interaction pathway through the regulation of HMMR, thus regulating the formation of HCC. Xue C et al20. reported that the down-regulation of lncRNA DGCR5 expression is associated with poor prognosis of HCC. Li X et al21. reported that MTLT-related genes can serve as diagnostic and prognostic biomarkers for HCC and new therapeutic targets, which may contribute to a further understanding of the molecular pathogenesis of HCC and provide potential therapeutic strategies for HCC. Recent advancements in miRNA-based biomarkers have provided valuable insights into the molecular mechanisms of hepatocarcinogenesis, diagnosis, and therapeutic strategies. Notably, the development of miRNA-derived risk signatures to predict HCC survival and recurrence has attracted significant attention22,23. These breakthroughs offer innovative frameworks for optimizing personalized treatment strategies in patients with HCC.
In this study, mRNA and miRNA expression profiles for HCC were obtained from the TCGA and GEO databases, respectively. Through a comprehensive analysis, four glycolysis-related miRNAs (hsa-mir-621, hsa-mir-454, hsa-mir-653, and hsa-mir-149) were identified and integrated into a prognostic model for HCC. The predictive efficacy of this model was rigorously validated using independent training and testing cohorts, showing robust performance in stratifying survival outcomes.
Current literature suggests that hsa-mir-621 primarily functions as a tumor suppressor. For example, in acute myeloid leukemia (AML), hsa-mir-621 inhibits cell proliferation by targeting the long non-coding RNA SNHG1024. Similarly, in colorectal cancer (CRC), hsa-mir-621 directly suppresses LEF1, disrupting oncogenic signaling25. However, the role of hsa-mir-621 in HCC remains poorly defined, and its mechanistic contributions to hepatocarcinogenesis are not fully understood. Yao Zhang et al.26 reported that downregulation of hsa-mir-621 promotes HCC cell proliferation by targeting CAPRIN1. Nevertheless, whether hsa-mir-621 participates in hepatocarcinogenesis through modulation of the glycolytic pathway remains unexplored. Our study demonstrated an inverse correlation between hsa-mir-621 expression and poor HCC prognosis, with its expression significantly lower in recurrent HCC samples compared to primary tumors, suggesting tumor-suppressive functions. Further pathway analysis linked hsa-mir-621 to glycolytic metabolism, indicating its potential role in regulating glycolysis-related genes to reprogram HCC metabolic dependencies.
hsa-mir-454 exhibits context-dependent roles across various malignancies. It functions as a tumor suppressor in AML and non-small cell lung cancer (NSCLC) by targeting oncogenic pathways27,28, yet paradoxically promotes tumorigenesis in HCC and CRC29,30. Our data support its oncogenic role in HCC, showing significant upregulation in recurrent tumors compared to primary tumors. Li N et al.31 demonstrated that hsa-mir-454 regulates STAT3-driven mesothelial-mesenchymal transition and glycolysis under hyperglycemic conditions. Identified as a glycolysis-associated miRNA in this study, hsa-mir-454 is postulated to facilitate HCC progression by activating glycolytic pathways, thereby exacerbating clinical outcomes. Through the detection of fresh tissue samples from HCC patients, we also found that the expression of hsa-mir-454 was up-regulated in HCC, which may suggest that hsa-mir-454 may play a role in promoting cancer in HCC.
Dysregulated expression of hsa-mir-653 has been linked to tumor aggressiveness and unfavorable prognosis across various malignancies32. For example, hypoxia-induced hsa-mir-653 accelerates CRC progression via the circSETD3/KLF6 axis33 and modulates chondrocyte senescence in osteoarthritis34. Despite accumulating evidence in other cancers, its mechanistic function in HCC remains elusive. The current analysis revealed marked overexpression of hsa-mir-653 in HCC, strongly associated with glycolytic activity, implicating its potential role as a metabolic modulator of disease prognosis. However, through the detection of clinical samples from HCC patients, our results showed that the expression level of hsa-mir-653 in HCC was not significantly different, which may be related to the limited number of samples. The clinical sample size should be further expanded for further detection and analysis.
hsa-mir-149 exhibits context-dependent roles, functioning either as an oncogene or tumor suppressor depending on cancer type35. It suppresses tumorigenesis in prostate and cervical cancers36,37 but promotes malignancy in lung cancer and acute lymphoblastic leukemia38,39. Although Lin C et al40. reported tumor-suppressive activity in HCC, the present findings correlate hsa-mir-149 with poor prognosis, highlighting a potential contradiction that underscores the need for further mechanistic clarification in HCC. In addition, our detection results showed that hsa-mir-149 expression was up-regulated in HCC, which also suggested that hsa-mir-149 may play a role in promoting cancer in HCC. However, in the comparison between primary HCC and recurrent HCC, there was no significant difference in the expression of hsa-mir-149. Therefore, its potential biological functions and mechanism of action in HCC need to be further studied.
Metascape-based pathway enrichment analysis revealed significant involvement of the model miRNAs in the RNA-induced silencing complex (RISC) pathway (adjusted p < 0.05). MicroRNAs, in concert with Argonaute (AGO) family proteins, assemble into the RISC to mediate gene silencing through transcriptional and post-transcriptional suppression41. Further, GSEA stratifying high- and low-risk groups uncovered key pathways enriched in the prognostic model, including the cytosolic DNA-sensing pathway, DNA replication, and Notch signaling. The cytosolic DNA-sensing pathway is primarily driven by the cGAS/STING signaling axis. Cyclic GMP-AMP synthase (cGAS), an innate immune sensor, binds to double-stranded DNA (dsDNA) in a sequence-independent manner, initiating its activation. This interaction induces conformational changes in cGAS, which in turn activates the cGAS/STING cascade, playing a pivotal role in DNA damage repair and innate immune responses42. Emerging evidence suggests that hyperactivation of the cGAS-STING DNA-sensing pathway plays a pivotal role in the pathogenesis of liver diseases, positioning it as a promising therapeutic target43. DNA replication, an essential process for duplicating genetic material prior to cell division, ensures accurate transmission to daughter cells and is conserved across all living organisms. In cancer cells, DNA replication is often compromised, leading to increased DNA damage due to defective replication machinery, elevated reactive oxygen species (ROS), loss of tumor suppressors, and/or constant oncogene activation44. These genomic instabilities contribute significantly to the molecular heterogeneity of HCC, emphasizing the critical need for targeting DNA repair pathways in personalized HCC treatments45. Dysregulation of Notch signaling, marked by aberrant gene regulatory networks, plays a pivotal role in oncogenesis and tumor progression. Notably, Notch signaling influences immune cell functional plasticity, modulating both anti-tumor and pro-tumor responses while altering tumor immunogenicity46. Montagner A et al47. demonstrated that disrupted Notch signaling in HCC hampers immune cell activation, reducing the effectiveness of cancer immunotherapies. Collectively, the enriched pathways within the high-risk group are integral to tumorigenesis and immune evasion, directly contributing to HCC progression. These findings offer a solid mechanistic framework for understanding HCC development and refining prognostic stratification approaches.
In the analysis of risk scores and their correlation with immune microenvironment alterations and immune cell profiles, the high-risk group exhibited higher levels of immune cells, such as M0 macrophages and Tregs, alongside a reduction in resting NK cells. Additionally, significant overexpression of chemokines, immunoinhibitors, and immunostimulators was observed in the high-risk group compared to the low-risk group. These alterations further highlight the profound differences in the immune microenvironment between the two risk groups. Drug sensitivity analysis revealed a strong association between risk scores and responsiveness to various chemotherapeutic agents, including Methotrexate and Paclitaxel. While Methotrexate and Paclitaxel are critical in HCC treatment, they are frequently associated with drug resistance48,49. In conclusion, these findings offer valuable guidance for optimizing therapeutic choices for patients with HCC, encompassing chemotherapy, targeted therapy, and immunotherapy. They also provide new insights for developing precision treatment strategies aimed at improving the prognosis of patients with HCC.
Limitations
Despite the successful establishment of a glycolysis-related miRNA-based risk model for assessing prognosis, immune microenvironment alterations, and therapeutic response in HCC, several limitations persist. These limitations include the number of research samples, the impact of altered expression of key miRNAs on glycolytic function indicators (e.g., glucose uptake, lactate production) in HCC, the role and efficacy of glycolysis-related key miRNAs and their associated chemotherapeutic drugs in HCC treatment, the mechanistic validation of interactions between model miRNAs and their predicted glycolysis-related target genes, and the proportion of key immune cells (such as M0 macrophages and regulatory T cells) in high-risk and low-risk groups detected by immunohistochemistry (IHC) to further investigate alterations in the TMB. Future research should prioritize large-scale, multi-center prospective studies and further explore the regulatory molecular mechanisms of key miRNAs in the development and progression of HCC through in vivo and in vitro experiments, so as to provide more robust references for the prognosis, recurrence, and clinical treatment of HCC.
Conclusion
This study constructed a robust risk model based on four glycolysis-related miRNAs (hsa-mir-621, hsa-mir-454, hsa-mir-653, and hsa-mir-149) through comprehensive bioinformatics analyses. Moreover, we further verified the expression levels of four miRNAs in HCC tissues by RT-qPCR. The model successfully evaluated prognosis, immune microenvironment changes, immunotherapy response, and chemotherapy sensitivity in HCC. These findings offer valuable clinical insights for early diagnosis, treatment strategies, and monitoring therapeutic efficacy in patients with HCC.
Data availability
All data generated or analysed during this study are included in this published article [and its supplementary information files].
References
Bray, F. et al. Global cancer statistics 2022: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 74, 229–263. https://doi.org/10.3322/caac.21834 (2024).
Wang, Y. & Deng, B. Hepatocellular carcinoma: molecular mechanism, targeted therapy, and biomarkers. Cancer Metastasis Rev. 42, 629–652. https://doi.org/10.1007/s10555-023-10084-4 (2023).
Llovet, J. M. et al. Molecular pathogenesis and systemic therapies for hepatocellular carcinoma. Nat. Cancer. 3, 386–401. https://doi.org/10.1038/s43018-022-00357-2 (2022).
Ye, W. et al. Association between higher expression of Vav1 in hepatocellular carcinoma and unfavourable clinicopathological features and prognosis. Protein Pept. Lett. 31, 706–713. https://doi.org/10.2174/0109298665330781240830042601 (2024).
Mu, R. et al. Analysis of the expression of PRDX6 in patients with hepatocellular carcinoma and its effect on the phenotype of hepatocellular carcinoma cells. Curr. Genomics. 25, 2–11. https://doi.org/10.2174/0113892029273682240111052317 (2024).
Sell, M. C., Ramlogan-Steel, C. A., Steel, J. C. & Dhungel, B. P. MicroRNAs in cancer metastasis: biological and therapeutic implications. Expert Rev. Mol. Med. 25, e14. https://doi.org/10.1017/erm.2023.7 (2023).
Dayakar, A., Shanmukha, K. D. & Kalangi, S. K. Spectrum of MicroRNAs and their target genes in cancer: intervention in diagnosis and therapy. Mol. Biol. Rep. 49, 6827–6846. https://doi.org/10.1007/s11033-021-07040-2 (2022).
Wang, X. et al. Harnessing glucose metabolism with nanomedicine for cancer treatment. Theranostics 14, 6831–6882. https://doi.org/10.7150/thno.100036 (2024).
Halma, M. T. J., Tuszynski, J. A. & Marik, P. E. Cancer metabolism as a therapeutic target and review of interventions. Nutrients 15 https://doi.org/10.3390/nu15194245 (2023).
Fukushi, A., Kim, H. D., Chang, Y. C. & Kim, C. H. Revisited metabolic control and reprogramming cancers by means of the Warburg effect in tumor cells. Int. J. Mol. Sci. 23 https://doi.org/10.3390/ijms231710037 (2022).
Bose, S., Zhang, C. & Le, A. Glucose metabolism in cancer: the Warburg effect and beyond. Adv. Exp. Med. Biol. 1311, 3–15. https://doi.org/10.1007/978-3-030-65768-0_1 (2021).
Koufaris, C., Papandreou, M. E., Ellis, J. K., Nicolaidou, V. & Keun, H. C. miR-22-enriched breast cancer cells display repressed glycolytic metabolism, increased glycogen synthesis, and reduced survival in low glucose conditions. Mol. Biol. Rep. 50, 5185–5193. https://doi.org/10.1007/s11033-023-08458-6 (2023).
Yuwanati, R. P. Jr., Sekaran, M., M, S. & S. & MiRNA associated with glucose transporters in oral squamous cell carcinoma: a systematic review. Cureus 15, e46057. https://doi.org/10.7759/cureus.46057 (2023).
Yao, Y. et al. Glycolysis related LncRNA SNHG3 / miR-139-5p / PKM2 axis promotes castration-resistant prostate cancer (CRPC) development and enzalutamide resistance. Int. J. Biol. Macromol. 260, 129635. https://doi.org/10.1016/j.ijbiomac.2024.129635 (2024).
Wei, Y. et al. Tumor-suppressive miR-323a inhibits pancreatic cancer cell proliferation and Glycolysis through targeting HK-2. Pathol. Int. 72, 617–630. https://doi.org/10.1111/pin.13289 (2022).
Zheng, Y., Wu, R., Wang, X. & Yin, C. Identification of a four-gene metabolic signature to evaluate the prognosis of colon adenocarcinoma patients. Front. Public. Health. 10, 860381. https://doi.org/10.3389/fpubh.2022.860381 (2022).
Toh, M. R. et al. Global epidemiology and genetics of hepatocellular carcinoma. Gastroenterology 164, 766–782. https://doi.org/10.1053/j.gastro.2023.01.033 (2023).
Lou, X. et al. The prognostic role of an optimal machine learning model based on clinical available indicators in HCC patients. Front. Med. (Lausanne). 11, 1431578. https://doi.org/10.3389/fmed.2024.1431578 (2024).
Wang, J., Sun, L., Liu, Y. & Zhang, Y. FIGNL1 promotes hepatocellular carcinoma formation via remodeling ECM-receptor interaction pathway mediated by HMMR. Curr. Gene Ther. 24, 249–263. https://doi.org/10.2174/0115665232274223231017052707 (2024).
Xue, C., Chen, C., Gu, X. & Li, L. Progress and assessment of LncRNA DGCR5 in malignant phenotype and immune infiltration of human cancers. Am. J. Cancer Res. 11, 1–13 (2021).
Li, X. et al. Mitochondrial lipid metabolism genes as diagnostic and prognostic indicators in hepatocellular carcinoma. Curr. Genomics. 24, 110–127. https://doi.org/10.2174/1389202924666230914110649 (2023).
Shi, Z. et al. Construction of a prognostic model based on genome-wide methylation analysis of MiRNAs for hepatocellular carcinoma. Epigenomics 16, 513–527. https://doi.org/10.2217/epi-2023-0365 (2024).
Zhang, X., Zhang, D., Bu, X., Zhang, X. & Cui, L. Identification of a novel miRNA-based recurrence and prognosis prediction biomarker for hepatocellular carcinoma. BMC Bioinform. 23, 479. https://doi.org/10.1186/s12859-022-05040-y (2022).
Xiao, S., Zha, Y. & Zhu, H. miR-621 May suppress cell proliferation via targeting LncRNA SNHG10 in acute myeloid leukemia. Cancer Manag Res. 13, 2117–2123. https://doi.org/10.2147/CMAR.S269528 (2021).
Chen, X., Tu, J., Liu, C., Wang, L. & Yuan, X. MicroRNA-621 functions as a metastasis suppressor in colorectal cancer by directly targeting LEF1 and suppressing Wnt/beta-catenin signaling. Life Sci. 308, 120941. https://doi.org/10.1016/j.lfs.2022.120941 (2022).
Zhang, Y. et al. Downregulated miR-621 promotes cell proliferation via targeting CAPRIN1 in hepatocellular carcinoma. Am. J. Cancer Res. 8, 2116–2129 (2018).
Wang, X. et al. MiR-454-3p promotes apoptosis and autophagy of AML cells by targeting ZEB2 and regulating AKT/mTOR pathway. Hematology 28, 2223874. https://doi.org/10.1080/16078454.2023.2223874 (2023).
Liao, H. et al. miR–454–3p inhibits non–small cell lung cancer cell proliferation and metastasis by targeting TGFB2. Oncol. Rep. 45 https://doi.org/10.3892/or.2021.8018 (2021).
Liu, H. et al. Mir-454-3p induced WTX deficiency promotes hepatocellular carcinoma progressions through regulating TGF-beta signaling pathway. J. Cancer. 13, 1820–1829. https://doi.org/10.7150/jca.67478 (2022).
Qian, X. L. et al. MiR-454-3p promotes oxaliplatin resistance by targeting PTEN in colorectal cancer. Front. Oncol. 11, 638537. https://doi.org/10.3389/fonc.2021.638537 (2021).
Li, N. et al. MiR-454-3p regulates high glucose-induced mesothelial-mesenchymal transition and Glycolysis in peritoneal mesothelial cells by targeting STAT3. Ren. Fail. 46, 2394635. https://doi.org/10.1080/0886022X.2024.2394635 (2024).
Omorou, M. et al. The emerging role of miR-653 in human cancer. Cancer Epidemiol. 79, 102208. https://doi.org/10.1016/j.canep.2022.102208 (2022).
Chen, Q., Deng, Q., Pan, Y., Ding, X. & Liu, J. Hypoxia-induced miR-653 enhances colorectal cancer progression by targeting circSETD3/KLF6 axis. J. Cancer. 14, 163–173. https://doi.org/10.7150/jca.78865 (2023).
Lin, Y. et al. MiR-653-5p drives osteoarthritis pathogenesis by modulating chondrocyte senescence. Arthritis Res. Ther. 26, 111. https://doi.org/10.1186/s13075-024-03334-5 (2024).
Ren, F. J. et al. MiR-149-5p: an important MiRNA regulated by competing endogenous RNAs in diverse human cancers. Front. Oncol. 11, 743077. https://doi.org/10.3389/fonc.2021.743077 (2021).
Ma, J., Wei, H., Li, X. & Qu, X. Hsa-miR-149-5p suppresses prostate carcinoma malignancy by suppressing RGS17. Cancer Manag Res. 13, 2773–2783. https://doi.org/10.2147/CMAR.S281968 (2021).
Zhou, L., Xu, X. L. & Long Non-Coding RNA ARAP1-AS1 Facilitates the progression of cervical cancer by regulating miR-149-3p and POU2F2. Pathobiology 88, 301–312, (2021). https://doi.org/10.1159/000507830
Li, H. et al. Long Non-coding RNA HOTAIR function as a competing endogenous RNA for miR-149-5p to promote the cell growth, migration, and invasion in non-small cell lung cancer. Front. Oncol. 10, 528520. https://doi.org/10.3389/fonc.2020.528520 (2020).
Zhu, Y. et al. Mechanism of circADD2 as CeRNA in childhood acute lymphoblastic leukemia. Front. Cell. Dev. Biol. 9, 639910. https://doi.org/10.3389/fcell.2021.639910 (2021).
Lin, C., Wu, J., Wang, Z. & Xiang, Y. Long non-coding RNA LNC-POTEM-4 promotes HCC progression via the LNC-POTEM-4/miR-149-5p/Wnt4 signaling axis. Cell. Signal. 124, 111412. https://doi.org/10.1016/j.cellsig.2024.111412 (2024).
Iwakawa, H. O. & Tomari, Y. Life of RISC: Formation, action, and degradation of RNA-induced Silencing complex. Mol. Cell. 82, 30–43. https://doi.org/10.1016/j.molcel.2021.11.026 (2022).
Ghosh, M., Saha, S., Li, J., Montrose, D. C. & Martinez, L. A. p53 engages the cGAS/STING cytosolic DNA sensing pathway for tumor suppression. Mol. Cell. 83 (e266), 266–280. https://doi.org/10.1016/j.molcel.2022.12.023 (2023).
Xu, D., Tian, Y., Xia, Q. & Ke, B. The cGAS-STING pathway: novel perspectives in liver diseases. Front. Immunol. 12, 682736. https://doi.org/10.3389/fimmu.2021.682736 (2021).
Gu, L., Hickey, R. J. & Malkas, L. H. Therapeutic targeting of DNA replication stress in cancer. Genes (Basel). 14. https://doi.org/10.3390/genes14071346 (2023).
Jia, X. et al. Comprehensive multi-omics analyses expose a precision therapy strategy that targets replication stress in hepatocellular carcinoma using WEE1 Inhibition. J. Adv. Res. https://doi.org/10.1016/j.jare.2025.02.016 (2025).
Li, X. et al. The Notch signaling pathway: a potential target for cancer immunotherapy. J. Hematol. Oncol. 16 https://doi.org/10.1186/s13045-023-01439-z (2023).
Montagner, A. et al. Notch signaling and PD-1/PD-L1 interaction in hepatocellular carcinoma: potentialities of combined therapies. Biomolecules 14 https://doi.org/10.3390/biom14121581 (2024).
Tang, B. et al. Diagnosis and prognosis models for hepatocellular carcinoma patient’s management based on tumor mutation burden. J. Adv. Res. 33, 153–165. https://doi.org/10.1016/j.jare.2021.01.018 (2021).
Yang, X., Tian, X., Zhao, P., Wang, Z. & Sun, X. Paclitaxel inhibits hepatocellular carcinoma tumorigenesis by regulating the circ_0005785/miR-640/GSK3beta. Cell. Biol. Int. 47, 1170–1182. https://doi.org/10.1002/cbin.11906 (2023).
Funding
This study was supported by Joint Projects of Applied Basic Research of Kunming Medical University and Yunnan Province Science and Technology Department (202201AY070001-070), The Clinical Key Specialty Project in Pathology of Yunnan Province (300061).
Author information
Authors and Affiliations
Contributions
Guomiao Su: Writing -original draft、Data curation、Validation and Visualization.Juan Wang: Writing -review & editing、Supervision and Resources.Pingrong Xu: Methodology and Formal analysis.Yu Zhang and Yuxin Gao: Data curation and Formal analysis.Fenglin Xue and Zhenhui Li: Project administration and Funding acquisition.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
This study involving human participants was reviewed and approved by the ethics committee of the First Affiliated Hospital of Kunming Medical University [(2023) Ethics L No. 205]. The patients/participants provided written informed consent to participate in this study.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Su, G., Wang, J., Xu, P. et al. Glycolysis-related MiRNA signature predicts prognosis, recurrence risk, and therapeutic responses in hepatocellular carcinoma. Sci Rep 16, 332 (2026). https://doi.org/10.1038/s41598-025-29846-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-29846-x
