
Abstract
The pro-inflammatory molecule leukotriene B4 binds to the leukotriene B4 receptors 1 and 2 (BLT1 and BLT2). Although BLT1 has potential as a target for treating inflammatory diseases, no selective BLT1 antagonist has been marketed to date. Most of the developed compounds bind to both BLT receptors, which are now considered to have some opposite functions. Our goal was to identify potential selective antagonists of BLT1 that could be advanced in future studies for the treatment of inflammatory conditions. We performed a robust virtual screening process using a library of bioactive compounds. The library was screened against BLT1 first, and the top hits were then screened against BLT2. The top eight candidates were selected based on several criteria for further studies, starting with predictions of various physicochemical, pharmacokinetic, and toxicity properties. Next, each selected compound underwent a 500 ns molecular dynamics simulation to evaluate ligand–protein complex stability and the interaction profile. Finally, the compounds were ranked based on predicted binding affinities by calculating their binding free energies. Three hits showed ligand–protein stability in MD simulations, maintained essential interactions, and showed lower binding free energies than the reference compound, indicating they may be more potent BLT1 antagonists. The three identified candidates have unique scaffolds and show potential as potent and selective BLT1 antagonists. These findings pave the way for future experimental evaluation to advance these compounds as anti-inflammatory agents.
Similar content being viewed by others
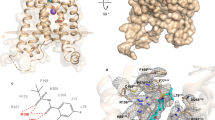


Introduction
Leukotrienes are a group of endogenous molecules with potent biological activities that function similarly to histamine, which mediates inflammatory effects often associated with asthma and edema. Leukocytes primarily synthesize leukotrienes in response to immunological and non-immunological stimuli. One leukotriene of interest is leukotriene B4 (LTB4), a lipid that promotes inflammation. LTB4 is biosynthesized from arachidonic acid, catalyzed by several enzymes, including cytosolic phospholipase A2, 5-lipoxygenase, and leukotriene A4 hydrolase1. This leukotriene has been extensively investigated, as it is one of the most potent chemoattractant agents known. LTB4 plays a vital role in promoting the accumulation of granulocytes2,3 and macrophages4, as well as neutrophil chemotaxis3.
Two receptors have been identified for LTB4, the leukotriene B4 receptor 1 (BLT1) and the leukotriene B4 receptor 2 (BLT2). Both receptors are members of class A G protein-coupled receptors (GPCRs) that adopt the canonical seven-transmembrane (7TM) helical structure typical of lipid receptors. The orthosteric binding pockets of BLT1 and BLT2 are located within the 7TM region and are primarily composed of hydrophobic residues that form a deeply buried hydrophobic core5. The pocket opens toward the extracellular surface, where polar residues contribute to ligand anchoring and receptor activation6,7. Notably, BLT1 has a unique feature among lipid receptors, an open pocket that lacks a cap (or lid) on the extracellular side5. Other lipid receptor 7TM regions have caps from the extracellular side to prevent solvent entry and non-specific binding5.
BLT1 and BLT2 share 42.22% sequence identity, mostly in the 7TM region. Despite moderate overall sequence identity, both receptors maintain conserved hydrophobic transmembrane regions that form the core of the orthosteric pocket (Fig. 1). In contrast, variations near the pocket entrance define their selectivity. In BLT1, His 94 and Arg 156 serve as critical polar anchors for the antagonist, whereas in BLT2, Thr 274, Arg 270, Gln 247, and His 172 form an interaction surface with different properties, providing a structural rationale for ligand selectivity between the two subtypes.

Sequence alignment of human BLT1 and BLT2 receptors. The TM regions are shown in boxes, and identical residues are highlighted in blue. Sequence alignment was performed using UniProt’s Align tool8.
BLT1 binds LTB4 with higher affinity compared to the promiscuous BLT2, which also binds to other eicosanoids9. BLT1 is the most specific receptor for LTB4 and has been implicated in the pathogenesis of several diseases, including asthma10,11,12, osteoporosis13,14, rheumatoid arthritis15,16, atherosclerosis17, aneurysm18, and spinal cord injury19, among others. On the other hand, the physiological and pathophysiological roles of BLT2 have only recently been studied in detail. Unlike BLT1, BLT2 exhibits anti-inflammatory actions20, supports the maintenance of skin21 and barrier22 functions, and promotes intestinal23, skin24, and corneal25 wound healing. Overall, the role of BLT1 is mainly pathogenic, while BLT2 appears to have protective functions.
BLT1 is expressed on the surface of most immune cells; therefore, targeting BLT1 with antagonists is a logical approach to combat inflammatory diseases. Many BLT1 antagonists have been developed for the treatment or management of various diseases, and their development has been extensively reviewed26,27. Growing evidence continues to support their ability to reduce inflammation and, consequently, their potential as anti-inflammatory agents. However, none of the developed antagonists have reached the market due to non-selectivity, toxicity, long half-life, and poor efficacy26,27. Interestingly, most of these compounds were antagonists of both BLT1 and BLT2, and we now know that these two receptors have some opposing functions. This can be a major drawback of non-selective BLT antagonists, as these opposing actions are likely to manifest in vivo, resulting in poor efficacy. Moreover, since BLT2 is expressed in many cell types and tissues, binding to the receptor may worsen the side-effect profile of a developed compound. The efficacy and safety of BLT1 antagonists may be improved by developing BLT1-selective agents that do not antagonize BLT2.
Recently, the first X-ray structure of BLT1 in its inactive conformation (antagonist-bound) was solved, laying the groundwork for research on BLT1 antagonism by revealing the binding behavior of a selective BLT1 antagonist6. The usefulness of antagonizing BLT1 has long been established, and new findings on the role of BLT2 in inflammation will enable more informed drug discovery projects. Here, we use a robust structure-based screening workflow to identify potential selective BLT1 antagonists that can be further developed in future studies for various inflammatory conditions. A variety of in silico methods were utilized in this work, including homology modeling, virtual screening, prediction of absorption, distribution, metabolism, excretion, and toxicity (ADME/T) properties, molecular dynamics (MD) simulations, and binding free energy calculations (Fig. 2).

The in silico screening workflow used to identify potential selective antagonists of BLT1.
Results and discussion
Homology modeling
We used homology modeling to fill in missing residues in BLT1 and to construct a BLT2 model, which lacks experimental structures. The X-ray crystal structure of BLT1 (PDB ID: 7K15, 2.88 Å)6 was selected as the template for both studies. This BLT1 structure shares 40% similarity with BLT2, covers 82% of the BLT2 sequence, and is in an inactive state. For each receptor, five homology models were initially generated, and the models with the lowest discrete optimized protein energy (DOPE) scores were selected for further loop modeling, yielding ten models per entry. For each receptor, the final model that had the lowest molecular probability density function (molpdf) score was chosen. The structural quality of both the BLT1 and BLT2 models was assessed using Ramachandran plot analyses (Fig. 3). As shown in the Ramachandran plots, most of the residues in both receptors were in the favored regions. Enrichment studies for both receptors and docking of the co-crystallized antagonist (MK-D-046) into BLT1 were used to validate model quality further.

Analyses of Ramachandran plots for the homology models of (A) BLT1 and (B) BLT2. Residues are shown in blue and red. Favored regions are highlighted in orange, allowed regions in yellow, and disallowed regions in white.
Enrichment studies
Homology model performance and the docking protocol can be evaluated through enrichment studies to determine whether the protein structures and protocol used can enrich active molecules from numerous decoy molecules. Enrichment is important to ensure that, when a set of unknown compounds is screened, the identified hits are likely to be active. Before performing virtual screening against BLT1 and BLT2, two enrichment studies were conducted, one for BLT1 and the other for BLT2. All compounds were prepared, and potent antagonists were identified from the literature (an antagonist was considered potent if its IC50 value was 1 μM or less). Using AutoDock Vina (version 1.2.5)28,29, potent BLT1 antagonists and decoy molecules were docked against the prepared model of BLT1 (Table S1), and potent BLT2 antagonists and decoy molecules were also docked against the prepared model of BLT2 (Table S2).
Enrichment curves are shown in Fig. 4. For the BLT1 enrichment study, the area under the curve of the receiver operating characteristic (AUC ROC) value was 0.73. In the BLT2 study, the AUC ROC was 0.67. These values indicate that the docking method used for both receptors can effectively distinguish between active and decoy molecules. AUC ROC values that are closer to 1 indicate better performance in separating active molecules from others.

ROC AUC curves for each of the enrichment studies. (A) BLT1 and (B) BLT2.
Additionally, the docked pose of the co-crystallized BLT1 antagonist MK-D-046 (docking score = − 11.31 kcal mol−1) aligned very well with its pose in the crystal structure (Fig. 5) (PDB ID: 7K15)6, with a root mean square deviation (RMSD) value of 1.12. This low RMSD value also validates the docking protocol used for BLT1. Moreover, MK-D-046 forms hydrogen bonds with the 3N-H tautomer of His 94 and with Arg 156 in the X-ray crystal structure of BLT1 (PDB ID: 7K15)6, and the same interactions are observed in the docked pose of MK-D-046 (Fig. 5). In the BLT1 enrichment study, the docking scores of active molecules were within the − 11.31 to − 7.56 kcal mol−1 range. In the BLT2 enrichment study, the docking scores of the active molecules ranged from − 10.73 to − 7.45 kcal mol−1. These values are important for understanding how to assess the binding of unknown ligands to either receptor.

Docking of MK-D-046, the BLT1 antagonist co-crystallized with the receptor. (A) Overlay of docked pose (cyan) with the X-ray crystal structure pose (silver), and (B) interactions of the docked MK-D-046 (cyan) with BLT1 residues (grey). The ball and stick representation is used for the residues and MK-D-046. Interactions are depicted as dashed lines. Hydrogen bonds are shown in yellow and π–π stacking interactions in blue.
Virtual screening
We used AutoDock Vina (version 1.2.5)28,29 for the virtual screening to identify potential selective antagonists of BLT1. First, the commercially available Bioactive Compound Library was retrieved from MedChemExpress30. This library comprises biologically active compounds with known targets, including clinical, natural, approved, and innovative compounds. 16,879 Prepared ligands from the library were docked into the BLT1 model. The range of the docking scores in this stage of the virtual screening was from − 14.20 to 196.20 kcal mol−1. Next, ligands with docking scores of − 7.56 kcal mol−1 or lower were retained for the next stage of screening. This cutoff value was set based on the docking scores of potent BLT1 antagonists observed in the enrichment studies, with − 7.56 kcal mol−1 as the highest score. This filtration criterion resulted in 11,954 ligands, which were docked into the BLT2 model.
For the second screening stage, the docking scores ranged from − 14.41 to 26.52 kcal mol−1. In a second filtration step, ligands that had docking scores of − 6.75 kcal mol−1 or lower (i.e., stronger predicted binding) against BLT2 were excluded, which resulted in 221 ligands. This cutoff value was derived from the docking scores observed for potent BLT2 antagonists in the enrichment study, with − 7.45 kcal mol−1 as the highest (least favorable) score. To lower the likelihood of retaining potent BLT2 binders, we applied a cutoff score of − 6.75 kcal mol−1 (higher than − 7.45 kcal mol−1 and indicative of less favorable predicted binding to BLT2), thereby enhancing the potential selectivity toward BLT1. The 221 ligands were examined in detail to assess their binding interactions with both receptors.
The critical roles of the BLT1 residues His 94 and Arg 156 in antagonist affinity, efficacy, and potency have been established through mutagenesis studies6. His 94 was also found to be important for ligand selectivity for BLT1 over BLT26. In the crystal structure of BLT1 (PDB ID: 7K15)6, MK-D-046 was found to form hydrogen bonds to these two key residues, the 3N-H tautomer of His 94 and Arg 156. Hydrogen bonding to these two residues was found to be critical for BLT1 antagonist potency6. Since MK-D-046 is a potent and selective BLT1 antagonist, we prioritized ligands that showed a similar interaction pattern with BLT1 to that of MK-D-046. Specifically, ligands that made hydrogen bonds with both His 94 and Arg 156 in BLT1 were selected.
Next, the chosen ligands were examined for their interactions with BLT2. In mutagenesis studies of BLT2 residues, Thr 274 was shown to be important for BLT2 antagonist potency and selectivity6. In another study, the potent and selective BLT2 antagonist LMT-2074-L was docked into BLT2, forming interactions with Arg 270, Gln 247, and His 1727. Therefore, ligands that formed one or more interactions with the BLT2 residues Thr 274, Arg 270, Gln 247, or His 172 were filtered out to improve the potential for the selective antagonism of BLT1. Eight ligands were selected for further assessment by prioritizing those with favorable binding to BLT1 based on the set cutoff docking score and key hydrogen bonding with His 94 and Arg 156, while excluding ligands that showed favorable binding to BLT2 based on the set cutoff docking score and interactions with Thr 274, Arg 270, Gln 247, or His 172.
The identified top eight hits represent a structurally diverse set of compounds (Fig. 6). This structural diversity may be advantageous for optimizing selectivity and compound properties in future design efforts. The docking scores of the eight hits and MK-D-046 are shown in Fig. 6 as well, while their binding interactions with BLT1 are shown in Fig. 7. Two-dimensional (2D) diagrams for the interactions between the eight hits with BLT1 and BLT2 are shown in the supplementary information Figs. S1 and S2, respectively.

Structures and docking scores for the eight hits and MK-D-046. Scores are in kcal mol−1.

Docking poses of the eight hits (compound numbers are shown in boxes). Ligands are shown in cyan, and residues in grey. Interactions are depicted as dashed lines. Hydrogen bonds are shown in yellow, π–cation interactions in green, and π–π stacking interactions in blue. The ball and stick representation is used for the ligands and residues.
ADME/T property prediction
The ADME/T data represent a detailed assessment of a range of pharmaceutically relevant properties of a series of compounds that potentially have utility as selective antagonists of BLT1 (Table 1). None of the compounds showed structural alerts associated with pan-assay interference compounds (PAINS). Compounds with high topological polar surface area (TPSA) tend to exhibit impaired absorption, as they are associated with reduced permeability. However, compounds with high predicted human intestinal absorption (HIA) may still be valuable oral candidates. Our compounds have TPSA values that range from 30.93 to 268.68 Å2, indicating that most of the compounds fall under the permissible range for membrane permeability; however, compounds with large TPSA values, like compounds 7 and 8 (TPSA = 212.89 and 268.68, respectively), can generally demonstrate reduced predicted absorption via poor membrane permeability. On the other hand, compounds 1–6 and MK-D-046 had high predicted HIA probabilities. The molecular weight (MW) values were also quite heterogeneous, with some compounds exceeding the Lipinski Rule of 5 (Ro5) limit of 500 Da, suggesting potential challenges with in vivo oral bioavailability. However, this is not a major concern at this stage, as our primary focus is on establishing activity and favorable overall compound properties.
Predicted human liver microsomal (HLM) stability varied among the compounds. For example, compounds 1, 5, and MK-D-046 had the highest probabilities of rapid metabolic clearance. The remaining compounds were predicted to be stable, i.e., not metabolized by the liver within 30 min. To assess the general safety profile of the compounds, the potential for non-selectivity was evaluated using promiscuity prediction. Except for compound 6, all compounds showed low probabilities of promiscuity, indicating a reduced risk of off-target interactions. The probability of causing human hepatotoxicity was evaluated. Compounds 1, 3, and 7 were predicted to be non-hepatotoxic and the safest, while others had variable predicted probabilities. Carcinogenicity was also assessed, with compounds 4 and 7 showing the highest probabilities of carcinogenicity, while compounds 3, 8, and MK-D-046 had the lowest. In general, our results reveal varying predicted carcinogenicity and human hepatotoxicity risks, which are particularly noteworthy given their importance in drug discovery and development.
Based on predicted ADME/T properties, the combination of PAINS-free structures, high HIA, favorable HLM stability, lack of promiscuity, and low to moderate risks of human hepatotoxicity and carcinogenicity identifies compounds 2 and 3 as the most promising candidates. Despite its poor HLM stability due to multiple ester groups, compound 1 remains a promising candidate. Overall, compounds 1, 2, and 3 are valuable candidates for the development of selective BLT1 antagonists. Our ADME/T predictions study demonstrates the importance of conducting an early and comprehensive evaluation of compound properties to prioritize potential drug candidates for further development.
Molecular dynamics simulations and binding free energy calculations
RMSD and RMSF analyses
For the top eight hits and the reference compound, MK-D-046, we evaluated the stability of the ligand–BLT1 complexes, docking poses, and essential hydrogen bonds using MD simulations. To assess the stability of the BLT1 protein backbone throughout the simulation in each ligand–BLT1 complex, we analyzed the RMSD of Cα atoms (Fig. 8). A rise in RMSD values was observed at the beginning of the simulation across all complexes. This rise is expected, as frame 0, corresponding to the initial protein structure, was used as the reference for the RMSD calculation. The increase likely reflects conformational adjustments as the system adapts to the simulation environment during the early stages. After 50 ns, the protein structure stabilizes in each of the complexes with MK-D-046 and compounds 1, 2, and 3. Each of the proteins in complex with compounds 4, 6, and 8 stabilizes after 125 ns, 75 ns, and 200 ns, respectively. The proteins in all complexes, except those with compounds 5 and 7, remained stable for the remainder of the simulation, with RMSD fluctuations within the acceptable range of approximately 1–3 Å, despite higher absolute RMSD values due to early conformational adaptation. Based on these findings, these proteins have reached equilibrium.

Analysis of RMSD for the Cα atoms of BLT1 in each complex. The average RMSD values are provided in parentheses.
On the other hand, the proteins in complexes with compounds 5 and 7 showed greater fluctuations than those in the other complexes, suggesting less stability. The RMSD values for the Cα atoms of BLT1 in complex with compound 5 increased continuously over the simulation, indicating that the protein did not reach equilibrium. The lowest average RMSD value for the BLT1 Cα atoms was 9.1 Å for the MK-D-046 complex, while the highest was 14.4 Å for the compound 4 complex.
RMSD analysis was also performed for ligand heavy atoms to assess conformational and positional stability within the BLT1 binding site (Fig. 9). Lower ligand RMSD values suggest a more stable binding mode. However, acceptable ranges can vary depending on compound size and flexibility. The lowest average ligand heavy-atom RMSD value was 2.6 Å for the MK-D-046 complex, while the highest was 5.6 Å for the compound 7 complex. The closest average RMSD value to the reference compound (MK-D-046) was that of compound 5.

Analysis of RMSD for the ligand heavy atoms in each complex. The average RMSD values are provided in parentheses.
Based on both RMSD analysis and visual inspection of the MD trajectories, all ligands except for compound 5 exhibited minor, acceptable fluctuations and remained stable throughout most of the simulation time. This stability suggests that these ligands bind efficiently within the BLT1 binding site. Compound 5 showed larger RMSD fluctuations, corresponding to positional shifts during the simulation. MK-D-046 and compounds 1, 2, and 3 had no major structural fluctuations compared to their docking poses. Compounds 1, 2, and 3 have larger molecular sizes compared to the others, so although some parts of their structures deviate from their initial conformations, the effect on the overall pose within the binding site is not as pronounced as the rest of the compounds. Indeed, the smaller compounds 4, 6, 7, and 8 exhibited structural deviations in certain regions of their structures relative to their docking poses, which altered their poses considerably and consequently changed their interaction patterns compared to those observed in docking. Compared to its docking pose, compound 5 showed positional shifts rather than conformational changes, as it has a more planar structure than the others. These results suggest that compounds 1–3, like MK-D-046, bind more consistently and stably within the BLT1 pocket.
Based on the root mean square fluctuation (RMSF) analysis, all ligand–BLT1 complexes show similar overall fluctuation patterns (Fig. 10). As expected, the largest fluctuations observed are in extracellular and intracellular loop regions. On the other hand, the TM helices and binding site residues show minimal fluctuations, with low RMSF values. These findings indicate that the helical structure was conformationally preserved, with no destabilization observed in any of the complexes throughout the simulations.

Per-residue RMSF analysis of Cα atoms of BLT1 in each complex over the 500 ns simulations. TM regions are shown in boxes.
Interaction analysis
Hydrogen bond occupancy is calculated from MD trajectories and represents the percentage of simulation time during which each hydrogen bond remains. Interactions that persist for ≥ 50% of the simulation time are considered strong and stabilizing. Notably, several strong hydrogen bonds are observed with key residues involved in antagonist anchoring and BLT1 selectivity, supporting their potential functional relevance.
Only MK-D-046 and compounds 1, 2, and 3 maintain the essential hydrogen bonds to His 94 and Arg 156 for most of the simulation, indicating stable binding to the two most important residues for potent BLT1 antagonism6 (Fig. 11). Such binding, especially to His 94, ensures a more selective antagonism toward BLT1 over BLT26. MK-D-046 forms additional strong interactions, including a hydrogen bond with Asn 268, a π–π stacking interaction with His 94, and other stable water-mediated interactions. Compound 1 forms another strong interaction with Ser 6, while compound 2 has an additional strong water-mediated hydrogen bond with Arg 156. Compound 3 has additional stable water-mediated interactions with several residues. Movies of the binding of compounds 1, 2, 3, and MK-D-046 are available as supplementary files; only strong interactions are shown.



Intermolecular interactions between the ligands and BLT1 residues during MD simulations. (A) MK-D-046, the reference compound, and (B–I) compounds 1–8. The diagrams show contacts that persisted for more than 30% of the 500 ns simulation time.
On the other hand, the remaining compounds do not form stable hydrogen bonds with the key residues His 94 and Arg 156. These findings indicate that although they may have performed well in docking, they do not bind to BLT1 stably in a dynamic, flexible environment and are not expected to act as potent antagonists based on the required interaction profile for BLT1 antagonism. Upon closer analysis, Compound 4 forms stable hydrogen bonds to Tyr 102, Tyr 237, and Glu 185. Compound 5 interacts only and strongly with a single residue, Tyr 102. The only strong interaction that compound 6 forms is a water-mediated hydrogen bond with Glu 185, while compound 7 forms several stable water-mediated and direct hydrogen bonds with multiple residues. The strongest hydrogen bonds that compound 8 makes are with Glu 185 and His 94. Taken together, these results divide the series into two groups: MK-D-046 and compounds 1–3, which sustain stable dual anchoring to His 94 and Arg 156, and compounds 4–8, which show only partial or transient anchoring and rely more heavily on other contacts.
Notably, compounds 1, 2, and 3 reveal stable and persistent hydrophobic interactions with pocket residues, including Phe 74, Phe 77, Phe 275, and Ile 271 (Fig. 12). Similarly, MK-D-046 forms stable hydrophobic interactions with Phe 74, Tyr 102, and Ile 271. These findings support the hydrophobic complementarity between these molecules and the BLT1 binding pocket, consistent with the scaffold analysis discussed below. For compounds 1, 2, 3, and MK-D-046, a detailed breakdown of interaction fraction by amino acid type is provided in the supplementary information (Figs. S3−S6).


Timeline representation of ligand–residue interactions during the 500 ns MD simulation for MK-D-046 and compounds 1–3. Each row represents a residue, and the color intensity reflects the number of interactions across the MD frames, including hydrogen bonds, hydrophobic interactions, ionic interactions, water bridges, and halogen bonds.
Binding free energy analysis
Binding free energies were calculated for MD simulation frames of the eight hits and MK-D-046 using the molecular mechanics with generalized Born and surface area (MM-GBSA) method (Fig. 13). This end-point method is commonly used to rank compounds based on predicted target affinity, which often correlates well with the corresponding affinity ranking in experimental assays. The reference compound MK-D-046 has an average ΔGbind value of –94.28 kcal mol−1. Compounds 4, 5, 6, 7, and 8 have considerably higher average ΔGbind values compared to the reference compound, indicating less efficient binding, especially for compound 6, which has the highest average ΔGbind value. On the other hand, compounds 1, 2, and 3 have average ΔGbind values of –105.56, –146.49, and –117.18 kcal mol−1, respectively. Compared to the potent and BLT1-selective antagonist MK-D-046 (IC50 = 2 nM31), the binding free energies for compounds 1, 2, and 3 are significantly lower. These findings suggest that compounds 1, 2, and 3 may be selective BLT1 antagonists with greater potency than the reference compound. Overall, the predicted affinity ranking for the all compounds is 2 (–146.49 kcal mol−1) > 3 (–117.18 kcal mol−1) > 1 (–105.56 kcal mol−1) > MK-D-046 (–94.28 kcal mol−1) > 7 (–80.08 kcal mol−1) > 4 (–68.39 kcal mol−1) > 5 (–67.18 kcal mol−1) > 8 (–63.56 kcal mol−1) > 6 (–54.20 kcal mol−1).

Average ΔGbind for the eight hits and MK-D-046 (in kcal mol−1).
Collectively, our MD simulations and subsequent binding free energy evaluations identify three promising candidates for further development as selective BLT1 antagonists. The protein–ligand complexes of compounds 1, 2, and 3 were stable and maintained the essential interactions required for potent and selective BLT1 antagonism throughout the majority of the simulation duration. These three compounds also had high predicted binding affinities, surpassing that of the reference compound. The overall compound selection process is summarized in Fig. 14, which shows the progression from the initial library to the final three MD-stable hits.

Stepwise compound selection workflow showing the progression from the initial library of 16,879 compounds to the top three candidates.
Scaffold analysis
To evaluate scaffold novelty of compounds 1, 2, and 3, we compared their core scaffolds to those of three known selective BLT1 antagonists, MK-D-046, CP-105696, and U-75302 (Fig. 15). Although the scaffolds of compounds 1, 2, and 3 are structurally distinct from those of established BLT1-selective antagonists, they were observed in our MD simulations to anchor to the essential His 94 and Arg 156 residues via hydrogen bonds, similar to MK-D-046. The scaffolds of compounds 1, 2, and 3 align their hydrogen-bonding groups in suitable locations and orientations for efficient interaction with these residues.

Scaffold comparison between (A) known selective BLT1 antagonists and (B) compounds 1–3. The scaffolds are highlighted in color.
A comparative analysis between the known selective BLT1 antagonists and the new scaffolds of compounds 1–3 further highlights both shared pharmacophoric elements and distinct structural features (Table 2). All compounds support dual anchoring to His 94 and Arg 156 through hydrogen bond acceptors (HBAs), which are essential interactions for BLT1 antagonism. MK-D-046 and CP-105696 have semi-rigid polyaromatic hydrophobic scaffolds with multiple polar groups that can act as HBAs for stable dual anchoring. In contrast, U-75302 and compounds 1–3 have mostly flexible structures containing HBAs and aliphatic or mixed aromatic-aliphatic hydrophobic groups. The new scaffolds have distinct hydrophobic moieties. Compounds 1 and 3 have a biphenyl core with methylene linkers, while compound 3 also features additional aliphatic chains, resulting in an overall higher hydrophobicity. Compound 2 has a steroid-like nucleus and a partially unsaturated chain.
The new scaffolds show a substantial hydrophobic character that complements the hydrophobic region of the BLT1 binding pocket. This hydrophobic region was previously characterized in the BLT1–LTB₄ and BLT1–MK-D-046 experimental structures5,6. They also contain the essential HBAs, which can maintain stable hydrogen bonds with His 94 and Arg 156, as observed in our MD simulation results. These findings demonstrate mechanistic consistency and structural diversity, presenting new chemotypes that are capable of reproducing the essential BLT1 anchoring motif and supporting the potential of compounds 1, 2, and 3 as BLT1-selective antagonists.
Conclusion
BLT1 binds LTB4 with high affinity and is an attractive target to inhibit for combating inflammation. Although a number of BLT1 antagonists have been identified, none have been marketed. Most of these compounds exhibited non-selective antagonism by targeting both BLT receptors. Recent research revealed that these two receptors have certain opposing functions, limiting the efficacy and safety of non-selective antagonists. We aimed to identify potential selective antagonists of BLT1 using in silico screening methods. These compounds may have improved safety and efficacy profiles and could be advanced in future studies to treat inflammatory conditions.
We performed enrichment studies to validate the docking protocols for BLT1 and BLT2 using sets of antagonists and decoys. Next, we virtually screened a commercial library of bioactive compounds against BLT1 and selected the best-scoring compounds for further screening against BLT2. We selected the top hits to move forward for additional studies based on selected docking score cutoff values, evaluation of the presence of hydrogen bonds with key residues in BLT1, absence of important interactions with BLT2, and assessment of ADME/T profiles.
For the top eight compounds, the stabilities of ligand–protein complexes and interaction profiles were evaluated by using MD simulations. Ligand–protein complexes for compounds 1, 2, and 3 remained stable for most of the simulation time and formed the required hydrogen bonds with key residues in BLT1 for the majority of the simulation. These three compounds are potentially potent and selective BLT1 antagonists based on their favorable and stable interaction profiles. Finally, we ranked the compounds based on predicted binding affinity by calculating their binding free energies. Remarkably, the same three hits had binding free energies lower than the co-crystallized antagonist, indicating they may be more potent BLT1 antagonists. Notably, these three hits also possess scaffolds distinct from known BLT1-selective antagonists, expanding the chemical space available for BLT1 antagonist design.
Our robust in silico approach has successfully identified three promising candidates as potential potent and selective antagonists of BLT1. These hits demonstrated favorable docking scores, selectivity potential, acceptable properties, stable binding with BLT1 that maintains essential hydrogen bonds, and exceptional binding free energies. These findings represent a starting point for experimental evaluation to advance these compounds as distinct anti-inflammatory agents.
Computational methods
Homology modeling
The antagonist-bound X-ray crystal structure of BLT1 (PDB ID: 7K15, 2.88 Å)6 was retrieved from the Protein Data Bank32, but it contains missing residues. Moreover, there are no experimental structures for BLT2 in the inactive state to date. To address these problems, MODELLER33 (version 10.4) was used to fill in missing residues in BLT1 (UniProtKB8 code: Q15722) and to construct the BLT2 model (UniProtKB8 code: Q9NPC1). The X-ray crystal structure of BLT1 (PDB ID: 7K15)6 was used as a template for both homology modeling studies. Compared to BLT2, this structure is 40% similar, has 82% coverage, and is in the inactive state. Water molecules and the sodium atom in the BLT1 binding pocket were conserved. The amino acid sequences of BLT1 and BLT2 were aligned with those in the X-ray crystal structure. Next, five homology models were generated for each receptor, and the models with the lowest DOPE scores were subjected to loop modeling. Ten homology models were generated for each initial model after loop modeling, and the final model with the lowest molpdf score was selected for use in this study. The Ramachandran plots for the models were generated using Maestro Viewer34 (version 2024-2). The homology model of BLT1 was further validated by docking of MK-D-046 using the process outlined below. For both BLT1 and BLT2, enrichment studies were used to validate the homology models as well.
Protein preparation
The Protein Preparation Workflow35 in Maestro Viewer34 (version 2024-2) was used for the initial preparation of the homology models for BLT1 and BLT2. The Chemical Component Dictionary (CCD) database was used to assign bond orders. Hydrogens were added, termini were capped, zero-order bonds to metals were created, and waters beyond 5 Å were deleted. The 3N-H tautomers of histidine (HID) were retained for BLT1, as they are important for capturing key hydrogen bonds to His 94. AutoDockTools36 (version 1.5.7) was used to add Kollman37 charges to the proteins, spread charge deficit over all atoms, and save the proteins in PDBQT format.
Ligand preparation
PyMol38 (version 3.0.2) was used to add missing hydrogens. 2D Ligands were cleaned in three-dimension (3D) using the fine build method in MarvinView39 (version 23.16.0). OpenBabel40 (version 3.1.1) was used to perform energy minimization for the ligands using the Merck Molecular Force Field (MMFF94)41,42. The files were saved in PDBQT format.
Enrichment studies
41 Potent BLT1 antagonists and 41 potent BLT2 antagonists (IC50 values of less than 1 μM) were retrieved from the PubChem43 database. 1000 Drug-like decoy molecules were obtained from the Schrödinger precompiled library, with an average molecular weight of approximately 400 Da. These decoy molecules are preprocessed to assign appropriate ionization and tautomeric states at physiological pH. The set of 42 active BLT1 antagonists (41 antagonists and MK-D-046) and the 1000 decoys were prepared and docked into BLT1, and the set of 41 active BLT2 antagonists and the 1000 decoys were prepared and docked into BLT2. These two separate enrichment studies were performed using the protocol described in the following section. The RMSD value between the docking pose of MK-D-046 and its X-ray crystal structure pose was calculated using PyMol38 (version 3.0.2). Screening Explorer44 was used to analyze enrichment study results and generate ROC curves.
Virtual screening
16,879 Prepared ligands from MedChemExpress30 Bioactive Compound Library were docked into the BLT1, and the best-scoring ones (11,954) were docked into the BLT2. AutoDock Vina (version 1.2.5)28,29 was used for the enrichment and virtual screening studies, following the protocol outlined below. The grid box was centered at X = 1.872, Y = − 9.602, Z = 17.298 with dimensions of 30 Å × 30 Å × 30 Å, defined around the co-crystallized antagonist MK-D-046 for BLT1. The same coordinates and dimensions were applied to BLT2 after structural superposition, since BLT1 and BLT2 have similar amino acid compositions in their binding sites (Fig. 16). The global searching exhaustiveness was set to 25, and an energy range cutoff of 5 kcal mol−1 was used. Ligands were treated as flexible, and one pose per compound was generated. The ligand interaction diagram in Maestro Viewer34 (version 2024-2) was used to visualize interactions between the ligands and receptors.

Grid box placement for BLT1 and BLT2 illustrating their orthosteric binding sites defined by structural superposition.
ADME/T property prediction
The physicochemical, pharmacokinetic, and toxicity profiles were predicted for the top eight compounds and MK-D-046 using ADMETlab 3.045, which provides comprehensive predictions of numerous molecular properties. The SMILES notation was used as the input for the ADMETlab 3.0 tool. We evaluated several properties, including MW, TPSA, log P, absorption, stability, and toxicity. Additionally, we assessed characteristics such as violations of Lipinski’s Ro5, promiscuity, and the presence of PAINS. This in silico approach can aid in selecting valuable drug candidates for further development as BLT1 antagonists for inflammatory diseases.
Molecular dynamics simulations
Molecular dynamics simulations were performed for MK-D-046 and the eight hits using Desmond in the Schrödinger suite46,47,48. To build the system, a lipid bilayer composed of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) at 300 K was added to the BLT1 structure, since it is a transmembrane GPCR. Next, the simple point charge (SPC)49 water model was selected, as it is the validated and recommended choice for simulations using the Optimized Potentials for Liquid Simulations (OPLS) force field in Desmond. An orthorhombic box of 10 Å3 was used, and placement of ion and salt was excluded within 20 Å of the ligand. To neutralize the system, counterions were added while maintaining a 0.15 M salt concentration, using sodium and chloride ions. The OPLS5 force field was selected50,51, and the built model was relaxed. For the 500 ns simulations, the temperature was set to 300 K and the pressure to 1 bar. The NPT ensemble was used, recording coordinates every 500 ps, resulting in 1000 frames. For thermal control, the Nose–Hoover chain coupling scheme was applied with a 1 ps coupling constant. For pressure control, the Martyna–Tobias–Klein coupling scheme was applied with a 2 ps coupling constant. The RESPA (Reference System Propagator Algorithm) was set to have a 2 fs time step, and Coulombic short-range interactions were set to have a 9 Å cutoff radius. Desmond’s Simulation Interaction Diagram tool46,47,48 was used to evaluate molecular dynamics, stability, and binding. The stabilities of ligand–protein complexes were evaluated using RMSD and per-residue RMSF.
Binding free energy calculations
Binding free energies (ΔGbind) of the eight hits and MK-D-046 against BLT1 were calculated using Schrödinger’s implementation of the MM-GBSA continuum solvation method52,53. For each ligand, ΔGbind was calculated based on MD simulation frames using Schrödinger’s thermal_mmgbsa script and a step size of 5. The average ΔGbind for each ligand is then calculated by using the equation below:
where Gligand is the ligand energy, Gtarget is the target energy, and Gcomplex is the energy of their bound state in the complex.
Data availability
Data will be made available upon reasonable request.
References
Yokomizo, T. Two distinct leukotriene B4 receptors, BLT1 and BLT2. J. Biochem. 157, 65–71 (2015).
Palmer, R. M. J., Stepney, R. J., Higgs, G. A. & Eakins, K. E. Chemokinetic activity of arachidonic acid lipoxygenase products on leuocytes of different species. Prostaglandins 20, 411–418 (1980).
Ford-Hutchinson, A. W., Bray, M. A., Doig, M. V., Shipley, M. E. & Smith, M. J. H. Leukotriene B, a potent chemokinetic and aggregating substance released from polymorphonuclear leukocytes. Nature 286, 264–265 (1980).
Smith, M. J. H., Ford-Hutchinson, A. W. & Bray, M. A. Leukotriene B: a potential mediator of inflammation. J. Pharm. Pharmacol. 32, 517–518 (1980).
Wang, N. et al. Structural basis of leukotriene B4 receptor 1 activation. Nat. Commun. 13, 1156 (2022).
Michaelian, N. et al. Structural insights on ligand recognition at the human leukotriene B4 receptor 1. Nat. Commun. 12, 1–12 (2021).
Kim, M. et al. Elucidation of mechanism for ligand efficacy at leukotriene B4 receptor 2 (BLT2). ACS Med. Chem. Lett. 11, 1529–1534 (2020).
The UniProt Consortium. UniProt: the Universal Protein Knowledgebase in 2025. Nucleic Acids Res 1–9 (2024).
Yokomizo, T. & Shimizu, T. The leukotriene B4 receptors BLT1 and BLT2 as potential therapeutic targets. Immunol. Rev. 317, 1–12 (2023).
Medoff, B. D. et al. Antibody-antigen interaction in the airway drives early granulocyte recruitment through BLT1. Am. J. Physiol. Lung Cell Mol. Physiol. 290, L170–L178 (2006).
Medoff, B. D. et al. BLT1-mediated T cell trafficking is critical for rejection and obliterative bronchiolitis after lung transplantation. J. Exp. Med. 202, 97–110 (2005).
Taube, C. et al. The leukotriene B4 receptor (BLT1) is required for effector CD8+ T cell-mediated, mast cell-dependent airway hyperresponsiveness. J. Immunol. 176, 3157–3164 (2006).
Hikiji, H., Ishii, S., Yokomizo, T., Takato, T. & Shimizu, T. A distinctive role of the leukotriene B4 receptor BLT1 in osteoclastic activity during bone loss. Proc. Natl. Acad. Sci. USA 106, 21294–21299 (2009).
Bouchareychas, L., Grössinger, E. M., Kang, M., Qiu, H. & Adamopoulos, I. E. Critical role of LTB4/BLT1 in IL-23 induced synovial inflammation and osteoclastogenesis via NF-κB. J. Immunol 198, 452–460 (2017).
Shao, W.-H., Del Prete, A., Bock, C. B. & Haribabu, B. Targeted disruption of leukotriene B4 receptors BLT1 and BLT2: A critical role for BLT1 in collagen-induced arthritis in mice. J. Immunol. 176, 6254–6261 (2006).
Mathis, S. P., Jala, V. R., Lee, D. M. & Haribabu, B. Nonredundant roles for leukotriene B4 receptors BLT1 and BLT2 in inflammatory arthritis. J. Immunol. 185, 3049–3056 (2010).
Heller, E. A. et al. Inhibition of atherogenesis in BLT1-deficient mice reveals a role for LTB4 and BLT1 in smooth muscle cell recruitment. Circulation 112, 578–586 (2005).
Ahluwalia, N. et al. Inhibited aortic aneurysm formation in BLT1-deficient mice. J. Immunol. 179, 691–697 (2007).
Asahara, M. et al. The absence of the leukotriene B4 receptor BLT1 attenuates peripheral inflammation and spinal nociceptive processing following intraplantar formalin injury. Mol. Pain 11, 1–12 (2015).
Iizuka, Y. et al. Protective role of the leukotriene B4 receptor BLT2 in murine inflammatory colitis. FASEB J. 24, 4678–4690 (2010).
Ishii, Y. et al. Leukotriene B4 receptor type 2 (BLT2) enhances skin barrier function by regulating tight junction proteins. FASEB J. 30, 933–947 (2016).
Hara, T. et al. The c-terminal region of BLT2 restricts its localization to the lateral membrane in a LIN7C-dependent manner. FASEB J 35, 1–11 (2021).
Matsumoto, Y. et al. Leukotriene B4 receptor type 2 accelerates the healing of intestinal lesions by promoting epithelial cell proliferation. J. Pharmacol Exp. Ther. 373, 1–9 (2020).
Liu, M. et al. 12-Hydroxyheptadecatrienoic acid promotes epidermal wound healing by accelerating keratinocyte migration via the BLT2 receptor. J. Exp. Med. 211, 1063–1078 (2014).
Iwamoto, S. et al. Non-steroidal anti-inflammatory drug delays corneal wound healing by reducing production of 12-hydroxyheptadecatrienoic acid, a ligand for leukotriene B4 receptor 2. Sci. Rep. 7, 1–10 (2017).
Bhatt, L., Roinestad, K., Van, T. & Springman, E. B. Recent advances in clinical development of leukotriene B4 pathway drugs. Semin Immunol 33, 65–73 (2017).
Hicks, A., Monkarsh, S. P., Hoffman, A. F. & Goodnow, R. Jr. Leukotriene B4 receptor antagonists as therapeutics for inflammatory disease: Preclinical and clinical developments. Expert Opin Investig Drugs 16, 1909–1920 (2007).
Trott, O. & Olson, A. J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 31, 455–461 (2010).
Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2.0: new docking methods, expanded force field, and Python bindings. J. Chem. Inf. Model 61:3891–8 (2021).
MedChemExpress. n.d. www.medchemexpress.com.
Han Y, Lim J, Tummanapalli S, Siliphaivanh P, Spencer K. Aryl acylsulfonamides as BLT1 antagonists. 10,336,733 (2019).
Research Collaboratory for Structural Bioinformatics Protein Data Bank (RCSB PDB). n.d.
Šali, A. & Blundell, T. L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 234, 779–815 (1993).
Schrödinger Release 2024-2: Maestro Viewer (2024).
Schrödinger Release 2024-2: Protein Preparation Workflow (2024).
Morris, G. M. et al. AutoDock4 and AutoDockTools4: automated docking with selective receptor flexibility. J. Comput. Chem. 30, 2785–2791 (2009).
Besler, B. H., Merz, K. M. Jr. & Kollman, P. A. Atomic charges derived from semiempirical methods. J. Comput. Chem. 11, 431–439 (1990).
The PyMOL Molecular Graphics System (2024).
MarvinView Version 23.16.0 (2023).
O’Boyle, N. M. et al. Open babel: An open chemical toolbox. J. Cheminform 3, 1–14 (2011).
Halgren TA. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 17:490–519 (1996).
Halgren TA. Merck molecular force field. II. MMFF94 van der Waals and electrostatic parameters for intermolecular interactions. J. Comput. Chem. 17:520–52 (1996).
Kim, S. et al. PubChem 2023 update. Nucleic Acids Res. 51, D1373–D1380 (2023).
Empereur-Mot, C., Zagury, J.-F. & Montes, M. Screening explorer–an interactive tool for the analysis of screening results. J. Chem. Inf. Model 56, 2281–2286 (2016).
Fu L, Shi S, Yi J, Wang N, He Y, Wu Z, Peng J, Deng Y, Wang W, Wu C. ADMETlab 3.0: an updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Res 52:W422–31 (2024).
Schrödinger Release 2025–1: Desmond Molecular Dynamics System (2025).
Schrödinger Release 2025–1: Maestro-Desmond Interoperability Tools (2025).
Bowers KJ, Chow DE, Xu H, Dror RO, Eastwood MP, Gregersen BA, Klepeis JL, Kolossvary I, Moraes MA, Sacerdoti FD, Salmon JK, Shan Y, Shaw DE. Scalable algorithms for molecular dynamics simulations on commodity clusters. Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), IEEE, p. 43–43 (2006).
Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. Interaction models for water in relation to protein hydration. Intermolecular Forces: Proceedings of the Fourteenth Symposium on Quantum Chemistry and Biochemistry, Springer, p. 331–42 (1981).
Schrödinger Release 2025–1: Force Fields (2025).
Jorgensen, W. L., Maxwell, D. S. & Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118, 11225–11236 (1996).
Kollman, P. A. et al. Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897 (2000).
Li, J. et al. The VSGB 2.0 model: a next generation energy model for high resolution protein structure modeling. Proteins Struct. Funct. Bioinf. 79, 2794–2812 (2011).
Acknowledgements
The authors acknowledge Ibrahim Alkuraiji for his participation during the early stages of this work. Funding for this study from the King Abdullah International Medical Research Center, Ministry of National Guard Health Affairs (SP23R/220/09) is acknowledged.
Funding
This work was supported by the King Abdullah International Medical Research Center, Ministry of National Guard Health Affairs [SP23R/220/09].
Author information
Authors and Affiliations
Contributions
R.A.A: Conceptualization, Writing—original draft, Writing—review and editing, Software, Resources, Project administration, Supervision, Methodology, Funding acquisition, Validation, Visualization, Investigation, Data curation, Formal analysis. S.A.A: Writing—original draft, Visualization, Investigation, Formal analysis. O.S.A: Writing—original draft, Visualization, Investigation, Formal analysis. M.D.A: Writing—original draft, Writing—review and editing, Investigation, Formal analysis.
Corresponding author
Ethics declarations
Competing interests
The authors have no relevant financial or non-financial interests to disclose.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Supplementary Material 2
Supplementary Material 3
Supplementary Material 4
Supplementary Material 5
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Alkhodier, R.A., Alshamrani, S.A., Almutlaq, O.S. et al. Structure-based identification of compounds with potential as selective BLT1 antagonists. Sci Rep 16, 1675 (2026). https://doi.org/10.1038/s41598-025-31192-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-31192-x
