
Abstract
Ecological speciation progresses along a continuum, beginning with population divergence and culminating in reproductive isolation. Defining species boundaries becomes particularly challenging when ecological divergence occurs despite morphological similarity, yet precise delineation is essential for biodiversity assessment and conservation. Phedimus aizoon and P. kamtschaticus, two perennial herbs, exemplify this challenge, exhibiting high morphological similarity despite distinct habitat preferences. To investigate their divergence, we genotyped 80 individuals from these two species, along with a reference species, using 5,051 SNPs. Integrating morphometric and phylogenetic analysis and environmental niche modeling, our study provides compelling evidence that ecological factors have driven species divergence in these taxa. While genetic analyses reveal clear differentiation, we also detected instances of gene flow, as indicated by admixture patterns in STRUCTURE, TreeMix, and f₄ statistics. Despite minimal morphological differences, substantial genetic divergence suggests that precipitation and temperature seasonality have played a critical role in their speciation.
Similar content being viewed by others

Introduction
Species are fundamental units of biological research, evolution, and biodiversity conservation, yet the concept itself remains a long-standing challenge in biology. A species is not a fixed entity but rather a hypothesis requiring rigorous testing1,2. Consequently, the accurate delimitation of species, especially within morphologically complex groups, requires the explicit application of a chosen species concept3,4. Over decades, numerous concepts have been proposed, including the Biological5, Evolutionary6, and Phylogenetic7 concepts. While no single concept is universally consistent, recent studies frequently integrate ecological and evolutionary data—including statistical analyses and Geographic Information Systems (GIS)—to test hypotheses under the Ecological Species Concept8,9.
Ecological speciation—the process by which divergent selection from contrasting environments drives reproductive isolation and speciation—is recognized as a major mechanism shaping biodiversity10,11,12. This process is often accompanied by genetic divergence and cessation of gene flow between populations12,13. Environmental differentiation can arise from biotic factors (e.g., interactions with predators, competitors, and pollinators)14,15 or abiotic factors (e.g., soil moisture and temperature)16,17,18,19. Changes in abiotic conditions may limit pollinator availability, potentially driving shifts in reproductive strategies, such as increased selfing20,21. Abrupt environmental changes can also drive rapid evolution in flowering phenology22, reinforcing postzygotic reproductive isolation through adaptive mechanisms23. For example, Mimulus species endemic to serpentine soils have been selected for early flowering to enhance reproductive success under harsh conditions24,25. Ecological speciation is thus a key evolutionary process, particularly in flowering plants where local adaptation to habitat conditions has been repeatedly demonstrated26,27.
Phedimus aizoon (L.) ′t Hart and P. kamtschaticus (Fisch.) ′t Hart illustrate an intriguing example of ecological speciation, where adaptation to distinct environments has likely driven the species divergence. Phedimus Raf. (Crassulaceae) is a genus of perennial herbs predominantly inhabiting rocky slopes and grasslands across Eurasia, comprising approximately 20 species28,29. Recent molecular and morphological studies have confirmed its separation from the sister genus Sedum, with key distinguishing traits found in leaf structure and testa morphology30,31,32,33. Within Phedimus, two subgenera, Phedimus and Aizoon, have been recognized based on differences in petal color, stem traits, and testa structure34. However, taxonomic classification within the genus remains challenging due to significant variation in vegetative morphology, while floral traits show comparatively minor differences30,35. Additionally, the widespread use of Phedimus in horticultural breeding programs has further obscured taxonomic boundaries, complicating species identification and classification36,37.
Phedimus aizoon and P. kamtschaticus represent the most widespread taxa in the genus, yet their precise relationship remains unclear. P. aizoon is broadly distributed across Eurasia, while P. kamtschaticus is confined to the Far East30. Morphological distinctions traditionally separate them: P. aizoon is characterized by its taller growth, erect stems, and lanceolate leaves, while P. kamtschaticus typically features numerous ascending stems and obovate leaves30. Crucially, however, P. kamtschaticus exhibits considerable infraspecific morphological variation, particularly in leaf shape, leading to persistent taxonomic ambiguity between the two taxa (Fig. 1).

Key morphological variation in stem leaves between (a) Phedimus kamtschaticus and (b) P. aizoon.
The current taxonomic ambiguity between P. aizoon and P. kamtschaticus is reinforced by genetic evidence. Prior molecular studies using ITS and chloroplast regions failed to resolve their relationship, as neither formed distinct monophyletic clades38. More recently, a genome-wide SNP analysis also yielded comparable results, further blurring the genetic boundaries between the species39. Given the lack of clear phylogenetic separation despite documented morphological and ecological differentiation, the current species status of these two taxa is tenuous and requires re-evaluation. Addressing this complexity, which is common in speciation-in-progress, necessitates an integrated approach that considers both population-level genetic divergence and ecological differentiation40,41. Recent advances in genomics provide powerful tools to support this process, offering greater precision and adaptability in conservation practices.
The Korean Peninsula serves as a critical geographic and taxonomic nexus for the genus Phedimus. This relatively small peninsula is one of evolutionary hotspots, harboring nine recorded Phedimus species belonging to subgenus Aizoon, which represents over half the known taxa in the subgenus worldwide. This high concentration includes four Korean endemics (e.g., P. takesimensis, P. latiovalifolius, P. daeamensis)38,39. The resultant taxonomic complexity—with historical treatments of subgenus Aizoon in Korea ranging from one to six species—is due to both high morphological variation and documented instances of habitat overlap and continuous gene flow between taxa42,43,44,45,46,47,48. While these endemic species are relatively rare, both P. aizoon and P. kamtschaticus are widespread in this key region, but their habitat preferences differ: P. kamtschaticus is predominantly found in montane environments, whereas P. aizoon, a habitat generalist, also thrives in coastal regions. This unique situation—where two geographically widespread, yet ecologically divergent taxa coexist alongside numerous endemics in a small area of high morphological complexity—makes the Korean Peninsula the ideal focal region to investigate the roles of ecological speciation and gene flow in species delimitation within Phedimus.
In the present study, we sought to define the species boundary between P. aizoon and P. kamstchaticus by investigating population-level divergences across the Korean Peninsula. Additionally, we compared the divergence patterns of these two widespread species with those of the narrow endemic P. latiovalifolius found in Korea. To achieve this, we genotyped 80 individuals from seven populations, including two populations of P. latiovalifolius, using high-density genome-wide data. First, we investigated whether P. aizoon and P. kamtschaticus harbor distinct gene pools to determine if they should be considered separate species. We then analyzed ecological niche differentiation between the two species. Finally, we compared the diversity patterns of these two common species with those of the narrow endemic species in Korea.
Materials and methods
Study system, sample collection and DNA extraction
Phedimus (x = 5, 6, 7, 8; Thiede & Eggli, 2007) is a self-incompatible, outcrossing species, though it can also propagate vegetatively via fragmentation of detached leaves or stems34. This perennial plant is mostly glabrous, emerging from a thin, woody rhizome34. Although the life history traits of the genus remain largely unexplored, its reproductive and dispersal strategies can be inferred from characteristics common to Crassulaceae. Fruits are follicles that release seeds immediately upon ripening, with dispersal primarily via wind and gravity34. Due to their relatively large size compared to dust-like wind-dispersed seeds, long-distance dispersal is unlikely.
For this study, we analyzed 80 DNA samples collected across the Korean Peninsula, encompassing three Phedimus species: P. aizoon, P. kamtschaticus, and the narrow endemic P. latiovalifolius (Table 1; Fig. 2). All required permits for sample collection were obtained. While P. aizoon and P. kamtschaticus were the focal species for testing ecological divergence, the endemic P. latiovalifolius was included for comparative analysis and to test for potential hybridization among taxa in subgenus Aizoon. This endemic species is narrowly restricted to a few high-altitude mountains (above 1,200 m). To ensure our data was representative of this rare taxon, we deliberately collected samples from two of the largest known populations found on different mountains. To minimize genetic redundancy across all species, sampled populations were spaced at least 30 km apart, and within each population, individual plants were selected at least 30 m apart. Fresh leaf tissues were immediately preserved in silica gel desiccant for subsequent DNA extraction. Taxonomic identities were determined based on morphological characters detailed in established regional floras30,35. Voucher specimens were collected from each sampled population and have been deposited in the Chosun University Herbarium (CHO) (Table 1). The taxonomic identification of all specimens was reviewed and confirmed by all authors.

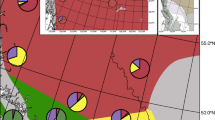
Genetic structure of seven Phedimus populations of three species (P. aizoon, P. kamtschaticus, and P. latiovalifolius). (a) Pie charts on the map illustrate the proportional genetic composition of each population, with chart sizes reflecting relative sample sizes. (b) STRUCTURE results for K = 3, showing genetic clustering patterns of the three species. Dashed vertical white lines indicate regional group boundaries, and colors represent inferred genetic clusters. Population acronyms are provided in Table 1.
Morphology analysis
To assess whether the key morphological characters traditionally used to differentiate Phedimus aizoon and P. kamtschaticus exhibit diagnostic value, we performed a quantitative morphometric analysis. Four continuous morphological traits—plant height, leaf length, leaf width, and the position of maximum leaf width—were measured for each specimen. In addition, two derived ratio variables, leaf length-to-leaf width (LL/LW) and leaf length-to-position of maximum leaf width (LL/PMW), were calculated to account for proportional differences in leaf shape. A total of 200 digitized herbarium specimens representing both taxa were examined from multiple institutional collections (A, GH, KH). Specimens were selected only if they were clearly in a fully mature vegetative stage to minimize ontogenetic variation. All measurements were taken directly from high-resolution specimen images using the ImageJ software49, following standardized protocols for morphometric measurements.
The resulting morphological dataset was analyzed using a multivariate statistical framework to explore patterns of morphological variation and to test for potential clustering between the two taxa. Principal Component Analysis (PCA) was performed using the PCA function in the R package FactoMineR v2.950,51, which allows for simultaneous analysis of quantitative and ratio variables. The ordination results were visualized using the factoextra package v1.0.752, enabling the identification of major axes of morphological variation and potential group separation among specimens.
RAD-seq library Preparation & Raw data processing
To genotype the collected samples, we employed the 3RAD approach53, a modified version of the widely used ddRADseq method54. 3RAD improves adapter ligation efficiency by incorporating a third restriction enzyme, which cleaves adapter dimers, thereby preventing chimera formation and enhancing amplicon yield. The adapter-ligated digested fragments were pooled and purified using a 1:1.8 ratio of AMPure XP beads, followed by a 70% ethanol wash. The purified libraries were PCR-amplified and size-selected for 500 bp fragments (± 10%) using a Pippin Prep system (Sage Science, Beverly, MA, USA). A final amplification step was then performed using the size-selected fragments. The final library quality was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Sequencing was performed at Macrogen Inc. (Korea) on the Illumina NovaSeq platform, generating 2 × 150 paired-end reads.
To process the raw sequence data, we first demultiplexed and trimmed reads using Stacks v2.4155. Low-quality reads (Phred score < 10) were filtered out using the process_radtags function to minimize sequencing errors. Due to the absence of a reference genome for the three Phedimus species, we employed a de novo approach for RAD loci assembly. We built sequence catalogs using ustacks with parameter settings -m 3 (minimum depth of coverage) and -M 3 (maximum mismatches allowed in a locus). The threshold -m 3 ensured that loci were supported by sufficient read depth to minimize inclusion of sequencing errors, while the -M 3 setting balanced allelic clustering and locus over-splitting. These settings have been commonly applied in comparable studies employing Stacks for de novo assembly of RAD data. To assemble the catalog across samples, we used cstacks, allowing up to one mismatch between loci (−n 1)56.
SNPs were called using the Populations module in Stacks, based on the assembled catalogs. We retained loci present in at least 80% of samples within each population and shared across at least 6 populations (-p 6, -r 0.8). To minimize linkage disequilibrium (LD), only the first SNP per locus was included (-write-single-snp). We removed loci showing significant deviation from Hardy–Weinberg equilibrium (HWE, p < 10−6)57,58 using Plink v1.959 to eliminate potential assembly errors leading to extreme heterozygosity. Further filtering was conducted in TASSEL 5.060, excluding genotypes with > 30% missing data and SNPs with a minor allele frequency (MAF) ≤ 0.05.
Genetic diversity and population structure
To assess genetic diversity, we analyzed 5,051 SNPs using Arlequin v. 3.561, estimating expected heterozygosity (He). Since the seven sampled populations had unequal sample sizes, we employed rarefaction curves62 to estimate the number of alleles (Na) using allelic.richness function in R package HIERFSTAT v. 0.5–11 (Table 1)63,64. Pairwise FST values were calculated in Arlequin, with significance assessed through 1,000 permutations. We also estimated effective population sizes of all collected populations using molecular coancestry method in NeEstimator v. 2.0165.
To infer population genetic structure, we conducted principal component analysis (PCA) on 5,051 unlinked SNPs using the ipyrad.pca tool from the ipyrad-analysis toolkit66. The results were visualized with a 3D scatter plot generated via plot_ly in R plotly package67. For clustering analysis, we estimated the number of genetic clusters (K) using STRUCTURE v2.3.468 within ipyrad, implementing an admixture model with 1,000,000 MCMC iterations after a 100,000-step burn-in. We tested K = 1 to 10, running 10 replicates per K to ensure stability. The ΔK method69 is generally unsuitable for determining the optimal number of clusters (ΔK) when the analysis includes comparisons between distinct species, as is the case in our study (three species). Therefore, we did not employ the Delta K method but instead focused our interpretation on ΔK = 3, which corresponds directly to the number of a priori recognized taxa (P. aizoon, P. kamtschaticus, and P. latiovalifolius). We then summarized ancestry coefficients with CLUMPP v1.1.270 and visualized the final results in DISTRUCT v1.171.
Phylogenetic relationships and admixture
We analyzed the phylogenetic relationships and assessed ongoing gene flow between the focal species. For gene flow assessment, we used TreeMix v. 1.1372. The dataset, consisting of 5,051 SNPs, was first converted into the TreeMix input format using the populations function in Stacks. We rooted the tree using the P. latiovalifolius samples via the -root function. To test for migration, we generated 500 bootstrap replicates with a SNP block size of 100 (using the -K flag) and tested migration models across a range of zero to five migration edges (m = 0–5). The optimal number of migration events was subsequently determined using the R package OptM73.
To robustly assess species distinction and potential hybridization signals, we also constructed a NeighborNet phylogenetic network using SplitsTree v. 4.17.174. To ensure that broader geographic sampling did not obscure the separation of the two focal species, we incorporated two additional samples of P. aizoon from China (GenBank accession Nos. SRR30607270 and SRX20242453). For this phylogenetic analysis, a dedicated dataset was prepared where SNPs (1,213 SNPs) were called separately. We minimized sampling bias by including only two samples from each population of P. aizoon and P. kamtschaticus. Uncorrected P-distances were estimated for the splits, and the resulting relationships were presented as a NeighborNet.
Given the admixture pattern observed in DA population (see Table 1 for the population acronym) from the STRUCTURE result, we further demonstrate admixture between the two species using f4-ratios implemented in R package, ADMIXTOOLS 251,75,76. We first converted the VCF file to PLINK format using PLINK v2.077. We then computed f2 statistics with the extract_f2 and f2_from_precomp functions to quantify genetic drift and relatedness among populations. To structure the analysis, we categorized populations into four distinct groups: (1) each parental species as separate groups, (2) putative hybrids (DA populations) forming the third group, and (3) an outgroup species as the fourth group. Using the derived f2 statistics, we calculated f3 and f4 statistics to detect admixture signals and assess population history.
We estimated the historical effective population sizes (Ne) for P. aizoon and P. kamtschaticus within the Korean Peninsula to investigate past demographic changes using the Stairway Plot method78. We first calculated the folded Site Frequency Spectrum (SFS) for each species using easySFS (available at https://github.com/isaac-overcast/easySFS). We ran Stairway Plot on 200 bootstrap replicates drawn from the calculated SFS, and the median Ne and its confidence intervals were derived from these 200 estimations. For model parameters, we used a per-site mutation rate (µ) of 7*10−9 mutations per year, derived from Arabidopsis thaliana79, acknowledging the absence of reliable data for the genus. To account for this uncertainty, we allowed a ± 10% deviation from this rate. Lacking species-specific data, we estimated the generation time based on the known life history of the Crassulaceae family. Given that sister genus Sedum taxa often have generation times of one to two years80,81 and perennial herbs generally exhibit a range of one to five years82, we conservatively applied a generation time of two years for the analysis.
Niche separation
We investigated ecological niche differentiation between P. kamtschaticus and P. aizoon by modeling their environmental niches. To build robust models, we gathered georeferenced occurrence data from the Global Biodiversity Information Facility (GBIF; http://www.gbif.org/), yielding 525 records for P. kamtschaticus and 3,525 for P. aizoon. To minimize potential misidentification, we retained only those records supported by herbarium specimen images or accompanying metadata that allowed verification of species identification. Environmental predictors were derived from 19 bioclimatic variables available in the WorldClim v. 2.1 database (http://www.worldclim.org/)83 at a 30-second resolution. The data were then refined and processed using raster v3.6-26 in R to prepare the input files for modeling.
We employed MaxEnt v. 3.4.484 to construct ecological niche models, using a standardized approach with 50 replicates and a maximum of 5,000 iterations. The modeling process incorporated the following settings: (1) enabling “Random seed” for variability, (2) selecting “Subsample” as the replicated run type, and (3) reserving 25% of occurrence data for validation. Model performance was evaluated using the Area Under the Receiver Operating Characteristic Curve (AUC), where values closer to 1.0 indicate higher predictive accuracy.
To quantify niche differentiation, we applied the R package ENMtools v. 1.0.451,85. Niche overlap was measured using Schoener’s D and Warren’s I indices86, while statistical significance was tested with niche equivalency and similarity analyses, each performed with 100 iterations. These analyses provided insights into whether the two species occupy distinct climatic spaces or if their niches exhibit significant overlap.
Results
SNP calling and genetic diversity
The 3RAD library generated approximately 230 million reads, averaging 2.9 million reads per sample, with a mean GC content of 36.1%. Initial SNP identification using the populations function in Stacks detected ~ 230,000 SNP loci, which were then filtered for quality. After the process, 5,051 high-quality SNPs remained for further analysis.
The mean genetic diversity values were 0.13 for expected heterozygosity (He) and 1.15 for the number of alleles (Na), with variation across species and populations (Table 1). Expected heterozygosity ranged from 0.09 in NH (P. kamtschaticus) to 0.16 in HB (P. latiovalifolius), while the number of alleles was highest in the DA population (a hybrid of P. aizoon and P. kamtschaticus) and lowest in HB (Table 1). Pairwise population differentiation (FST) varied widely, ranging from nearly zero to 0.78 across population pairs (Table 2). Overall, FST values were considerably lower within species compared to those between different species (Table 2).
Genetic and morphometric structure
PCA analysis identified at least three distinct clusters, each representing one of the three species (Fig. 3). The first three principal components (PCs) explained 62%, 20%, and 7% of the total variability, respectively. The first PC primarily separated the three species, while PCs 2 and 3 distinguished the DA population from the other P. kamtschaticus populations. The DA population’s genotypes clustered between P. aizoon and P. kamtschaticus, suggesting admixture. Since our analysis included all three species, we did not determine the optimal K value but instead present results for K = 3. Consistent with the PCA results, STRUCTURE analysis clearly differentiated the three species, with the DA population exhibiting admixture between P. aizoon and P. kamtschaticus (Fig. 2). STRUCTURE results also did not indicate any spatial structure within individual species. Despite the molecular differentiation between P. aizoon and P. kamtschaticus, morphometric analysis revealed no clear separation: the first two morphological PCs explained 45% and 23% of the total variance but did not form distinct clusters in the PCA plot (Fig. S2).

Plot of Principle Coordinate Analysis of seven Phedimus populations of three species (P. aizoon, P. kamtschaticus, and P. latiovalifolius).
Phylogenetic relationships and admixture
To further investigate the admixture pattern suggested by the STRUCTURE analysis (Fig. 2), we conducted a TreeMix analysis. The resulting phylogeny identified three well-defined clades, each corresponding to one of the three species, consistent with the clustering patterns observed in STRUCTURE and PCA (Fig. 4). The relatively long branch lengths between clades suggest a strong genetic drift effect since their initial divergence. Introducing a single migration edge substantially improved the model’s fit, increasing the explained genetic covariance from 99.3% (no migration) to 99.9% (Fig. S5). The population graph indicated a migration event (migration weight = ~ 0.4) from P. aizoon (ADA) to one of the P. kamtschaticus populations (DA), mirroring the admixture signal detected in STRUCTURE (Fig. 4).

Phylogenetic relationships among Phedimus populations of the two focal species (P. aizoon and P. kamtschaticus) and the outgroup (P. latiovalifolius), inferred using TreeMix. The maximum-likelihood tree includes one inferred gene flow event, explaining 99.9% of the variance in the model. Arrow color indicates the weight of gene flow, representing the proportion of ancestry contributed by the migration event.
The NeighborNet analysis further confirmed that Korean samples of the two focal species were correctly identified. In the network, P. kamtschaticus samples from Korea formed a distinct clade, while Korean and Chinese P. aizoon samples grouped together in a separate, well-supported clade (Fig. S3). Additionally, f4 statistics provided strong support for gene flow, as the admixture test among P. aizoon, P. kamtschaticus, and the admixed population (DA), with P. latiovalifolius as an outgroup, showed a significant deviation from neutrality (|Z-score| > 3; P < 0.001). These findings collectively indicate historical gene flow between P. aizoon and P. kamtschaticus. Furthermore, the Stairway Plot analysis based on folded site frequency spectra (SFS) revealed notable demographic fluctuations in both species, with two pronounced bottlenecks in effective population size occurring between approximately 40 and 100 thousand years ago (kya) (Fig. S4).
Ecological niche distinction
We generated ENMs for the two species with taxonomic delineation problems to determine if clear ecological niche distinction is present between the two. The mean AUC values calculated for the ENMs of P. aizoon and P. kamtschaticus, averaged across all 50 runs, were 0.911(± SD 0.003) and 0.963 (± SD 0.003), respectively showing robust model support (Fig. S1). The ENM models exhibited clear differences in the ecological niches of the two species (Fig. 4). In the ENMs, high habitat suitability was predicted throughout East Asia in P. aizoon (Fig. 4a), while P. kamtschaticus showed high suitability across Korea, northern Japan and Kamchatka Peninsula (Fig. 4b). The key environmental variables contributing to habitat suitability for both species were B18 (51.5%- P. aizoon; 41.6%- P. kamtschaticus, precipitation during the warmest quarter) and B4 (26.7%- P. aizoon; 29.3%- P. kamtschaticus, temperature seasonality). We further explored niche distinctness by measuring D (0.29, p < 0.01) and I (0.11, p < 0.01) indices, which were significantly lower than expected under the null hypothesis (Fig. 4c). The results demonstrated the significant differentiation between the ecological niches of the two species.
Discussion
Species serve as fundamental units for measuring biodiversity3,4, yet delineating species boundaries remains complex, requiring an integrated approach that considers both species-level and infraspecific diversity. The process of speciation is inherently complex, often shaped by spatial genetic heterogeneity across populations10,41,87. By employing population genomics and environmental niche modeling, our study provides evidence of species divergence driven by ecological factors in two morphologically similar Phedimus species (P. aizoon and P. kamtschaticus). Our analyses confirm their distinction, revealing both molecular and ecological differentiation. Despite the divergence, we also detected occasional gene flow, as indicated by admixture patterns in STRUCTURE, TreeMix, and f₄ statistics. Given the substantial genetic divergence despite minimal morphological differences, our findings suggest that ecological factors, particularly precipitation and temperature seasonality, have played a critical role in their speciation. Empirical studies like ours, which elucidate speciation mechanisms, can inform more effective conservation strategies.
Perennial species, including Phedimus, generally maintain higher genetic diversity than annual or short-lived plants due to the extended lifespan, which slows the loss of genetic variation caused by reduced gene flow88,89,90. Additionally, genetic diversity in long-lived plants tends to be greater in species with broad geographic distributions and outcrossing reproductive systems88. However, our genomic analysis revealed only low to moderate genetic variation within each Phedimus population—lower than typically observed in other long-lived perennials89. The pattern aligns with findings from a previous SNP-based study on Phedimus39. Furthermore, we found no clear relationship between genetic diversity and geographic range width among the three Phedimus species examined (Table 1). Despite the outcrossing reproductive strategy and widespread distribution, all seven populations exhibited limited genetic variability. The reduced genetic variation may be a consequence of recent population bottlenecks or severe inbreeding. However, given the low inbreeding coefficient values (FIS < 0.2), inbreeding is unlikely to be the primary cause. Instead, population bottlenecks appear to be the most plausible explanation, as effective population sizes were consistently small across all populations (Table 1). To further test this hypothesis, future studies should incorporate larger sample sizes and conduct direct tests for population bottlenecks.
Ecological speciation occurs along a continuum, beginning with population divergence and eventually leading to cessation of gene flow over time10,11,12,91. Our genetic analyses revealed clear molecular divergence between P. aizoon and P. kamtschaticus (Figs. 2, 3 and 5), despite the morphological similarities. However, phylogenetic analyses based on DNA sequence data and GBS markers did not recover the two species as distinct evolutionary lineages. Instead, the montane species P. kamtschaticus was nested within the more widespread generalist P. aizoon38,39. This pattern, coupled with strong evidence of gene flow, suggests ongoing hybridization and weak reproductive isolation. Further investigation using the f4 ratio confirmed interspecific gene flow, and the observed admixture patterns reinforce the idea of limited reproductive barriers. Our morphological observations revealed no apparent physical barriers to reproduction, and both species share similar phenology, further supporting the possibility of ongoing hybridization. Nevertheless, the pronounced molecular divergence between the two species suggests that other factors, such as ecological differentiation, may be driving genetic differentiation. To test the role of ecological divergence, we analyzed their habitat preferences and key environmental variables.


Ecological niche modeling (ENM) results for Phedimus aizoon and P. kamtschaticus, highlighting their predicted habitat distributions and niche differentiation. (a) Predicted distribution of P. aizoon. (b) Predicted distribution of P. kamtschaticus. (c) Identity test results comparing the ecological niches of the two species. Grey and black bars represent the null distributions of Schoener’s D and Warren’s I statistics, respectively, while arrows denote the observed D (grey) and I (black) values from MaxEnt analyses, indicating the degree of niche overlap.
Our ENM analysis highlights the critical role of precipitation during the warmest quarter and temperature seasonality in shaping the distinct ecological niches of P. kamtschaticus and P. aizoon. Phedimus kamtschaticus is primarily restricted to montane regions of northeastern Asia, where temperature fluctuations are more pronounced and precipitation is lower during the warmest months. In contrast, P. aizoon has a broader distribution across East Asia, including coastal regions, suggesting greater climatic tolerance (Fig. 4). Mountainous environments, with the steep elevation gradients, orographic precipitation effects, and complex topography, often exhibit extreme climatic variability over short distances92,93. Such heterogeneity can promote ecological specialization and, in some cases, functionally mimic allopatric conditions that drive species divergence94. Increasing evidence supports the role of mountain systems as centers of rapid plant diversification, particularly in the Andes and Himalayas95. In this context, the ecological speciation of P. kamtschaticus and P. aizoon may reflect broader biogeographic patterns observed in mountain-adapted floras, where climatic specialization plays a key role in species distribution and evolutionary trajectories.
Beyond ecological factors, the two species experienced severe population bottlenecks during the Late Pleistocene, as inferred from demographic history analysis. During this period, harsh environmental conditions, including extreme cold and dramatic climate fluctuations, caused significant habitat fragmentation96,97. This fragmentation is a classic driver of Pleistocene diversification, leading to population bottlenecks, subsequent genetic drift, local adaptation, and ultimately speciation98,99,100. Given this dramatic demographic decline, it is highly likely that the two species were isolated in separate glacial refugia, resulting in allopatric speciation and the establishment of their current, distinct habitat preferences.
Despite the wide horticultural use, species boundaries within Phedimus remain puzzling due to porous taxonomic limits and high morphological variability. However, eco-evolutionary studies of the group have been lacking. Our study is the first to delineate taxonomic boundaries between P. kamtschaticus and P. aizoon using an integrated approach that considers both species-level and infraspecific diversity. By analyzing over 5,000 SNPs distributed across the genomes, we achieved a more precise resolution of species differentiation. Future research should expand taxon sampling and increase overall sample sizes to refine species delimitation and clarify phylogenetic relationships, ultimately informing conservation strategies for this taxonomically challenging group.
Data availability
The raw DNA sequences obtained for this study are available in the GenBank accession number PRJNA1284615 (https://www.ncbi.nlm.nih.gov/sra/PRJNA1284615).
References
Hey, J., Waples, R. S., Arnold, M. L., Butlin, R. K. & Harrison, R. G. Understanding and confronting species uncertainty in biology and conservation. Trends Ecol. Evol. 18, 597–603 (2003).
Galtier, N. Delineating species in the speciation continuum: A proposal. Evol. Appl. 12, 657–663 (2019).
Avise, J. C. A role for molecular genetics in the recognition and conservation of endangered species. Trends Ecol. Evol. 4(9), 279–281. https://doi.org/10.1016/0169-5347(89)90203-6 (1989).
Coates, D. J., Byrne, M. & Moritz, C. Genetic diversity and conservation units: dealing with the species-population continuum in the age of genomics. Front. Ecol. Evol. 6, 1–13 (2018).
Mayr, E. Systematics and the Origin of Species from the Viewpoint of a Zoologist (Columbia University, 1942).
Wiley, E. O. The evolutionary species concept reconsidered. Syst. Zool. 27, 17–26 (1978).
De Queiroz, K. Different species problems and their resolution. BioEssays 27, 1263–1269 (2005).
Andersson, L. The driving force: species concepts and ecology. Taxon 39, 375–382 (1990).
De Queiroz, K. Species Concepts and Species Delimitation (Oxford Academic, 2007).
Schluter, D. Introduction to the symposium: Species interactions and adaptive radiation. American Naturalist 156 (2000).
Coyne, J. A. & Orr, H. A. Speciation (Sinauer Associates, 2004).
Nosil, P. Ecological Speciation (Oxford University Press, 2012).
Roux, C. et al. Shedding light on the grey zone of speciation along a continuum of genomic divergence. PLoS Biol. 14, e2000234. https://doi.org/10.1371/journal.pbio.2000234 (2016).
Turkington, A. R. & Harper, J. L. The Growth, distribution and neighbour relationships of trifolium repens in a permanent pasture: IV. Fine-Scale biotic differentiation. J. Ecol. 67, 245–254 (1979).
Parker, M. A. Local population differentiation for compatibility in an annual legume and its host-specific fungal pathogen. Evolution 39, 713–723 (1985).
McNeilly, T. & Antonovics, J. Evolution in closely adjacent plant populations IV. Barriers to gene flow. Heredity 23, 205–218 (1968).
Davies, M. S. & Snaydon, R. W. Rapid population differentiation in a mosaic environment. III. Measures of differentiation between populations. Heredity 36, 79–89. https://doi.org/10.1038/hdy.1976.9 (1976).
Doust, L. L. Population dynamics and local specialization in a clonal perennial (Ranunculus repens). I. The dynamics of Ramets in contrasting habitats. J. Ecol. 69, 743–755. https://doi.org/10.2307/2259634 (1981).
Galen, C. & Stanton, M. L. Consequences of emergence phenology for reproductive success in ranunculus adoneus (Ranunculaceae). Am. J. Bot. 78, 7978–7988. https://doi.org/10.1002/j.1537-2197.1991.tb14503.x (1991).
Berry, P. E. & Calvo, R. N. Wind pollination, self-incompatibility, and altitudinal shifts in pollination systems in the high Andean genus Espeletia (Asteraceae). Am. J. Bot. 76, 1602–1614. https://doi.org/10.1002/j.1537-2197.1989.tb15149.x (1989).
Weller, S. G., Sakai, A. K. & Straub, C. Self-compatibility in the rare endemic schiedea globosa (Caryophyllaceae). Am. J. Bot. 85, 1601–1607. https://doi.org/10.2307/2446404 (1998).
Carey, K. Breeding System, genetic Variability, and response to selection in plectritis (Valerianaceae). Evolution 37, 947–956 (1983).
Campbell, C. R. Population Genetics and Ecological Speciation in Vertebrates (University of Georgia, 2014).
Macnair, M. R. & Gardner, M. The evolution of edaphic endemics. In (eds Baskin, S. J. & Baskin, C. C.) Population Biology of Rare Plants 157–171 (Academic, 1998).
Sianta, S. A. & Kay, K. M. The impact of geographic isolation on speciation in alpine plants. Evolution 75, 2281–2295. https://doi.org/10.1111/evo.14264 (2021).
Keller, I. & Seehausen, O. Thermal adaptation and ecological speciation. Mol. Ecol. 21, 782–799 (2012).
Kadereit, J. W. & Abbott, R. J. Plant speciation in the quaternary. Plant. Ecol. Divers. 14, 105–142 (2021).
Fu, K., Ohba, H. & Gilbert, M. G. Crassulaceae. In Flora of China (eds Wu, P. H. & Raven, Z.Y.) 202–268 (2001)
’t Hart, H. & Bleij, B. Phedimus. In (ed Eggli, U.) Illustrated Handbook of Succulent Plants: Crassulaceae (196–203). Springer-, Berlin/Heidelberg. (2003).
Ohba, H. Phedimus. In: (eds Iwatsuki, K., Boufford, D. E. & Ohba, H.) Flora of Japan, vol. 2b. 19–21. Kodansha, Tokyo, Japan (2001).
Mayuzumi, S. & Ohba, H. The phylogenetic position of orostachys and related genera of crassulaceae from Asia. Taxon 53(2), 337–346. https://doi.org/10.2307/4135607 (2004).
Gontcharova, S. B., Gontcharov, A. A., Yakubov, V. V. & Kondo, K. Phylogeny of the genus sedum (Crassulaceae) inferred from the ITS region of nuclear rDNA. Biologia 61, 1–9. https://doi.org/10.2478/s11756-006-0001-9 (2006).
Gontcharova, S. B. & Gontcharov, A. A. Molecular phylogeny and systematics of the genus sedum L. (Crassulaceae) inferred from ITS rDNA sequence data. Russian J. Genet. 45, 574–582. https://doi.org/10.1134/S1022795409050070 (2009).
Thiede, J. & Eggli, U. Crassulaceae. In (ed Kubitzki, K.) The Families and Genera of Vascular Plants, Vol. IX: Flowering Plants – Eudicots (83–118). Springer, Berlin/Heidelberg (2007).
Park, K. R. (2018) Crassulaceae. In: Flora of Korea Editorial Committee (Eds.) The Genera of Vascular Plants of Korea. 513–520. Hongreung Science Publishing, Seoul.
Stephenson, A. G. & Harris, D. L. Cytoplasmic male sterility in plants: its origin, development and exploitation. Theor. Appl. Genet. 82, 633–640. https://doi.org/10.1007/BF00227384 (1991).
Han, S. K., Kim, T. H. & Kim, J. S. A molecular phylogenetic study of the genus Phedimus for tracing the origin of Tottori Fujita cultivars. Plants 9 (2020).
Choi, T. Y., Son, D. C., Shiga, T. & Lee, S. R. Phedimus daeamensis (Crassulaceae), a new species from Mt. Daeam Korea PhytoKeys. 212, 57–71 (2022).
Cho, M. S. et al. Phylogenetic relationships and genetic diversity of the Korean endemic Phedimus latiovalifolius (Crassulaceae) and its close relatives. Sci. Rep. 14, 1–15 (2024).
Mayr, E. Speciat. Macroevolution Evol. 36: 1119–1132. (1982).
Stankowski, S. & Ravinet, M. Defining the speciation continuum. Evolution 74, 2669–2682. https://doi.org/10.1111/evo.14065 (2020).
Lee, T. B. Illustrated flora of Korea 404–408 (Hyangmunsa, 1980).
Lee, T. B. A natural hybrid of the genus Sedum. (S. aizokamtschatica hyb. Nov). Nat. Plant. 50, 5–6 (2000).
Lee, Y. N. New taxa on Korean flora (4). Korean J. Pl Taxon. 22, 7–11 (1992).
Park, K. R. (2007) Crassulaceae. In: Flora of Korea Editorial Committee (Eds.) The Genera of Vascular Plants of Korea. 513–520. Academy Publishing Co., Seoul.
Oh, S. Y. The phytogeographical studies of family crassulaceae in Korea. Res. Rev. Kyungpook Nat. Univ. 39, 123–159 (1985).
Yoo, Y. G. & Park, K. R. A test of the hybrid origin of Korean endemic Sedum latiovalifolium (Crassulaceae). Korean J. Pl. Taxon. 46(4), 378–391 (2016).
Moon, A. & Jang, C. Taxonomic study of genus sedum and Phedimus (Crassulaceae) in Korea based on external morphology. Korean J. Plant. Reources. 33, 116–129 (2020).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH image to imageJ: 25 years of image analysis. Nat. Methods. 9, 671–675 (2012).
Lê, S., Josse, J. & Husson, F. FactoMineR: an R package for multivariate analysis. J. Stat. Softw. 25, 1–18 (2008).
R Core Team. R: A language and environment for statistical computing (R Foundation for Statistical Computing, 2023). https://www.R-project.org/
Kassambara, A. & Mundt, F. factoextra: Extract and Visualize the Results of Multivariate Data Analyses. R package version 1.0.7 (2020). https://CRAN.R-project.org/package=factoextra
Bayona-Vásquez, N. J. et al. Adapterama III: Quadruple-indexed, double/triple-enzyme RADseq libraries (2RAD/3RAD). PeerJ 7, e7724. https://doi.org/10.7717/peerj.7724 (2019).
Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S. & Hoekstra, H. E. Double digest radseq: an inexpensive method for de Novo SNP discovery and genotyping in model and non-model species. PLoS ONE. 7, e37135. https://doi.org/10.1371/journal.pone.0037135 (2012).
Rochette, N. C., Rivera-Colón, A. G. & Catchen, J. M. Stacks 2: analytical methods for paired-end sequencing improve RADseq-based population genomics. Mol. Ecol. 28, 4737–4754. https://doi.org/10.1111/mec.15253 (2019).
Paris, J. R., Stevens, J. R. & Catchen, J. M. Lost in parameter space: A road map for stacks. Methods Ecol. Evol. 8, 1360–1373. https://doi.org/10.1111/2041-210X.12775 (2017).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter Estimation from sequencing data. Bioinformatics 27(21), 2987–2993 (2011).
Hess, J. E., Campbell, N. R., Close, D. A., Docker, M. F. & Narum, S. R. Population genomics of Pacific lamprey: adaptive var- Iation in a highly dispersive species. Mol. Ecol. 22, 2898–2916 (2012).
Purcell, S. et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. https://doi.org/10.1086/519795 (2007).
Glaubitz, J. C. et al. TASSEL-GBS: A high capacity genotyping by sequencing analysis pipeline. PLoS ONE. 9, e90346. https://doi.org/10.1371/journal.pone.0090346 (2014).
Excoffier, L. & Lischer, H. E. L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and windows. Mol. Ecol. Resour. 10, 564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x (2010).
Kalinowski, S. T. Counting alleles with rarefaction: private alleles and hierarchical sampling designs. Conserv. Genet. 5, 539–543. https://doi.org/10.1023/B:COGE.0000041021.91777.1a (2004).
Goudet, J. hierfstat, a package for R to compute and test hierarchical F-statistics. Mol. Ecol. Notes. 5, 184–186. https://doi.org/10.1111/j.1471-8286.2004.00828.x (2005).
Goudet, J. & Jombart, T. Hierfstat: Estimation and tests of hierarchical F-statistics (R package version 0.5–11. R package. (2022). https://cran.r-project.org/package=hierfstat
Do, C. et al. NeEstimator v2: Re-implementation of software for the Estimation of contemporary effective population size (Ne) from genetic data. Mol. Ecol. Resour. 14, 209–214. https://doi.org/10.1111/1755-0998.12157 (2014).
Eaton, D. A. R. & Overcast, I. Ipyrad: interactive assembly and analysis of RADseq datasets. Bioinformatics 36, 2592–2594. https://doi.org/10.1093/bioinformatics/btz966 (2020).
Sievert, C. Interactive web-based Data Visualization with R, plotly, and Shiny (CRC, 2020).
Pritchard, J. K., Stephens, M. & Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000).
Evanno, G., Regnaut, S. & Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 14, 2611–2620 (2005).
Jakobsson, M. & Rosenberg, N. A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806. https://doi.org/10.1093/bioinformatics/btm233 (2007).
Rosenberg, N. A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes. 4, 137–138. https://doi.org/10.1046/j.1471-8286.2003.00566.x (2004).
Pickrell, J. K. & Pritchard, J. K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8, e1002967. https://doi.org/10.1371/journal.pgen.1002967 (2012).
Fitak, R. R. OptM: estimating the optimal number of migration edges on population trees using TreeMix. Biology Methods Protocols. 6, bpab017. https://doi.org/10.1093/biomethods/bpab017 (2021).
Huson, D. H. & Bryant, D. Application ofphylogenetic networks in evolutionary studies. Mol. Biol. Evol. 23, 254–267 (2006).
Patterson, N. et al. Ancient admixture in human history. Proc. Natl. Acad. Sci. 109, E360–E367. https://doi.org/10.1073/pnas.1200567109 (2012).
Maier, R. et al. On the limits of fitting complex models of population history to f-statistics. eLife 12, 1–62 (2023).
Purcell, S. & Chang, C. C. PLINK 2.0: Next-generation genome-wide association studies and research. GigaScience Database. https://doi.org/10.5524/100125 (2015).
Liu, X. & Fu, Y. X. Exploring population size changes using SNP frequency spectra. Nat. Genet. 47, 555–559 (2015).
Ossowski, S. et al. The rate and mo- lecular spectrum of spontaneous mutations in Arabidopsis Thaliana. Science 327, 92–94 (2010).
Olfelt, J., Furnier, G. P. & Luby, L. Reproduction and development of the endangered sedum integrifolium ssp. Leedyi (Crassulaceae). Amer J. Bot. 85, 346–351 (1998).
Pezzi, G. et al. Phenological and genetic characterization of sedum Hispanicum (Crassulaceae) in the Italian Peninsula at the Western margin of its distribution. Plant. Ecol. Evol. 150, 293–303 (2017).
Bennett D, M. Nuclear DNA content and minimum generation time in herbaceous plants. Proc. Royal Soc. B: Biol. Sci. 181, 109–135 (1972).
Fick, S. E. & Hijmans, R. J. WorldClim 2: new 1- Km Spatial reso- Lution climate surfaces for global land areas. Int. J. Climatol. 37, 4302–4315. https://doi.org/10.1002/joc.5086 (2017).
Phillips, S. J., Dudík, M. & Schapire, R. E. Maxent software for modeling species niches and distributions (Version 3.4.1) (2018). https://biodiversityinformatics.amnh.org/open_source/maxent/
Warren, D. L., Glor, R. E. & Turelli, M. ENMTools: a toolbox for comparative studies of environmental niche models. Ecography 33, 607–611. https://doi.org/10.1111/j.1600-0587.2009.06142.x (2010).
Warren, D. L., Glor, R. E. & Turelli, M. Environmental niche equivalency versus conservatism: quantitative approaches to niche evolution. Evolution 62, 2868–2883. https://doi.org/10.1111/j.1558-5646.2008.00482.x (2008).
Feder, J. L., Egan, S. P. & Nosil, P. The genomics of speciation-with-gene-flow. Trends Genet. 28, 342–350. https://doi.org/10.1016/j.tig.2012.03.009 (2012).
Hamrick, J. L. & Godt, M. J. W. Effects of life history traits on genetic diversity in plant species. Philosophical Trans. Royal Soc. B: Biol. Sci. 351, 1291–1298. https://doi.org/10.1098/rstb.1996.0112 (1996).
Nybom, H. Comparison of different nuclear DNA markers for estimating intraspecific genetic diversity in plants. Mol. Ecol. 13, 1143–1155. https://doi.org/10.1111/j.1365-294X.2004.02141.x (2004).
De Kort, H. et al. Landscape genomics and the evolutionary response of plants to climate change. Ecol. Lett. 24, 258–272. https://doi.org/10.1111/ele.13641 (2021).
Bertel, C., Schierup, M. H. & Hobolth, A. Ancestral recombination graphs for diploid populations. Genetics 202, 1113–1124. https://doi.org/10.1534/genetics.115.183814 (2016).
Rangwala, H. (ed, D.) Machine learning for cancer prediction using microarray gene expression data: an overview. Proc. IEEE 90 1763–1776 https://doi.org/10.1109/JPROC.2012.2202211 (2012).
Meng, H. H., Su, T., Gao, X. Y., Li, J. & Sun, H. Geographical isolation and environmental heterogeneity contributed to the Spatial genetic patterns of rheum nobile (Polygonaceae). J. Biogeogr. 46, 1502–1514. https://doi.org/10.1111/jbi.13591 (2019).
Barthlott, W., Lauer, W. & Placke, A. Global distribution of species diversity in vascular plants: towards a world map of phytodiversity. Erdkunde 50, 317–327. https://doi.org/10.3112/erdkunde.1996.04.03 (1996).
Hughes, C. E. & Atchison, G. W. The ubiquity of alpine plant radiations: from the Andes to the Hengduan mountains. New Phytol. 207, 275–282. https://doi.org/10.1111/nph.13230 (2015).
Lawing, A. M. & Polly, P. D. Pleistocene climate, phylogeny, and climate envelope models: an integrative approach to better understand species’ response to climate change. PLoS ONE 6 (2011).
Cabanne, G. S. et al. Effects of pleistocene climate changes on species ranges and evolutionary processes in the Neotropical Atlantic forest. Biol. J. Linn. Soc. 119, 856–872 (2016).
Duminil, J. et al. Late pleistocene molecular dating of past population fragmentation and demographic changes in African rain forest tree species supports the forest refuge hypothesis. J. Biogeogr. 42, 1443–1454 (2015).
Benítez-Benítez, C., Escudero, M., Rodríguez-Sánchez, F. & Martín-Bravo, S. and P. Jiménez-Mejías. 2018. Pliocene–Pleistocene ecological niche evolution shapes the phylogeography of a mediterranean plant group. Mol. Ecol. 27, 1696–1713 .
Wang, R. H. et al. Plio-Pleistocene Climatic change drives allopatric speciation and population divergence within the scrophularia Incisa complex (Scrophulariaceae) of desert and steppe subshrubs in Northwest China. Front. Plant Sci. 13, 1–18 (2022).
Funding
This work was supported by the Korea National Arboretum (KNA), project Establishment of plant seed production system for forest ecological restoration (KNA-1-2-46-23-4) and the research fund from the Chosun University, 2023.
Author information
Authors and Affiliations
Contributions
S.R.L., B.K.P. and D.C.S conceived ideas and designed the study. Field sampling and the laboratory work was planned and performed by T.Y.C., D.C.S and B.K.P. T.Y.C. and S.R.L. designed and performed the genetic and statistical analysis. S.R.L. wrote the manuscript. All authors edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Choi, TY., Park, B.K., Son, D.C. et al. Unraveling species diversification and niche separation in Phedimus Kamtschaticus and P. aizoon using RAD-seq data and ecological niche modeling. Sci Rep 16, 2145 (2026). https://doi.org/10.1038/s41598-025-31925-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-31925-y
