
Abstract
Neuroinflammation is closely associated with activation of NLRP3 inflammasome after acute ischemic stroke. Our previous study preliminarily found that remimazolam mitigated cerebral ischemia–reperfusion (I/R) injury in MCAO rats, possibly by inhibiting the expression of the NLRP3 inflammasome pathway. Previous studies showed that the prime and activation of NLRP3 inflammasome are regulated by NF-κB. Therefore, the exact mechanism of the effect of remimazolam on I/R injury needs further study. Rat MCAO I/R injury model and primary cultured rat cortical neurons OGD/R injury model were used to investigate the effect of remimazolam on reducing neuronal pyroptosis. The neurological deficit score assessed neurological function. Cerebral infarct volume was measured using TTC staining. Cell viability and injury were assessed by CCK-8 and LDH. Cell pyroptosis was evaluated using TEM. The mRNA levels of the NF-κB/NLRP3 inflammasome signaling pathway were assessed using qPCR. Western blotting and immunofluorescence staining detected the protein expression of the NF-κB/NLRP3 inflammasome signaling pathway. Remimazolam alleviates cerebral infarct volume and neurological deficit in MCAO rats, increases cell viability, and decreases LDH release of OGD/R cortical neurons. TEM showed that after remimazolam treatment, a significant reduction in the pyroptosis of neurons both in vivo and in vitro. Furthermore, remimazolam down-regulated the mRNA levels and protein expression of NF-κB, NLRP3, ASC, and Caspase-1, and reduced the release of IL-1β. Remimazolam may attenuate cortical neuronal pyroptosis by inhibiting NF-κB mediated activation of NLRP3 inflammasome after acute I/R injury.
Similar content being viewed by others


Introduction
Ischemic stroke is the second leading cause of preventable deaths in human beings1. The high morbidity and mortality caused by stroke are a huge financial burden for the government. Timely and successful vascular reperfusion is the most effective method to treat acute ischemic stroke and reduce post-stroke complications such as epilepsy, pain, cognitive problems, and depression2. However, vascularization can lead to reperfusion injuries, such as edema and hemorrhage, and further neuronal death3. General anesthesia is becoming the preferred treatment for endovascular therapy in patients with acute ischemic stroke4,5. Compared with local anesthesia and conscious sedation, general anesthesia has no negative effect on clinical outcome, and even better outcomes, higher recanalization rates, and fewer procedural complications6. In addition, general anesthetics also inhibit stress and inflammation7,8. However, general anesthetic often causes hypotension and may exacerbate ischemic damage to the brain9,10. Therefore, the selection of appropriate general anesthesia drugs is particularly important.
Neuroinflammation is increasingly important in ischemic stroke11,12,13. In addition, neuroinflammation can activate the NLRP3 inflammasome after acute ischemic stroke14. After that, the GSDMD N-terminus especially binds to the cell membrane to form the permeable pores whose inner diameter is 10–14 nm, which causes water influx, and then the cell swelling, cell membrane rupture, and the mass release of inflammatory factors like IL-1β, IL-18. This kind of cell death is called pyroptosis15,16,17. We know that the priming and activation of NLRP3/ASC/Caspase-1 inflammasome are regulated by NF-κB18. However, it is not clear whether general anesthetics can regulate NLRP3/ASC/Caspase-1 inflammasome through the NF-κB signaling pathway to reduce neuroinflammation and pyroptosis.
Remimazolam is a novel benzodiazepine ultra-short-acting compound, which can activate the GABAA receptor and has the characteristics of rapid induction, rapid awakening, stable hemodynamics, and light respiratory inhibition19,20,21,22,23. As a result, it is more and more widely used in clinical practice. A study showed that the expression of GABAA receptors in the cerebral cortex decreased after ischemia24. Furthermore, higher GABAA receptor expression and functions could be protective for acute cerebral ischemia injury25. However, whether remimazolam, as a GABAA receptor agonist, has a protective effect on acute ischemic stroke, and what the specific mechanism remains unclear.
Thus, the rat MCAO I/R injury model and primary cultured rat cortical neurons OGD/R injury model were used in the study to investigate the effect of remimazolam on reducing neuronal pyroptosis, possibly by inhibiting the NF-κB/NLRP3 inflammasome signaling pathway.
Materials and methods
Experimental animals
Male, specific pathogen-free, Sprague–Dawley rats (7 ~ 8 weeks, 280 ± 20 g), and neonatal rats less than one day old were obtained from the laboratory animal center of Guangxi Medical University. Rats were allowed to acclimate for 1 week before modeling. Neonatal rats were immediately used for the primary culture of cortical neurons. The animal experiment was conducted with the approval of the ethics committee of Guangxi Medical University (Animal Experimental Ethical Inspection No.202006009), strictly following The Guide for Care and Use of Laboratory Animals released by the National Institutes of Health.
Model establishment
The rat model of MCAO was constructed following the method described previously26. In brief, after the intraperitoneal injection of sodium pentobarbital took effect, the right common carotid artery (CCA), external carotid artery (ECA), and internal carotid artery (ICA) were isolated by dissection. A special line (RWD Life Science, 907-00018-01, China) was placed from ECA to ICA, passing through the bifurcation of CCA. The special line reached the intracranial part of the ICA to block the right MCA blood flow, with the strand-forward length as 18 ± 2 mm from the bifurcation of CCA. After 2 h of occlusion, the special line was pulled out for reperfusion. When the neurological deficit scores of rats were greater than 1, the MCAO model was regarded as successful27 (Fig. 1a,b).

Remimazolam alleviates cerebral infarct volume and neurological deficit in MCAO rat. (a, b) Left forelimb weakness after right MCAO in rats. (c) The representative picture of TTC staining. (d) The cerebral infarct volume after 24 h of reperfusion (n = 5 per group). (e, f) The neurological deficit score at 2 h of ischemia and after 24 h of ischemia–reperfusion (n = 6 per group). Data are presented as Mean ± SEM. ****p < 0.0001 and ***p < 0.001: significant as compared to the Sham group; ###p < 0.001: significant as compared to MCAO group.
Experimental groups and drug treatment in vivo
Rats were randomly divided into 8 groups: Group Sham, Group MCAO, Group R5, Group R10, Group R20, Group DMSO, Group MCC950, and Group JSH-23. In the sham group, the procedure was the same except that the special line was not inserted. The MCAO group underwent the right middle cerebral artery occlusion. The R5, R10, and R20 groups underwent the right middle cerebral artery occlusion, and immediately after reperfusion, remimazolam was administered 5 mg/kg (1*ED50)28, 10 mg/kg (2*ED50), and 20 mg/kg (4*ED50) through the tail vein, respectively. MCC950 (Selleck, S7809, USA), a select NLRP3 inhibition, was administered as the group MCC95029,30. JSH-23 (Selleck, S7351, USA), a selective NF-κB inhibitor, was administered as the group JSH-2331,32. The vehicle group was treated with equivalent normal saline or dimethyl sulfoxide (DMSO) (Fig. 1). Each experiment was independently repeated 5 times.
Neurological deficit score
Five-grade (0 ~ 4 points) Longa method was applied to confer a neurological deficit score at 2 h of ischemia and 24 h of reperfusion, respectively. Grade I (0-point): normal behavior without any neurological deficit; Grade II (1-point): the left front paw fails to fully extend; Grade III (2-point): turn to the left (paralyzed side) while walking; Grade IV (3-point): tip to the left (paralyzed side) while walking; Grade V (4-point): unable spontaneous walking with impairment of consciousness. Models scored 1–3 points were included, they were excluded from the study27.
Cerebral infarct volume measurement
Briefly, following model establishment, the rats were decapitated under deep anesthesia, and the brain tissue, excluding the cerebellum, olfactory bulb, and lower brain stem, was extracted and frozen at −20℃ for 20 min. The tissue block was cut to obtain coronal brain slices (2 mm). Being exposed to 2% TTC solution (Solarbio, G3005, China), the slices were water-bathed at 37℃ in the dark for 30 min. Shaking at 5-min intervals was performed to ensure uniform staining. Subsequently, the stained tissue samples were fixed overnight with 4% paraformaldehyde and dried on the following day for photography. Image J software was run to obtain the infarct area (white) on the tail side, and the infarct volume was calculated according to the slice thickness33.
Cortical neuron culture
Primary cortical neurons were cultured from the cerebral cortex of newborn SD rats according to a previously described protocol34. Briefly, in a sterile environment, the brain tissue of newborn SD rats was quickly removed after anesthesia, and then the cortical tissue was isolated in a culture medium on ice. The cerebral cortex was cut into pieces with sterile scissors and digested with 0.25% trypsin–EDTA (Gibco, 15,050–057, USA) for 10 min at 37 °C. The same amount of medium was added to neutralize trypsin and then centrifuged at 1000 rpm for 5 min, and the supernatant was discarded. The planting medium was added to the precipitate for re-suspension and screening (200 and 400 mesh) to obtain single-cell suspensions, which were then planted on poly-lysine-coated plates (Solarbio, P2100, China) containing the planting medium. The plating medium contained 88% DMEM/F12 (Gibco, G4610, USA), 10% fetal bovine serum (FBS, Gibco, 10,099-141C, USA), 1% glutamine (Solarbio, G0200, China), and 1% penicillin/streptomycin (Solarbio, P1400, China), then transferred into a cell incubator of 5% CO2 at 37 °C. After three hours in the planting culture medium, the culture medium was replaced with a serum-free maintenance medium consisting of 1% glutamine, 1% penicillin/streptomycin, 96% neurobasal medium (Gibco, 21,103,049, USA), and 2% B27 (Gibco, 17,504,044, USA). The maintenance medium was replaced in half every three days.
Establishment of the OGD/R model and experimental group
After 7 days in primary culture, the cortical neurons’ maintenance medium was changed to glucose-free DMEM (Gibco, 11966-025, USA) and transferred to a modular incubator chamber (Billups-Rothenberg, MIC-101, USA) filled with 95% N2 and 5% CO2 at 37 °C. After 2 h of oxygen and glucose deprivation, cortical neurons were transferred into a normal cell incubator for reoxygenation for 12 h. The primary cortical neurons in different Petri dishes were randomly divided into 13 groups: Group C, Group CR1, Group CR10, Group CR20, Group CR50, Group OGD, Group R1, Group R10, Group R20, Group R50, Group DMSO, Group MCC950, and Group JSH-23 (Fig. 2). In the control group, the neurons were cultured with a fresh maintenance medium. In CR1, CR10, CR20, and CR50 groups, the cells were incubated with different concentrations of remimazolam (1, 10, 20, and 50 µM, respectively) alone for 12 h. In the OGD group, the neurons underwent OGD for 2 h and reoxygenation for 12 h. In R1, R10, R20, and R50 groups, the cells underwent OGD for 2 h and then, during reoxygenation, immediately with different concentrations of remimazolam (1, 10, 20, 50 µM, respectively) post-treatment for 12 h. In MCC950, JSH-23, and DMSO groups, neurons were pre-incubated for 30 min before OGD, then OGD for 2 h, and reoxygenation for 12 h with 10 µM MCC950, 10 µM JSH-23, and 0.25% DMSO, respectively (Fig. 2).

Remimazolam increased cell viability of OGD/R cortical neurons in vitro. (a) The purity of cortical neurons. The cortical neurons were stained for NeuN as neuronal marker (red staining). Nuclei were stained with DAPI. Bar: 20 μm. (b) Neurons were treated with 1, 10, 20, and 50 μM remimazolam. Morphology of cortical neuron. Scale bars: 20 μm. (c) The cell viability of neurons was incubated with different concentrations of remimazolam alone for 12 h by CCK-8. (d) The viability of OGD/R cortical neurons after different concentrations of remimazolam post-treatment. (e) Relative release of LDH in cortical neurons culture medium. All tests were repeated independently three times. Data are presented as Mean ± SEM. ****p < 0.0001: significant as compared to the control group; ##p < 0.01: significant as compared to OGD group.
Transmission electron microscopy
Cerebral cortex tissues and cortical neuron preparation were performed as previously described35,36. Rats were transcardially perfused with 50 ml of 2.5% glutaraldehyde after anesthesia. Samples were obtained from cortical infarcts as cubes measuring approximately 1 mm3. Cerebral cortex tissues and cortical neurons were fixed with 2.5% glutaraldehyde. After that, they were dehydrated by concentration gradient ethanol and acetone, embedded in acetone and embedding solution, cured in an oven, sliced by the ultra-thin slicer, and dyed with 3% uranium acetate and lead citrate. Then, cortical neuron morphology and pyroptosis were observed with an H-7650 TEM (Hitachi, Japan).
Quantitative real-time PCR (qPCR)
RNA extraction, reverse transcription, and qPCR were performed strictly according to the instructions of TRIzol Reagent (Invitrogen, 15,596–026, USA), PrimeScript RT reagent kit (Takara, RR047A, Japan), and SYBR Premix Ex Taq TM II kit (Takara, RR820A, Japan), respectively. β-actin was used as a reference for gene expression. The primers used in the RT-PCR were as follows:
β-actin, forward 5′-AGAAATGGTGCCTGGACACCTCAT-3′
and reverse 5′-GCTCAGTAACAGTCCGCCTAGA-3′;
NF-κB, forward 5′-CCAGGCGGACATCTACAA-3′
and reverse 5′-CAAGGCCAAATGAAAGGA-3′;
NLRP3, forward 5′-GGTGACCTTGTGTGTGCTTG-3′
and reverse 5′-ATGTCCTGAGCCATGGAAGC-3′;
ASC, forward 5′-GTGGGTGGCTTTCCTTGATT-3′
and reverse 5′-TTGTCTT GGCTGGTGGTCTCT-3′;
GSDMD, forward 5′-TTGAGTGTCTGGTGCTCGAC-3′
and reverse 5′-ATGGGGTGCTCTGTTCCAAG-3′;
Caspase-1, forward 5′-TTATCAGGGTTGACCCCTTGG-3′
and reverse 5′-TTGCCCTCAGGATCTTGTCAG-3′. Each sample was measured thrice.
Immunofluorescence
The preparation of cell slivers and the implementation of immunofluorescence were similar to our previous study36. After 7 days of culture on slides, the cortical neurons were fixed with 4% paraformaldehyde (Solarbio, P1110, China) at room temperature for 30 min and then washed 3 times with PBS for 5 min each. After permeabilization with 0.1% Triton X-100 (Solarbio, P1080, China), cell slides were blocked with 1% BSA (Solarbio, SW3015, China) for 1 h at room temperature. Subsequently, cell slides were incubated overnight with the primary antibody at 4 °C. The primary antibodies used for immunofluorescence were: rabbit anti-NF-κB p65 antibody (1:500, CST, 8242, USA), rabbit anti-NLRP3 antibody (1:200, Wanleibio, WL02635, China), rabbit anti-GSDMD antibody (1:200, ABclonal, A18281, China), rabbit anti-ASC antibody (1:200, Abcam, ab180799, USA), rabbit anti-Caspase-1 p20 antibody (1:200, Invitrogen, PA5-9584, USA), mouse anti-NeuN (1:10,000, Abcam, ab104224, USA). The cell slides were then incubated with goat anti-rabbit IgG H&L (1:500, Alexa Fluor® 488 conjugation, Abcam, ab150077, USA) and goat anti-mouse IgG H&L (1:500, Alexa Fluor® 594 conjugation, Abcam, ab150116, USA) for 1 h. Lastly, the cell slides were observed with a fluorescence microscope (EVOS, Japan).
Cell viability assay
Cortical neuron viability was assessed by CCK-8 (Solarbio, CK04, China) assay and performed strictly according to the manufacturer’s instructions as the same as described in our previous research36. The results were read in a 450 nm wavelength absorbance test (Thermo Fisher Scientific Inc., USA).
Lactate dehydrogenase assay
LDH released in the culture medium was detected for the evaluation of cell injury by the LDH kit (Beyotime, C0016, China) and performed strictly according to the instructions.
Western blotting
Cerebral cortex tissues and cortical neuron preparation were performed as our previous studies described36,37. The cerebral cortex was homogenized in a cryogenic homogenizer (LUKYM-1, China) after adding the proper amount of lysis buffer (RIPA buffer, protease inhibitor, and phosphatase inhibitor, 100:1:1). For cells, after washing with PBS 3 times, an appropriate amount of lysate into the culture plate, and neuronal cells were scraped off and homogenized with sonication. After the samples were lysed on ice for 20 min, they were centrifuged at 4 °C at 12000 g for 20 min, then the supernatant was transferred into a new EP tube. The protein contents of tissues and cells were determined by the BCA kit (Beyotime, P0010S, China). Then, each protein sample was added to 8 ~ 12% sodium dodecyl sulfate–polyacrylamide (SDS-PAGE) gels and separated by electrophoresis, followed by wet-transfer to polyvinylidene fluoride (PVDF) membrane (0.22 μm, Merck Millipore, GER). After blocked with 5% BSA (Solarbio, China) for 1 h, all PVDF membranes were incubated overnight at 4 °C with primary antibodies which listed as follows: rabbit anti-β-actin (1:1000, CST, 4970, USA), rabbit anti-NLRP3 (1:1000, Abcam, ab214185, USA), rabbit anti-p65 (1:1000, CST, 8242, USA), rabbit anti-GSDMS antibody (1:1000, Abcam, ab219800, USA), rabbit anti-ASC antibody (1:1000, Abcam, ab180799, USA), rabbit anti-Caspase-1 p20 antibody (1:1000, Invitrogen, PA5-99390, USA). Then, the membranes were incubated with fluorescent dye-coupled secondary antibody (Invitrogen, 31460, USA) at room temperature for 1 h. Images were captured by the Odyssey system (LI-COR Biosciences) and analyzed on ImageJ software.
Enzyme-linked immunosorbent assay (ELISA)
After the homogenized tissues and cell culture mediums were centrifuged at 4 °C, 1000*g for 20 min, the supernatant was transferred to a new EP tube. IL-1β levels released in cortex samples and neuronal culture medium were analyzed using ELISA kits (Elabscience, China) and performed as in previous studies38.
Statistical analysis
All data are presented as Mean ± SD or SEM. Statistical analyses were performed using SPSS 22.0 (IBM, USA) and GraphPad Prism 9 (Origin Lab, USA). After Normality and Lognormality Tests, multiple comparisons were performed with one-way ANOVA or the Kruskal–Wallis Test. P-values < 0.05 were considered statistically significant.
Results
Remimazolam alleviates cerebral infarct volume and neurological deficit score in MCAO rats
Immediately after reperfusion, remimazolam was injected into the tail vein of rats and given 5, 10, and 20 mg of R5, R10, and R20 groups, respectively. TTC staining was used to measure the infarct volume of MCAO rats after 24 h of ischemia–reperfusion (Fig. 1c). The infarct volumes in the MCAO, R5, R10, and R20 groups (28.84 ± 1.731, 19.86 ± 2.039, 16.04 ± 1.055, and 20.18 ± 2.255, Mean ± SEM, %, respectively) were significantly higher than those in the Sham group (Fig. 1d). The infarct volume in the R10 group was significantly lower than that in the MCAO group (Fig. 1d). A neurological deficit score was used to evaluate the neurological function of MCAO rats at 2 h of ischemia and after 24 h of ischemia–reperfusion. The neurological deficit scores in the MCAO, R5, and R20 group (2.700 ± 0.1528, 2.300 ± 0.1528, and 2.400 ± 0.1623, Mean ± SEM, respectively) were significantly higher than that in the Sham group after 24 h of ischemia–reperfusion. The neurological deficit score in the R10 group (1.600 ± 0.1623) was significantly lower than that in the MCAO group (Fig. 1e,f).
Remimazolam increased cell viability of OGD/R cortical neurons of primary culture in vitro
Primary cortical neurons cultured for 7 days were subjected to identification and purity analyses using immunofluorescence with neuronal marker NeuN (Neuronal Nuclei) antibody, a neuron-specific nuclear protein. Under the fluorescent microscope, the nucleus of the cortical neurons was dyed red. The purity of cortical neurons was 96.2 ± 1.2% (Fig. 2a). The morphological changes of cortical neurons in primary culture for 7 days (Control group), incubation with different concentrations of remimazolam alone for 12 h (C + R group), oxygen–glucose deprivation for 2 h and reoxygenation for 12 h (OGD/R group), and during reoxygenation immediately with different concentrations of remimazolam post-treatment (R groups) were observed under a light microscope. Under the light microscope, the number of neurons began to decrease when the concentration of remimazolam exceeded 20 µM. The number of neurons in the OGD group was significantly reduced, and the synaptic structure was broken. After remimazolam (concentration < 20 µM) post-treatment, the number of neurons and the synaptic structure were significantly improved, especially in the R10 group (Fig. 2b). The results of CCK-8 showed that remimazolam post-treatment significantly increased the viability of OGD/R cortical neurons by 25.66%. Moreover, when the concentration of remimazolam was lower than 20 µM, it did not affect the viability of normal cortical neurons (Fig. 2c,d). LDH assays revealed that compared with the control group, the relative release of LDH from cortical neurons in the OGD/R group was significantly increased (1.00 ± 0.051, 2.08 ± 0.039, Mean ± SD, respectively). The relative release of LDH was reduced by 15.72% in the R10 group compared with the control group (Fig. 2e).
Remimazolam alleviates cortical neuronal pyroptosis both in MCAO rats and in the OGD/R model of Primary cultured cortical neurons in vitro
TEM showed that the cell membrane of the cortical neuron was perforated and ruptured, displaying a typical feature of pyroptosis membrane destruction in the MCAO group after 24 h of ischemia–reperfusion and in the OGD/R cortical neurons. After remimazolam post-treatment, the pore formation of the neuron cell membrane was reduced both in vivo and in vitro and was observed by TEM (Fig. 3a,b).

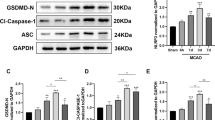
Remimazolam reduces cortical neuronal NLRP3/ASC/Caspase-1 inflammasome-dependent pyroptosis. (a, b) The cell membrane of the cortical neuron was perforated and ruptured, displaying a typical feature of pyroptosis membrane destruction. After remimazolam post-treatment, the pore formation of the neuron cell membrane was reduced both in vivo and in vitro and was observed by transmission electron microscope (TEM). (c, e) In vivo experiments, compared with the sham group, the mRNAs and proteins expression of the NLRP3, ASC, GSDMD, and Caspase-1 were significantly increased in the MCAO group after 24 h of ischemia–reperfusion. After remimazolam post-treatment, the mRNAs and protein expression of the NLRP3, ASC, GSDMD, and Caspase-1 were significantly lower in the remimazolam group (R10) than those in the MCAO group. (d, f) In vitro studies, the mRNAs and proteins expression of the NLRP3, ASC, GSDMD, and Caspase-1 p20 was significantly increased in the OGD/R group, as compared with the control group. After remimazolam post-treatment, the mRNAs and protein expressions of the NLRP3, ASC, GSDMD, and Caspase-1 p20 mRNAs were significantly lower in the R10 group than those in the OGD/R group. (g) Levels of IL-1β in cortex samples and cortical neuron culture supernatants were measured by ELISA. All tests were repeated independently three times. Data are presented as Mean ± SEM. ns: no significant; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001: significant as compared to Sham group or control group; #p < 0.05, ##p < 0.01, and ####p < 0.0001: significant as compared to MCAO group or OGD group.
Remimazolam reduces cortical neuronal NLRP3/ASC/Caspase-1 inflammasome pyroptosis
To investigate whether remimazolam alleviates NLRP3/ASC/Caspase-1 inflammasome pyroptosis induced by cerebral ischemia–reperfusion injury, we conducted both in vivo and in vitro studies. In vivo experiments, compared with the sham group, the mRNAs and proteins expression of the NLRP3, ASC, GSDMD, and Caspase-1 were significantly increased in the MCAO group after 24 h of ischemia–reperfusion (Fig. 3c,e). After remimazolam post-treatment, the mRNAs and protein expression of the NLRP3, ASC, GSDMD, and Caspase-1 were significantly lower in the remimazolam group (R10) than those in the MCAO group (Fig. 3c,e). ELISA indicated levels of IL-1β in the cortex sample were significantly increased in the MCAO group compared with the sham group after 24 h of ischemia–reperfusion. After remimazolam post-treatment, the levels of IL-1β were significantly lower in the remimazolam group (R10) than in the MCAO group (Fig. 3g). In vitro studies, the mRNAs and proteins expression of the NLRP3, ASC, GSDMD, and Caspase-1 p20 was significantly increased in the OGD/R group, as compared with the control group. After remimazolam post-treatment, the mRNAs and protein expressions of the NLRP3, ASC, GSDMD, and Caspase-1 p20 mRNAs were significantly lower in the R10 group than those in the OGD/R group (Fig. 3d,f). ELISA assays revealed that the secretion levels of IL-1β from cortical neurons in the OGD/R group were significantly increased, as compared with the control group. The levels of IL-1β were significantly lower in the remimazolam group (R10) than that in the OGD/R group (Fig. 3g).
Remimazolam reduces cortical neuronal NLRP3/ASC/Caspase-1 inflammasome pyroptosis by inhibiting NF-κB pathway activation
NF-κB is a key initiator of inflammatory response and plays an important role in the inflammatory process of various diseases. To clarify the connections among NF-κB pathway, NLRP3/ASC/Caspase-1 inflammasome pyroptosis, and the effect of remimazolam treatment, we used JSH-23 (an inhibitor of transcriptional activity of NF-κB, Selleck Chemicals, USA) and MCC950 (an inhibitor of transcriptional activity of NLRP3, Selleck Chemicals, USA) as the positive control. We also tested them in vivo and in vitro studies. Our study showed that, after 24 h of ischemia–reperfusion, the protein expressions of the NF-κB p65, NLRP3, and ASC were significantly reduced in the JSH-23 group, however, the protein expression of the NF-κB p65 was not significantly changed in the MCC950 group compared with the MCAO group (Fig. 4a). In vitro studies, immunofluorescence results showed that, as compared with the OGD/R group, the fluorescence intensity of NF-κB p65, NLRP3, GSDMD, and Caspase-1 p20 was markedly decreased in the JSH-23 group, while NF-κB p65 had no significant change in the MCC950 group (Fig. 4b). Compared with the OGD/R group, the mRNAs of NF-κB p65, NLRP3, ASC, GSDMD, and caspase-1 p20 and the protein expressions of NF-κB p65, NLRP3, ASC, and caspase-1 p20 were significantly reduced in the JSH-23 group, while mRNA and protein of NF-κB p65 showed no significant difference in the MCC group (Fig. 4c,d). Similar results were also shown in ELISA assays, both in vitro and in vivo studies, IL-1β secretion in the JSH-23 group and MCC950 group was significantly lower than that in the model group (Fig. 4e).

Remimazolam reduces cortical neuronal NLRP3/ASC/Caspase-1 inflammasome-dependent pyroptosis by inhibiting NF-κB pathway activation. (a) The protein expressions of the NF-κB p65, NLRP3, and ASC were significantly reduced in the JSH-23 group, however, the protein expression of the NF-κB p65 was not significantly changed in the MCC950 group compared with the MCAO group. (b) Fluorescence intensity (green staining) of NF-κB p65, NLRP3, GSDMD, and Caspase-1 p20. Nuclei were stained with DAPI. Bar: 20 μm. (c, d) the proteins expressions of NF-κB p65, NLRP3, ASC, and caspase-1 p20 and the mRNAs of NF-κB p65, NLRP3, ASC, GSDMD, and caspase-1 p20 in vitro. (e) IL-1β secretion by ELISA assays both in vitro and in vivo studies. All tests were repeated independently three times. Data are presented as Mean ± SEM. ns: no significant; *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001: significant as compared to Sham group or control group; #p < 0.05, ##p < 0.01, ###p < 0.001, and ####p < 0.0001: significant as compared to MCAO group or OGD group.
Discussion
This study evaluated the protective effect of remimazolam on cortical neurons after acute ischemic stroke using a rat MCAO/I/R model (in vivo) and a primary cultured rat cortical neuron OGD/R model (in vitro) (Fig. 5). The results demonstrated that cortical neurons undergo NLRP3/ASC/Caspase-1 inflammasome-mediated pyroptosis following I/R injury. Furthermore, post-treatment with remimazolam downregulates the transcriptional expression of NF-κB, inhibits the activation of the NLRP3/ASC/Caspase-1 inflammasome, and thereby alleviates cortical neuronal pyroptosis—providing new insights into the mechanism of remimazolam in cerebral I/R injury.

Diagram of the mechanism of remimazolam alleviating cerebral ischemia–reperfusion injury of rats by inhibiting NF-κB/NLRP3 inflammasome-dependent pyroptosis.
The incidence of ischemic stroke is increasing globally, with a growing trend toward younger adults (≤ 55 years old)39. A rising number of these patients require endovascular therapy under general anesthesia to restore cerebral blood flow. Beyond physical dysfunction, stroke survivors often face psychological sequelae such as depression, which significantly impacts quality of life and requires equal clinical attention40. General anesthetics, traditionally used for perioperative sedation and analgesia, are now being explored for therapeutic potential in conditions like sleep disorders and depression41,42. A large body of evidence indicates that anesthetics exhibit dual properties of neurotoxicity and neuroprotection: under normal physiological conditions, they may induce mild neuronal damage, but in pathological states (e.g., traumatic intracerebral hemorrhage, ischemic stroke), they exert neuroprotective effects—primarily by mitigating perioperative stress and excessive inflammatory responses43,44,45. Remimazolam, a novel ultra-short-acting benzodiazepine intravenous anesthetic, possesses advantages including rapid sedation, anxiolysis, anterograde amnesia, quick onset of action, and rapid recovery. Our previous randomized controlled trial showed that remimazolam induction causes less hemodynamic fluctuation in patients undergoing cardiac valve replacement surgery compared to propofol46 —a critical advantage for stroke patients, who are highly sensitive to hypotension-induced cerebral hypoperfusion. Additionally, multiple studies have reported that propofol exhibits dose-dependent neurotoxicity, leading to neuronal apoptosis in the developing brain or under ischemic conditions36,47; thus, remimazolam may represent a safer alternative to propofol for anesthesia in stroke-related procedures48.
Our prior study preliminarily observed that remimazolam mitigates cerebral I/R injury in MCAO rats, potentially by inhibiting the NLRP3 inflammasome pathway26. To further delineate the underlying molecular mechanism, we established a stable rat MCAO/I/R model (2 h of ischemia followed by 24 h of reperfusion) and a primary cortical neuron OGD/R model (2 h of OGD followed by 12 h of reoxygenation) in the current work—protocols consistent with well-validated standards in the field27,49. In vivo experiments showed that remimazolam post-treatment reduced cerebral infarct volume and improved neurological deficit scores, while in vitro assays demonstrated increased neuronal viability and decreased LDH release. A key observation was the non-dose-dependent neuroprotective effect of remimazolam: the R10 group (10 mg/kg in vivo, 10 μM in vitro), which uses a clinically relevant concentration, exhibited the optimal efficacy, whereas the R5 (lower dose) and R20 (higher dose) groups were less effective. This phenomenon requires in-depth mechanistic interpretation beyond mere description. First, the subtherapeutic effect of the R5 group (5 mg/kg in vivo, 1 μM in vitro)—equivalent to 1 × effective dose for 50% of animals (ED50) for remimazolam sedation—can be attributed to insufficient target engagement. Our data showed that R5 reduced infarct volume and neurological deficits relative to the MCAO/OGD group but to a lesser degree than R10 (Figs. 1d,e, 2d). Molecular analyses further revealed that R5 only decreased NF-κB p65 and NLRP3 expression by ~ 30–40%, whereas R10 achieved a ~ 60–70% reduction (Fig. 4c,d). This aligns with pharmacokinetic principles: subtherapeutic doses of agonists fail to saturate target receptors (e.g., GABAA receptors) or signaling pathways (e.g., NF-κB), resulting in incomplete inhibition of pyroptosis.
Second, the reduced efficacy of the R20 group (20 mg/kg in vivo, 20 μM in vitro)—4 × ED50, exceeding the clinically recommended concentration range18,44—stems from two dose-dependent phenomena: GABAA receptor desensitization and off-target toxicity. Benzodiazepines are known to induce rapid desensitization of GABAA receptors at high concentrations50; as remimazolam concentrations exceed 10 μM, the number of functional GABAA receptors on neuronal membranes declines, limiting downstream inhibition of NF-κB. This is supported by our immunofluorescence data, which show no further reduction in NF-κB p65 fluorescence intensity in the R20 group compared to R10 (Fig. 4b). Additionally, high concentrations of remimazolam may activate off-target pathways (e.g., mitochondrial metabolic disruption) that counteract its neuroprotective effect. A prior study by Freyer et al.51 demonstrated that remimazolam concentrations > 20 μM disrupt metabolic activity in primary human hepatocytes; our in vitro observation of reduced neuronal numbers at > 20 μM (Fig. 2b) suggests a similar toxic effect in cortical neurons, which offsets the anti-pyroptotic benefits of NF-κB inhibition. Collectively, these factors explain the non-monotonic dose–response curve, underscoring the importance of selecting clinically relevant doses to balance efficacy and safety.
To contextualize remimazolam’s uniqueness, it is critical to compare it with other benzodiazepines and commonly used anesthetics for stroke patients. Among benzodiazepines, midazolam and diazepam have been investigated for neuroprotection in cerebral I/R injury, but differ substantially from remimazolam. Midazolam reduces neuroinflammation via inhibition of the toll-like receptor 4 (TLR4)/myeloid differentiation factor 88 (MyD88) pathway but does not target the NLRP3 inflammasome—whereas our data clearly show that remimazolam directly suppresses the expression of NLRP3, ASC, and Caspase-1 (Fig. 3c–f) and reduces interleukin-1β (IL-1β) release (Fig. 3g), specifically targeting pyroptosis15. Diazepam, meanwhile, exhibits weak neuroprotective effects and causes prolonged sedation35,36, increasing the risk of pneumonia or delirium in stroke patients—limitations avoided by remimazolam’s ultra-short-acting pharmacokinetics48. Compared to propofol (a widely used anesthetic for stroke endovascular therapy), remimazolam also offers distinct advantages: propofol induces neuronal apoptosis via inhibition of the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) pathway52, while remimazolam (≤ 20 μM) does not reduce the viability of normal cortical neurons (Fig. 2c), demonstrating a superior safety profile. Additionally, propofol’s anti-inflammatory effect is non-specific, whereas remimazolam targets the NF-κB/NLRP3/pyroptosis axis—critical for mitigating the excessive inflammatory response that drives poor stroke outcomes53. Recent studies have further supported remimazolam’s favorable pharmacology in neurocritical care, including its ability to maintain hemodynamic stability while exerting anti-inflammatory effects—reinforcing its potential as a preferred anesthetic for stroke patients48.
Pyroptosis is a pro-inflammatory programmed cell death pathway distinct from apoptosis and necrosis, and neuroinflammation plays a pivotal role in the pathogenesis and prognosis of ischemic stroke15. Thus, modulating inflammation has become a key therapeutic target for post-stroke intervention. Multiple studies have confirmed that the NLRP3/ASC/Caspase-1 inflammasome regulates neuroinflammation and neuronal death in ischemic stroke14,26. Upon activation, the GSDMD inserts into the neuronal membrane, forming pores (10–14 nm in inner diameter) that trigger water influx, cell swelling, and membrane rupture—subsequently releasing large quantities of inflammatory cytokines (e.g., IL-1β, IL-18) that amplify the inflammatory cascade and exacerbate neuronal damage16. These cytokines are critical mediators of the intense inflammatory response following ischemic stroke, directly influencing disease progression and functional outcomes. In the present study, we observed reduced mRNA and protein expression of NLRP3, ASC, GSDMD, and Caspase-1, as well as decreased IL-1β secretion, in remimazolam-treated groups (Fig. 3c–g). Transmission electron microscopy (TEM) further confirmed that remimazolam attenuated neuronal membrane perforation and rupture—hallmark features of pyroptosis (Fig. 3a,b). These results collectively demonstrate that acute ischemic stroke induces NLRP3/ASC/Caspase-1 inflammasome-dependent neuronal pyroptosis and that remimazolam alleviates this process by inhibiting inflammasome activation.
A critical question raised is the causal relationship between remimazolam’s effect and the NF-κB/NLRP3 pathway, as well as the precise mechanism by which remimazolam inhibits NF-κB. While our study does not employ reverse genetics (e.g., gene knockout) or reverse pharmacology (e.g., mutant protein overexpression) to directly prove causality, we have provided multiple lines of correlative and functional evidence to support the proposed “remimazolam → inhibit NF-κB → suppress NLRP3 inflammasome → alleviate pyroptosis” axis. First, we used two specific pathway inhibitors (JSH-23 for NF-κB, MCC950 for NLRP3) as positive controls. Our results showed that remimazolam (10 mg/kg in vivo, 10 μM in vitro) reduced the expression of NF-κB p65, NLRP3, ASC, and Caspase-1 to a similar extent as JSH-23 (Fig. 4a,c,d). In contrast, MCC950 did not affect NF-κB p65 expression (Fig. 4a,b), confirming that NF-κB acts upstream of NLRP3 in this context. Both remimazolam and JSH-23 also reduced IL-1β release (Fig. 4e) and neuronal pyroptosis (Fig. 3a,b)—consistent with NF-κB being a key mediator of remimazolam’s neuroprotective effect.
Regarding the mechanism of NF-κB inhibition, we acknowledge that the current study cannot definitively distinguish between direct and indirect regulation by remimazolam. However, indirect evidence—combined with established literature—favors a GABAA receptor-dependent pathway: remimazolam is a well-characterized selective GABAA receptor agonist19, and prior studies have shown that enhancing GABAA receptor activity suppresses NF-κB activation to reduce neuroinflammation in cerebral ischemia. Our in vitro data further support this: remimazolam (≤ 20 μM) exerts no toxic effect on normal cortical neurons (Fig. 2c) but only protects against OGD/R-induced injury—consistent with GABAA receptors mediating context-dependent (pathological state-specific) regulation of NF-κB, rather than non-specific cellular effects. Recent work has also reported that GABAA receptor α2 subunits mediate remimazolam’s anti-inflammatory effects in the brain, providing additional support for this upstream pathway. Nevertheless, we explicitly recognize that definitive confirmation requires GABAA receptor blocking experiments (e.g., using bicuculline) or conditional knockout models—work we plan to pursue in future studies to fill this mechanistic gap.
Despite its contributions, this study has several limitations that warrant acknowledgment, particularly regarding clinical translation. First, our animal model uses healthy young Sprague–Dawley rats without comorbidities (e.g., hypertension, diabetes), whereas clinical stroke patients are often elderly (≥ 65 years) with multiple comorbidities that alter neuroinflammatory responses. For example, diabetes enhances NLRP3 activation in cerebral I/R injury, and it remains unknown whether remimazolam’s protective effect would be preserved in such models. Second, our in vitro experiments focus exclusively on primary cortical neurons, but cerebral I/R injury involves a complex interplay of multiple cell types (e.g., microglia, astrocytes, endothelial cells). Microglia are major producers of the NLRP3 inflammasome, and our failure to address remimazolam’s effect on microglial pyroptosis limits our understanding of its impact on the global brain inflammatory microenvironment. Third, a critical gap is the lack of in vivo toxicity assessment: we focused solely on remimazolam’s protective effect against cerebral I/R injury and did not evaluate its impact on other organs (e.g., liver, kidneys) or the overall physiological state of rats (e.g., body weight, behavior, hematological parameters). While the doses used (5–20 mg/kg) were selected based on prior studies demonstrating short-term safety in rats and translate to clinically relevant human doses (~ 0.1–0.4 mg/kg), long-term or high-dose systemic toxicity requires further investigation. Future studies should address this by: (1) measuring serum biomarkers of organ injury (e.g., alanine transaminase for liver, creatinine for kidneys); (2) monitoring long-term physiological parameters (e.g., body weight, locomotor activity) for 7–14 days post-treatment; and (3) performing histopathological analysis of non-brain organs to rule out structural damage.
To address these limitations and deepen mechanistic understanding, future studies should adopt conditional knockout mouse models. First, neuronal-specific NF-κB p65 knockout mice (Nestin-Cre; p65flox/flox) could verify whether remimazolam’s effect depends on neuronal NF-κB—if remimazolam no longer reduces pyroptosis in these mice, it would confirm neuronal NF-κB as a key target. Second, microglial-specific NLRP3 knockout mice (CX3CR1-Cre; NLRP3flox/flox) would clarify whether remimazolam affects microglial pyroptosis, addressing the current cell-type limitation. Third, GABAA receptor subunit-specific knockout mice (e.g., α2 subunit deletion) could validate the “remimazolam → GABAA → NF-κB” pathway, defining the specific receptor subunits involved. Additionally, future work should include aged or comorbidity-induced animal models and long-term outcome assessments (3–6 months) to better reflect clinical reality and evaluate remimazolam’s impact on cognitive function and functional independence.
In conclusion, remimazolam may attenuate cortical neuronal pyroptosis after acute cerebral I/R injury by inhibiting NF-κB-mediated activation of the NLRP3/ASC/Caspase-1 inflammasome. This study provides experimental evidence for the clinical application of remimazolam as a neuroprotective anesthetic in acute ischemic stroke, while also identifying key directions for future mechanistic and translational research to address remaining gaps in safety and mechanism.
Conclusions
Remimazolam may attenuate cortical neuronal pyroptosis after acute cerebral I/R injury by inhibiting NF-κB-mediated activation of the NLRP3/ASC/Caspase-1 inflammasome.
Data availability
The data and materials supporting the conclusions of this study are available from the corresponding author upon reasonable request.
Abbreviations
- NLRP3:
-
Nucleotide-binding oligomerization domain‐like receptors pyrin domain containing 3
- GABAA :
-
γ-aminobutyric acid A
- MCAO:
-
Middle cerebral artery occlusion
- I/R:
-
Ischemia–reperfusion
- OGD/R:
-
Oxygen–glucose deprivation reperfusion
- TTC:
-
2, 3, 5-triphenyl tetrazolium chloride
- CCK-8:
-
Cell counting kit-8
- LDH:
-
Lactate dehydrogenase
- TEM:
-
Transmission electron microscope
- CCA:
-
Common carotid artery
- ECA:
-
External carotid artery
- ICA:
-
Internal carotid artery
- DMSO:
-
Dimethyl sulfoxide
- ELISA:
-
Enzyme-linked immunosorbent assay
- NeuN:
-
Neuronal nuclei
References
Wu, S. et al. Stroke in China: advances and challenges in epidemiology, prevention, and management. Lancet Neurol. 18(4), 394–405 (2019).
Huang, J. et al. Intra-arterial tenecteplase following endovascular reperfusion for large vessel occlusion acute ischemic stroke: the POST-TNK randomized clinical trial. JAMA 333(7), 579–588 (2025).
Rabinstein, A. A. Update on treatment of acute ischemic stroke. Continuum (Minneapolis Minn.) 26(2), 268–286 (2020).
Pop, R. et al. Local anesthesia versus general anesthesia during endovascular therapy for acute stroke: a propensity score analysis. J. Neurointervent. Surg. 13(3), 207–211 (2021).
Mao, M. et al. Vialinin A alleviates oxidative stress and neuronal injuries after ischaemic stroke by accelerating Keap1 degradation through inhibiting USP4-mediated deubiquitination. Phytomed. Int. J. Phytother. Phytopharmacol. 124, 155304 (2024).
Baker, J. R. et al. Cerebral blood flow dynamics in neurogenic orthostatic hypotension: a systematic review and meta-analysis. Hypertension (Dallas Tex 1979) 82, 106–117 (2024).
Zhou, Z., Yang, Y., Wei, Y. & Xie, Y. Remimazolam attenuates LPS-derived cognitive dysfunction via subdiaphragmatic vagus nerve target α7nAChR-mediated Nrf2/HO-1 signal pathway. Neurochem. Res. 49(5), 1306–1321 (2024).
Odeh, D. et al. The impact of the combined effect of inhalation anesthetics and iron dextran on rats’ systemic toxicity. Int. J. Mol. Sci. 25(12), 6323 (2024).
Thomasius, M. A., Menghini, M. & Breckwoldt, J. Brainstem anesthesia after retrobulbar block under brief analgosedation: Evidence for the underlying patho-mechanism. Eur. J. Ophthalmol. 35(4), Np62–Np67 (2025).
Lussier, G. et al. Compact arterial monitoring device use in resuscitative endovascular balloon occlusion of the aorta (REBOA): a simple validation study in swine. Cureus 16(10), e70789 (2024).
Alsbrook, D. L. et al. Neuroinflammation in acute ischemic and hemorrhagic stroke. Curr. Neurol. Neurosci. Rep. 23(8), 407–431 (2023).
Li, Q. et al. Melatonin alleviates neuroinflammation in ischemic stroke by regulating cGAS-mediated microglial pyroptosis signaling. Neural Regen. Res. https://doi.org/10.4103/NRR.NRR-D-24-01070 (2025).
Zhang, M. et al. IFP35, a novel DAMP, aggravates neuroinflammation following acute ischemic stroke via TLR4/NF-κB/NLRP3 signaling. J. Neuroinflamm. 22(1), 164 (2025).
Akif, A. et al. Targeting NLRP3 signaling with a novel sulfonylurea compound for the treatment of vascular cognitive impairment and dementia. Fluids Barriers CNS 22(1), 55 (2025).
Broz, P. Pyroptosis: molecular mechanisms and roles in disease. Cell Res. 35(5), 334–344 (2025).
Wu, J. et al. Atypical sulfur-containing physalin from Physalis minima and protective effect against ischemia-reperfusion injury. Phytochemistry 235, 114478 (2025).
Liu, L. et al. Transient receptor potential vanilloid 4 blockage attenuates pyroptosis in hippocampus of mice following pilocarpine-induced status epilepticus. Acta Neuropathol. Commun. 13(1), 73 (2025).
Sun, R. et al. Low-density lipoprotein receptor (LDLR) regulates NLRP3-mediated neuronal pyroptosis following cerebral ischemia/reperfusion injury. J. Neuroinflamm. 17(1), 330 (2020).
Pingel, L. et al. Remimazolam for procedural sedation: A systematic review with meta-analyses and trial sequential analyses. Eur. J. Anaesthesiol. 42(4), 298–312 (2025).
Song, J. L. et al. Remimazolam vs propofol for induction and maintenance of general anesthesia: A systematic review and meta-analysis of emergence agitation risk in surgical populations. J. Clin. Anesth. 103, 111815 (2025).
Fang, Y. B. et al. Safety and efficacy of remimazolam tosilate for general anaesthesia in paediatric patients undergoing elective surgery: a multicentre, randomised, single-blind, controlled trial. Anaesthesia 80(3), 259–268 (2025).
Ripoll, J. G. et al. Remimazolam in cardiac anesthesia: analysis of recent data. J. Cardiothorac. Vasc. Anesth. 39(1), 273–285 (2025).
Sneyd, J. R. Remimazolam – current status, opportunities and challenges. Anesthesiol. Perioperat. Sci. 1(3), 25 (2023).
Mordyl, B. et al. Preferential synaptic type of GABA-A receptor ligands enhancing neuronal survival and facilitating functional recovery after ischemic stroke. J. Med. Chem. 67(24), 21859–21889 (2024).
Li, P. et al. Intranasal delivery of engineered extracellular vesicles promotes neurofunctional recovery in traumatic brain injury. J. Nanobiotechnol. 23(1), 229 (2025).
Shi, M. et al. Protective effects of remimazolam on cerebral ischemia/reperfusion injury in rats by inhibiting of NLRP3 inflammasome-dependent pyroptosis. Drug Des. Dev. Ther. 16, 413–423 (2022).
Longa, E. Z., Weinstein, P. R., Carlson, S. & Cummins, R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke 20(1), 84–91 (1989).
Liu, Y. et al. Design, synthesis, and biological evaluation of novel CNS 7056 derivatives as sedatives in rats and rabbits. Chem. Biol. Drug Des. 88(1), 38–42 (2016).
Zhang, M. et al. MCC950 suppresses NLRP3-dependent neuroinflammation and ameliorates cognitive decline in a rat model of cerebral small vessel disease. Neural Regen. Res. https://doi.org/10.4103/NRR.NRR-D-24-01055 (2025).
Liu, L. et al. Molecular hydrogen mitigates traumatic brain injury-induced lung injury via NLRP3 inflammasome inhibition. BMC Chem. 19(1), 138 (2025).
Wang, J. Y. et al. Astrocyte-specific activation of sigma-1 receptors in mPFC mediates the faster onset antidepressant effect by inhibiting NF-κB-induced neuroinflammation. Brain Behav. Immun. 120, 256–274 (2024).
Gao, J. et al. ITIH1 suppresses carcinogenesis in renal cell carcinoma through regulation of the NF-κB signaling pathway. Exp. Ther. Med. 28(3), 368 (2024).
Benedek, A. et al. Use of TTC staining for the evaluation of tissue injury in the early phases of reperfusion after focal cerebral ischemia in rats. Brain Res. 1116(1), 159–165 (2006).
Robert, F., Cloix, J. F. & Hevor, T. Ultrastructural characterization of rat neurons in primary culture. Neuroscience 200, 248–260 (2012).
Shi, C. X. et al. Sevoflurane attenuates brain damage through inhibiting autophagy and apoptosis in cerebral ischemia-reperfusion rats. Mol. Med. Rep. 21(1), 123–130 (2020).
Tu, Y. et al. Dexmedetomidine attenuates the neurotoxicity of propofol toward primary hippocampal neurons in vitro via Erk1/2/CREB/BDNF signaling pathways. Drug Des. Dev. Ther. 13, 695–706 (2019).
Lv, J. et al. Dexmedetomidine attenuates propofol-induce neuroapoptosis partly via the activation of the PI3k/Akt/GSK3β pathway in the hippocampus of neonatal rats. Environ. Toxicol. Pharmacol. 52, 121–128 (2017).
Chen, Y. et al. Dexmedetomidine reduces the neuronal apoptosis related to cardiopulmonary bypass by inhibiting activation of the JAK2-STAT3 pathway. Drug Des. Dev. Ther. 11, 2787–2799 (2017).
Putaala, J. Ischemic stroke in young adults. Continuum (Minneapolis Minn.) 26(2), 386–414 (2020).
Barnes, S. C., Beishon, L. C., Hasan, M. T., Robinson, T. G. & Minhas, J. S. Cerebral haemodynamics, anaesthesia and the frail brain. Anesthesiol. Perioperat. Sci. 1(3), 19 (2023).
Zhang, Q.-w, Wang, X., Wang, Z.-y & Sun, H.-l. Low-dose esketamine improves acute postoperative pain in patients undergoing thoracoscopic surgery. Anesthesiol. Perioperat. Sci. 2(1), 5 (2024).
Ni, Y., Yu, M. & Liu, C. Sleep disturbance and cognition in the elderly: a narrative review. Anesthesiol. Perioperat. Sci. 2(3), 26 (2024).
Hausburg, M. A. et al. Effects of propofol on ischemia-reperfusion and traumatic brain injury. J. Crit. Care 56, 281–287 (2020).
Lee, Y. M., Song, B. C. & Yeum, K. J. Impact of Volatile Anesthetics on Oxidative Stress and Inflammation. Biomed. Res. Int. 2015, 242709 (2015).
Wang, K. et al. Effects of dexmedetomidine on perioperative stress, inflammation, and immune function: systematic review and meta-analysis. Br. J. Anaesth. 123(6), 777–794 (2019).
Liu, T. et al. Effect of remimazolam induction on hemodynamics in patients undergoing valve replacement surgery: A randomized, double-blind, controlled trial. Pharmacol. Res. Perspect. 9(5), e00851 (2021).
Xiao, Y. et al. Dexmedetomidine attenuates the propofol-induced long-term neurotoxicity in the developing brain of rats by enhancing the PI3K/Akt signaling pathway. Neuropsychiatr. Dis. Treat. 14, 2191–2206 (2018).
Liu, T. et al. Remimazolam and propofol combination in ischemic cerebrovascular disease endovascular treatment: a randomized study. Sci. Rep. 15(1), 21083 (2025).
Cai, F. et al. Theaflavin ameliorates cerebral ischemia-reperfusion injury in rats through its anti-inflammatory effect and modulation of STAT-1. Mediators Inflamm. 2006(5), 30490 (2006).
Prokic, E. J. et al. Cortical oscillatory dynamics and benzodiazepine-site modulation of tonic inhibition in fast spiking interneurons. Neuropharmacology 95, 192–205 (2015).
Freyer, N. et al. Metabolism of remimazolam in primary human hepatocytes during continuous long-term infusion in a 3-D bioreactor system. Drug Des. Dev. Ther. 13, 1033–1047 (2019).
Doi, M. et al. Efficacy and safety of remimazolam versus propofol for general anesthesia: a multicenter, single-blind, randomized, parallel-group, phase IIb/III trial. J. Anesth. 34(4), 543–553 (2020).
Lammerding, L., Slowik, A., Johann, S., Beyer, C. & Zendedel, A. Poststroke inflammasome expression and regulation in the peri-infarct area by gonadal steroids after transient focal ischemia in the rat brain. Neuroendocrinology 103(5), 460–475 (2016).
Acknowledgements
All authors made substantial contributions to this study. We are grateful to the staff involved in this research.
Funding
This work was funded by the Youth Science Foundation of Guangxi Medical University (No. GXMUYSF202309), National Natural Science Foundation of China (No. 82460239), Guangxi Science and Technology Base and Talent Special Project (No. AD25069060), Special Fund of Neurotoxicity of General Anesthetics and Its Prevention and Treatment Innovation Team of the First Affiliated Hospital of Guangxi Medical University (No. YYZS2022001), and Guangxi Key Research and Development Program (No. AB24010066).
Author information
Authors and Affiliations
Contributions
Tianxiao Liu, Jing Chen, Min Shi, and Yubo Xie designed the research, wrote the manuscript, and revised the paper. Tianxiao Liu, Jing Chen, Min Shi, Weixin Dai, Cuihua Chen, and Chunlai Li carried out the experiments. Tianxiao Liu, Yubo Xie, Jing Chen, Min Shi, and Chunlai Li performed data analysis. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
All the animal experiments were conducted with the approval of the ethics committee of Guangxi Medical University (Animal Experimental Ethical Inspection No.202006009), strictly following The Guide for Care and Use of Laboratory Animals released by the National Institutes of Health. In this study, all the experimental animals were euthanized by excessive anesthesia after completing the experiments. The study was reported in accordance with ARRIVE guidelines (see Supplementary Information file).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, T., Chen, J., Shi, M. et al. Remimazolam alleviates cerebral ischemia–reperfusion injury of rats by inhibiting NF-κB/NLRP3 inflammasome pyroptosis. Sci Rep 16, 1671 (2026). https://doi.org/10.1038/s41598-025-31205-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-31205-9
