
Abstract
The preferential accumulation of anthracyclines in the mitochondrial matrix has been proposed to trigger a self-perpetuating vicious cycle with mitochondrial DNA (mtDNA) alteration and redox stress enhancing one each other to lead to a progressive mitochondrial impairment. To test this hypothesis, we monitored oxygen consumption rate (OCR) and mt-DNA copy number (mtDNA-CN) of peripheral blood mononuclear cells (PBMCs) harvested from 23 patients with Hodgkin lymphoma (HL) submitted to adriamycin-bleomycin-vinblastine-dacarbazine (ABVD) for the whole treatment, according to a Deauville score ≤ 3 after two cycles of chemotherapy at the interim PET/CT. PBMCs were isolated before treatment (baseline), at interim and one month after End of Therapy (EoT). Baseline data were compared with 23 healthy subjects selected according to a case-control criterion. OCR was estimated under control condition, after blockade of ATP-synthase, and of mitochondrial Complexes I and III. mtDNA-CN was assayed by droplet digital PCR and normalized against nuclear DNA. Mitochondrial DNA mutational status was assayed by next generation sequencing and alignment to reference genome after sequencing depth equalization. At diagnosis, mitochondrial OCR was lower in HL PBMCs than in controls, despite a preserved mitochondrial asset, testified by the mtDNA-CN. In the 18 subjects with complete remission at EoT PET/CT, both variables decreased back to the baseline values. By contrast, in the 5 patients with persistent disease, both mitochondrial OCR and mtDNA-CN remained elevated. ABVD therapy alters mitochondrial function and biogenesis of normal PBMCs whose metabolic pattern might represent a possible marker of treatment effectiveness.
Similar content being viewed by others


Background
Chemotherapy with anthracyclines is associated with a considerable risk of serious consequences. Indeed, survival after complete remission is threatened by an increased risk of new independent neoplasms or heart failure, while quality of life may be impaired by the occurrence of cognitive impairment or reproductive failure1,2.
Involved tissues and organs can be different. Nevertheless, these complications share a common feature: their incidence increases with elapsing time for months and years after treatment discontinuation3. This characteristic indicates an “irreversible” nature of anthracycline toxicity possibly related to a genetic damage. However, the diffuse nature of the toxic damage and its high incidence in tissues, the myocardium and the brain, characterized by slow proliferation is also compatible with the activation of a self-perpetuating vicious cycle leading to a progressive functional deterioration.
Both concepts fit with the notion that anthracyclines preferentially accumulate in the mitochondrial matrix4,5, where they can interact with both cardiolipin and mitochondrial DNA (mtDNA)6. The consequent acceleration in the generation of reactive oxygen species7,8 is thought to trigger a self-perpetuating vicious cycle in which mtDNA and oxidative damage reinforce each other, leading to progressive impairment of mitochondrial bioenergetic performance9.
Doxorubicin (DXR) has been found to cause a persistent impairment in mitochondrial dynamics characterized by upregulation of GTPase dynamin-related protein 1 (DRP-1), the “fission promoter”10, combined with downregulation of the fusion factors mitofusin 1 and 2 and optic atrophy 111. Altogether, these considerations indicate that anthracycline treatment might impair the mitochondrial network causing an accumulation of fragmented mitochondria in both cancer and innocent bystander host cells for a long time12.
An accessible approach to test this hypothesis is to analyze peripheral blood mononuclear cells (PBMCs). The support of immune function by this heterogeneous cell population has already prompted a wide literature on the metabolic properties and mitochondrial respiration of these cells in various conditions, including chemotherapy13,14. Accordingly, the aim of the present study was to examine the response of mitochondrial respiration to DXR. These functional analyses were thus complemented with the evaluation of mtDNA copy number (mtDNA-CN) to verify whether mitochondrial dynamics and function of “normal” PBMCs are affected by chemotherapy and whether their response is related to treatment effectiveness. This evaluation was performed in patients with Hodgkin lymphoma (HL) as a clinical model allowing a serial evaluation of DXR effect, and standard criteria to define the treatment effectiveness using positron emission tomography/computed tomography (PET/CT) scanning at well-established time-points.
Methods
Patient population
From June 2020 up to January 2023, a total of 38 patients were admitted to our institute for suspected, and subsequently confirmed, HL; 15 of them were subsequently excluded due to the escalation of therapy after two cycles of doxorubicin, bleomycin, vinblastine, and dacarbazine (ABVD), leaving a study cohort of 23 patients. Control subjects (n = 23) participating in the Preventive Medicine Program of our Institute were enrolled according to a case-control criterion. Exclusion criteria were positivity for HBV, HCV and HIV or any other coexistent disease asking for pharmacological therapy. All participants provided their written informed consent to participate in this study that was approved by the Ethical Committee of Regione Liguria (50/20-DB id 10306).
According to current guidelines15, all patients were submitted to a staging PET/CT imaging of 18F-Fluorodeoxyglucose (FDG) uptake just before the start of treatment with ABVD, administered at standard doses. FDG imaging was repeated after two ABVD cycles (interim-PET) as well as at three weeks-one month after the end of therapy (EoT-PET).
The interim PET was evaluated according to the Deauville criteria16 to identify the 23 patients with a score 1 to 3 in whom the maintenance of ABVD regimen was indicated. Finally, the EoT-PET was analyzed according to the Lugano criteria to identify responders and non-responders17.
PBMCs isolation
Blood cell count was assayed according to the same routine procedure of our Institute for both HL patients and control subjects. Sampling was performed at the scheduled time in control subjects. In HL patients, samples were harvested at the three time points of imaging, just before the FDG administration, and thus before treatment started, at interim- and EoT-PET/CT evaluation, respectively.
At the time of blood sampling, further 15 mL of blood were collected and analyzed within 24 h. According to our validated procedure18, PBMCs were isolated using lympholyte gradients (Cedarlane), washed three times with Ca2+/Mg2+ free phosphate-buffered saline and resuspended at 5 × 106 cells/mL. Obtained cells were divided and dedicated to the different experimental evaluations.
Seahorse analysis
PBMCs were seeded in Seahorse XFp Extracellular Flux Analyzer (Agilent Technologies, Santa Clara, CA, US) cell plates (105 cells/well) and gently centrifuged with no brake at 40 × g for 3 min. The plate was then rotated 180° before a further centrifugation at 80× g for 3 min to encourage adhesion to the plate and to obtain an evenly dispersed monolayer19. Cells were then incubated at 37 °C for 45 min in a no-CO2 incubator with Agilent Seahorse DMEM, pH 7.4, enriched with glucose (11 mM). OCR and extracellular acidification rate (ECAR) were monitored according to the manufacturer instructions. Briefly, three measurements were taken under control conditions and after sequential injections of 1.5 µM oligomycin (ATP-synthase inhibitor) and 0.5 µM rotenone (Complex I inhibitor) plus 0.5 µM antimycin A (Complex III inhibitor).
ECAR was converted to proton efflux rate using WAVE software (Version 2.4, Agilent), after evaluating the buffer factor of the medium. PER value was thus divided by 2, to estimate glucose flux through glycolysis (nanomoles × min− 1/million cells).
Flow cytometry analysis
To assess the redox status, 5 × 105 PBMCs were stained for 20 min at 37 °C with 5 µM MitoSOX Red and 10 µM 2′,7′-dichlorodihydrofluorescein (DCF) (Thermo Fisher Scientific, Waltham, MA, USA). The cells were then washed twice with PBS, centrifuged for 5 min at 290× g and resuspended in PBS + 1% BSA for flow cytometry analysis. Data were acquired on a FACSCan (Becton Dickinson, Milan, Italy) and data analysis was performed with FlowJo software (Version 9.96).
Mitochondrial DNA extraction and copy number assessment
Total DNA, including mtDNA, was extracted from the PBMCs using QIAamp DNA Kit (Qiagen, Hilden, Germany). DNA concentration was assessed by Qubit Fluorometer using dsDNA HS Assay Kit (ThermoFisher Scientific, San Jose, CA, USA). The mean mtDNA-CN per cell was determined by QX200 droplet digital polymerase chain reaction (ddPCR) system (Bio-Rad, Hercules, CA, USA) using a previously described multiplexing strategy20.
We employed two custom FAM-labeled assays targeting mtDNA genes (i.e., mitochondrially encoded NADH dehydrogenase 1, ND1 (dHsaCNS669425578) and DN6 (dHsaCNS941916401)) normalized against two HEX-labeled assays targeting nuclear DNA (i.e., ribonuclease P/MRP subunit p30, RPP30 (dHsaCP2500350) and argonaute RISC component 1, AGO1 (dHsaCP1000002)).
Due to the higher mtDNA-CN compared to nuclear DNA, the two mitochondrial assays (ND1 and ND6) were evaluated in two separate reactions and then averaged. Briefly, 0.9 ng of total DNA was amplified using the ddPCR Supermix for Probes (No dUTP) (Bio-Rad) with the following TaqMan® assays: ND1 or ND6 (900 nM), RPP30 (900 nM), and AGO1 (450 nM) at an annealing/extension temperature of 58 °C. Each sample was run in duplicate with a negative control (no template) and the data analysis was conducted using QuantaSoft™ Analysis Pro software v.1.0.596 (Bio-Rad). The mtDNA-CN assessment was performed by calculating the ratio between the concentration (copies/µl) of the ND1 or ND6 locus and the mean concentration of RPP30 and AGO1. This ratio was then multiplied by two to account for the presence of two copies of nuclear genes in diploid cells, and the ND1 and ND6 copy numbers were averaged.
Mitochondrial DNA mutational status
Mitochondrial DNA sequencing was performed using KAPA HyperCap v3.3 workflow (Roche Diagnostics, Mannheim, Germany), according to the manufacturer’s instructions, starting from 100 ng of total DNA. The amplified libraries were quantified by Qubit Fluorometer (dsDNA HS Assay Kit) and combined into sets of 8 libraries (187.5 ng for each library).
These pooled libraries were then hybridized at 55 °C for 18 h using KAPA HyperCap Target Enrichment probes (KAPA HyperCap DS Human mtDNA Design, 0.5 Mb T1, 1000005603). After hybridization, the captured pools were amplified through 16 PCR cycles, followed by purification and quantification. Then, library pools were run on a NextSeq500 (Illumina Inc., San Diego, CA, USA) instrument using 2 × 150pb paired-end sequencing.
Raw fastq files were processed for UMI extraction and consensus reads generation using fgbio (Fulcrum Genomics). Consensus reads were then aligned to GRCh38 human genome assembly using BWA-mem. Coverage of mitochondrial chromosome was equalized by trimming BAM files to the highest possible common coverage (about 150 UMIs). Variant calling and annotation were performed by Mutect2 (GATK) and ANNOVAR respectively. Variants retrieved in a single consensus read were discarded.
Statistics
All data were reported as means of ± SD. Unpaired t test was used to compare data between control subjects and HL patients at the first examination. Response of sample cell composition, energy metabolism, mtDNA-CN at the tree time point was tested according to one-way or two-way ANOVA as appropriate. All statistical analyses were carried out by using a dedicated software package, GraphPad Prism version 8 (GraphPad, San Diego, CA, USA).
Results
Patient population
As shown in Table 1, control subjects and HL patients showed comparable age and similar prevalence of female gender. On routine peripheral blood examination, red blood cell counts, and hemoglobin levels were similar in both groups. However, HL patients had significantly higher white blood cell counts, which mostly included both neutrophils and monocytes, while lymphocyte counts were comparable in both groups.
Data are mean ± standard deviation, and number (percentage). Abbreviations: HL, Hodgkin lymphoma; ns, not significant.
HL and energy metabolism of “Normal” PBMCs
At the baseline study, PBMCs sampled from HL patients had almost twice the rate of glycolytic flux compared to control subjects (Fig. 1A). This “Warburg effect” activation paralleled a measurable decrease in overall OCR in HL PBMCs (Fig. 1B) that explained by a selective impairment in mitochondrial function. Indeed, PBMCs harvested from HL patients showed a lower rate of oxygen usage in both its ATP-linked (Fig. 1C) and ATP-independent (Fig. 1D) fractions. By contrast, non-mitochondrial (rotenone/antimycin-insensitive) oxygen usage was unchanged in patients compared to controls (Fig. 1E), as well as the extra-mitochondrial/mitochondrial OCR ratio (Fig. 1F).
These results suggest that PBMCs from HL patients have a measurable decrease in mitochondrial OCR. Thus, the marked acceleration of lactate release might be compatible with a compensatory role of glycolysis in supporting the energy demands of HL PBMCs.

Baseline metabolic characteristics of peripheral blood mononuclear cells in controls and patients with Hodgkin lymphoma. (A) Glycolytic flux. (B) Overall oxygen consumption rate (OCR). (C) ATP-linked and (D) ATP-independent OCR. (E) Extra-mitochondrial OCR and (F) extra-mitochondrial/mitochondrial OCR ratio. *p < 0.05; ***p < 0.001; ****p < 0.0001; ns: not significant.
Chemotherapy and energy metabolism of PBMCs
Mean glucose consumption (Supplementary Fig. 1 A) and overall OCR (Supplementary Fig. 1B) remained unchanged from baseline to interim and up to the end of the observation period. This finding suggests that the energy requirements of PBMCs exposed to chemotherapy remained unchanged. However, this interpretation was challenged by the OCR behavior. Indeed, although overall OCR did not show significant changes, its ATP-linked fraction progressively increased during treatment (Supplementary Fig. 1 C) as opposed to mitochondrial ATP-independent OCR that remained substantially invariant (Supplementary Fig. 1D).
Oxygen consumption of PBMC and response to chemotherapy
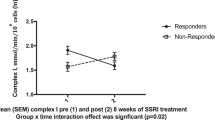
According to the Lugano criteria, 18 of 23 (78%) patients showed a complete response to ABVD therapy and were thus defined as “responders”. Despite the similar Deauville score in the interim study16, the remaining five patients were classified as “non-responders” (Fig. 2). No difference was found in age (54 ± 18 vs. 52 ± 21 in non-responders vs. responders, p = ns) or in female gender prevalence (40% vs 33%, respectively).

Representative PET/CT images of two patients with Hodgkin lymphoma. Representative Maximum Intensity Projection (MIP) and PET/CT fused images of baseline, interim, and end of therapy (EoT) scans. On the left (blue), the figure shows a patient which responded to treatment (Deauville score at interim, and complete metabolic response after therapy according to Lugano criteria). On the right (red), the figure presents a patient showing an initial response at interim evaluation (Deauville score 1) but classified as non-responder at the final assessment (progressive disease according to Lugano criteria).
Glucose consumption was not related to treatment effectiveness at any of the three time points studied (Fig. 3A). In contrast, the OCR response to ABVD treatment (Fig. 3B) was different in the two groups. It did not change in responders, whereas it progressively increased in non-responders and became significantly higher after the end of treatment than in patients with complete remission.
In non-responder patients, this behavior largely involved the mitochondria, as both respiratory ATP-linked (oligomycin-sensitive) and ATP-independent (oligomycin-insensitive) OCR progressively increased from baseline to interim and final phases of treatment (Fig. 3C and D). On the other hand, the non-mitochondrial (rotenone/antimycin-insensitive) OCR fraction slightly (though significantly) increased up to the EoT time-point, again only in non-responders (Fig. 3E). Finally, the different response of mitochondrial function was not related to a change in the composition of PBMCs, as the monocyte/lymphocyte ratio was similar in both groups at all time points examined (Fig. 3F).
The different metabolic pattern was not matched by the redox status. This analysis was performed in a subgroup of patients whose size was relatively small in the interim phase and definitely not adequate at the EoT, that was thus not considered. DCF staining reported a comparable cell redox status in the PBMCs harvested from either responders or non-responders (Supplementary Fig. 2 A). This similarity extended to the oxidative stress of mitochondria, since Mitosox staining did not report any significant difference between the two cohorts of patients (Supplementary Fig. 2B).

Metabolic and mitochondrial genetic characteristics of peripheral blood mononuclear cells in patients with Hodgkin lymphoma. Baseline, interim, and end of therapy (EoT) metabolic and mitochondrial genetic characteristics of peripheral blood mononuclear cells in responder (blue) and non-responder (red) patients with Hodgkin lymphoma. (A) Glycolytic flux. (B) Overall oxygen consumption rate (OCR). (C) ATP-linked, (D) ATP-independent, and (E) extra-mitochondrial OCR. (F) Monocyte/lymphocyte ratio. Mitochondrial DNA copy number (mtDNA-CN) in (G) responder and (H) non-responder patients. *p < 0.05; #p < 0.05 vs. corresponding baseline; ns: not significant.
Mitochondrial DNA analysis
The value of mtDNA-CN did not differ significantly between control subjects (mean: 290.4; range: 193.4–428.8.4.8) and patients at baseline (mean: 254.9; range: 131.6–697.2.6.2) (Supplementary Fig. 3 A, Supplementary Table 1). However, the first two ABVD cycles significantly increased the mtDNA copy number at the interim time point in the entire HL cohort. This effect was apparently transient, as this value returned to baseline on EoT-PET examination (Supplementary Fig. 3B; Supplementary Table 2). It is noteworthy that this was not due to a change in the cells populating the analyzed sample, whose composition did not change throughout the course of treatment (Supplementary Fig. 3 C). Similarly, mt-DNA copy numbers were largely independent of the ratio of monocytes to lymphocytes, regardless of the phase of disease analyzed (Supplementary Fig. 3D).
The behavior of mtDNA-CN was not homogeneously distributed. In responder patients, the first two ABVD cycles did not modify the mtDNA-CN that instead showed a significant decrease in the following phase, up to the EoT time-point (Fig. 3G). By contrast, non-responder patients showed a completely different pattern: mtDNA-CN significantly increased from baseline to Interim and remained elevated thereafter, up to one month after treatment discontinuation (Fig. 3H).
In a subset of 18 out of 23 patients (i.e., 15 responders and 3 non-responders), mtDNA mutational status was also investigated at all three time points. The number of variants was then quantified after equalizing mtDNA-CN coverage among all samples. Notably, a high degree of heterogeneity in the number of mtDNA variants was observed at baseline, spanning from a median value of 32 to a median value of 347 (data not shown) that, however, was not associated with treatment response. Furthermore, no significant changes in the number of variants were detected during treatment between responders and non-responder patients. Similarly, despite the increase in mtDNA-CN, comprehensive analysis of mtDNA deletions revealed no significant change during therapy, and no difference between responders and non-responders.
Discussion
In the present study, the PBMCs obtained from HL patients were characterized by an acceleration of the incomplete glucose degradation to lactate compared to control subjects. The association of the high glycolytic rate with the invariance of baseline oxygen consumption may suggest that the presence of HL increases the energy requirements of PBMCs despite the lack of contamination by neoplastic elements. However, this assumption is contradicted by the evidence that HL decelerates the respiratory function of these same PBMCs and decreases the amount of oxygen consumed by the mitochondria, either linked to ATP regeneration or independent of energy asset.
This metabolic pattern was observed in treatment-naïve patients and cannot be explained by a loss of mitochondria, as shown by the mtDNA-CN content, which was comparable to that of PBMCs harvested from the control subjects. Thus, these data suggest that the presence of HL triggers a metabolic reprogramming that activates aerobic glycolysis in the “normal” cells populating the peripheral blood.
In responder patients, ABVD therapy only slightly modified overall mtDNA-CN of PBMCs that remained virtually invariant from baseline to EoT time-point. This trend was reproduced by all fractions of OCR, that remained stable at all the three time-points of evaluation. The same “normal” blood cells of patients with persistent disease showed a different behavior. Their mtDNA-CN significantly increased after the first two ABVD cycles, and remained elevated thereafter, at least until the late EoT control. This trend was also nicely reproduced by mitochondrial function, with both ATP-related and ATP-independent OCR fractions significantly increasing from baseline to interim, and up to the EoT assessment. Finally, this effect was apparently not explained by any effect of therapy on mtDNA mutational burden.
Overall, these data suggest that ABVD effectiveness is associated with a specific behavior of function and number of the mitochondria populating the PBMCs, as an epitome of normal host cells. Complete remission is associated with a negligible and non-significant response of both parameters. By contrast, the persistence of HL lesions after the same chemotherapy is associated with an increase in the mitochondrial function and mtDNA-CN that persists at least one month after chemotherapy.
Metabolic assets in the studied populations.
The number of HL patients enrolled in the present study was relatively low. However, this limitation was motivated by the need to avoid the interference of any variation in the administered chemotherapy regimen on PBMCs metabolic features. Accordingly, the analysis was limited to those patients submitted to the same ABVD regimen for the whole observation period, from the staging up to the end of therapy. This selection criterion forced us to limit the enrollment according to a Deauville score at the interim evaluation ranging 1 to 316.
The large acceleration of the glycolytic flux of HL patients compared with control subjects was not related to any bias related to age, according to the case-control criterion used for the selection21. Similarly, it was not attributable to a significant difference in the composition of sampled PBMCs, since the relative prevalence of monocytes and lymphocytes were similar in the two groups at the baseline examination. Likewise, the different behavior of OCR and mtDNA-CN in responders and non-responder patients cannot be explained by a possible alteration of the PBMCS composition. Indeed, the ratio between monocytes and lymphocytes did not significantly change during the three time points, although monocyte prevalence showed a slight and not significant increase at the interim phase.
Accordingly, the variation of mitochondrial number and respiratory function most likely reflects the metabolic response of studied cells to ABVD. This same response implied a progressive acceleration of mitochondrial number and respiratory activity in non-responders as opposed to responder ones whose PBMCs showed an only transient increase in mtDNA-CN associated with a trivial and not significant alteration in mitochondrial OCR.
Mitochondrial asset and anthracycline toxicity.
So far, several studies have already evaluated the effect of anthracyclines on mitochondrial function and number. In agreement with our observation in normal PBMCs, an increase in mtDNA-CN has been found to predict the resistance against anthracycline toxicity of cultured cell lines derived from breast22, head and neck or lung carcinomas23. Similarly, Kusao et al. reported that a marked increase in mtDNA-CN identifies chemo-resistant cells harvested from pediatric patients treated with doxorubicin for Burkitt lymphoma24. Unfortunately, the limited amount of harvested PBMCS did not permit to couple our functionals with the assessment of proteins related to either pro- or anti-apoptotic signals as well as mitochondrial dynamics. Nevertheless, Malik et al. proposed that the increased mtDNA asset might reflect an upregulated mitochondrial biogenesis in the presence of a high redox stress25. Similarly, the preliminary administration of dexrazoxane has been shown to almost fully prevent this response in a pediatric cohort of survivors after diagnosis of acute lymphoblastic leukemia13. The relevance of these in vitro studies has been verified in the clinical setting by Hsu and coworkers who documented that high mtDNA-CN in the surgical specimen is associated with a poor outcome in patients with breast cancer treated with anthracyclines26.
The previously quoted studies focused on cancer, our approach aimed instead to characterize the metabolic effect of ABVD regimen on normal cells epitomized by the circulating PBMCs of HL patients. This decision was motivated by the notion that the mitochondrial function is a primary promoter of anthracycline toxicity on the myocardium27,28. Nevertheless, a relatively lower experience focused on the hypothesis that the cytotoxic effect of anthracyclines might reflect the activation of the same mechanisms in cancer cells as in normal ones. Christensen et al. reported an increased respiration coupled with an increase in mtDNA-CN in the PBMCs harvested from patients treated with adjuvant therapy for early-stage breast cancer29. Similarly, Qu et al. already reported that a high mtDNA-CN in leucocytes populating the peripheral blood predicts a poor outcome in patients with colorectal carcinoma30. The present data extends this previous observation documenting that the anthracycline interference on mitochondrial balance is not limited to cancer and is rather shared with normal host cells.
Defining the mechanisms underlying this response would have asked for a more precise definition of mitochondrial biology to verify the role of an increased number and an augmented mass due to the synthesis of mitochondrial proteins, and the activity of key transcriptional regulators. These analyses could not be performed in our study. Nevertheless, the PBMCs harvested from HL patients with incomplete response to ABVD combined the progressive increase in OCR and the persistent elevation of mtDNA-CN. This observation corroborates the concept that the mitochondrial response of normal cells might predict the cytotoxic effect of chemotherapy on lymphoma lesions.
Conclusions
The present data indicate that the therapeutic effectiveness of anthracyclines on HL is associated with a specific behavior of PBMCs whose mtDNA-CN and respiratory function are slightly, if any, altered by chemotherapy. By contrast, the resistance to ABVD treatment is associated with a progressive acceleration of mitochondrial function and mtDNA-CN that persists after treatment discontinuation.
Previous studies already suggested the cancer and its generating ecosystem (the host) share a similar response to anthracycline toxicity27. The present study extends this concept by documenting that this sharing involves the regulation of mitochondrial function and biogenesis, identifying the analysis of PBMCs as a possible marker of treatment effectiveness, at least in HL patients.
Data availability
The datasets generated and/or analysed during the current study are not publicly available but are available from the corresponding author on reasonable request.
Abbreviations
- ABVD:
-
Doxorubicin, bleomycin, vinblastine, and dacarbazine
- ddPCR:
-
Droplet digital polymerase chain reaction
- DRP-1:
-
Dynamin-related protein 1
- DXR:
-
Doxorubicin
- ECAR:
-
Extracellular acidification rate
- EoT:
-
End of therapy
- FDG:
-
Fluorodeoxyglucose
- HL:
-
Hodgkin lymphoma
- mtDNA:
-
Mitochondrial DNA
- mtDNA-CN:
-
Mitochondrial DNA copy number
- OPA1:
-
Optic atrophy 1
- PBMCs:
-
Peripheral blood mononuclear cells
- PET/CT:
-
Positron emission tomography / computed tomography
References
Kamińska, K. & Cudnoch-Jędrzejewska, A. A review on the neurotoxic effects of doxorubicin. Neurotox. Res. https://doi.org/10.1007/s12640-023-00652-5 (2023).
Mohan, U. P., P B TP, Iqbal, S. T. A. & Arunachalam, S. Mechanisms of doxorubicin-mediated reproductive toxicity - A review. Reprod. Toxicol. https://doi.org/10.1016/j.reprotox.2021.04.003 (2021).
Steinherz, L. J., Steinherz, P. G., Tan, C. T., Heller, G. & Murphy, M. L. Cardiac toxicity 4 to 20 years after completing anthracycline therapy. JAMA 266 (12), 1672–1677 (1991).
Sokolove, P. M. Interactions of adriamycin aglycones with mitochondria May mediate adriamycin cardiotoxicity. Int. J. Biochem. https://doi.org/10.1016/0020-711x(94)90176-7 (1994).
Tokarska-Schlattner, M. et al. Reduced creatine-stimulated respiration in doxorubicin challenged mitochondria: particular sensitivity of the heart. Biochim. Biophys. Acta. https://doi.org/10.1016/j.bbabio.2007.08.006 (2007).
Lebrecht, D., Setzer, B., Ketelsen, U. P., Haberstroh, J. & Walker, U. A. Time-dependent and tissue-specific accumulation of MtDNA and respiratory chain defects in chronic doxorubicin cardiomyopathy. Circulation https://doi.org/10.1161/01.CIR.0000093196.59829.DF (2003).
Deng, S. et al. Gp91phox-containing NAD(P)H oxidase increases superoxide formation by doxorubicin and NADPH. Free Radic Biol. Med. https://doi.org/10.1016/j.freeradbiomed.2006.11.013 (2007).
Huang, J. et al. Understanding anthracycline cardiotoxicity from mitochondrial aspect. Front. Pharmacol. https://doi.org/10.3389/fphar.2022.811406 (2022).
Lebrecht, D., Kokkori, A., Ketelsen, U. P., Setzer, B. & Walker, U. A. Tissue-specific MtDNA lesions and radical-associated mitochondrial dysfunction in human hearts exposed to doxorubicin. J. Pathol. https://doi.org/10.1002/path.1863 (2005).
Catanzaro, M. P. et al. Doxorubicin-induced cardiomyocyte death is mediated by unchecked mitochondrial fission and mitophagy. FASEB J. https://doi.org/10.1096/fj.201802663R (2019).
Marques-Aleixo, I. et al. Exercise and doxorubicin treatment modulate cardiac mitochondrial quality control signaling. Cardiovasc. Toxicol. https://doi.org/10.1007/s12012-017-9412-4 (2018).
Kraus, F., Roy, K., Pucadyil, T. J. & Ryan, M. T. Function and regulation of the divisome for mitochondrial fission. Nature https://doi.org/10.1038/s41586-021-03214-x (2021).
Lipshultz, S. E. et al. Impaired mitochondrial function is abrogated by Dexrazoxane in doxorubicin-treated childhood acute lymphoblastic leukemia survivors. Cancer https://doi.org/10.1002/cncr.29872 (2016).
Mohd Khair, S. Z. N., Abd Radzak, S. M. & Mohamed Yusoff, A. A. The uprising of mitochondrial DNA biomarker in cancer. Dis. Markers. https://doi.org/10.1155/2021/7675269 (2021).
Hoppe, R. T. et al. Hodgkin Lymphoma, version 2.2020, NCCN clinical practice guidelines in oncology. J. Natl. Compr. Canc Netw. https://doi.org/10.6004/jnccn.2020.0026 (2020).
Biggi, A. et al. Predictive value on advance hodgkin lymphoma treatment outcome of end-of treatment FDG PET/CT in the HD0607 clinical trial. Hematol. Oncol. https://doi.org/10.1002/hon.3117 (2023).
Cheson, B. D. et al. Recommendations for initial evaluation, staging, and response assessment of hodgkin and non-Hodgkin lymphoma: the Lugano classification. J. Clin. Oncol. https://doi.org/10.1200/JCO.2013.54.8800 (2014).
Carta, S. et al. Cell stress increases ATP release in NLRP3 inflammasome-mediated autoinflammatory diseases, resulting in cytokine imbalance. Proc. Natl. Acad. Sci. U S A. https://doi.org/10.1073/pnas.1424741112 (2015).
Jones, N. et al. Bioenergetic analysis of human peripheral blood mononuclear cells. Clin. Exp. Immunol. https://doi.org/10.1111/cei.12662 (2015).
Truini, A. et al. Downregulation of miR-99a/let-7c/miR-125b MiRNA cluster predicts clinical outcome in patients with unresected malignant pleural mesothelioma. Oncotarget https://doi.org/10.18632/oncotarget.19800 (2017).
Wong, J. et al. Mitochondrial DNA content in peripheral blood monocytes: relationship with age of diabetes onsetand diabetic complications. Diabetologia https://doi.org/10.1007/s00125-009-1424-6 (2009).
Zhuang, F., Huang, S. & Liu, L. PYCR3 modulates MtDNA copy number to drive proliferation and doxorubicin resistance in triple-negative breast cancer. Int. J. Biochem. Cell. Biol. https://doi.org/10.1016/j.biocel.2024.106581 (2024).
Mei, H. et al. Reduced MtDNA copy number increases the sensitivity of tumor cells to chemotherapeutic drugs. Cell. Death Dis. https://doi.org/10.1038/cddis.2015.78 (2015).
Kusao, I., Agsalda, M., Troelstrup, D., Villanueva, N. & Shiramizu, B. Chemotoxicity recovery of mitochondria in non-Hodgkin lymphoma resulting in minimal residual disease. Pediatr. Blood Cancer. https://doi.org/10.1002/pbc.21545 (2008).
Malik, F. et al. Reactive oxygen species generation and mitochondrial dysfunction in the apoptotic cell death of human myeloid leukemia HL-60 cells by a dietary compound Withaferin A with concomitant protection by N-acetyl cysteine. Apoptosis https://doi.org/10.1007/s10495-007-0129-x (2007).
Hsu, C. W., Yin, P. H., Lee, H. C., Chi, C. W. & Tseng, L. M. Mitochondrial DNA content as a potential marker to predict response to anthracycline in breast cancer patients. Breast J. https://doi.org/10.1111/j.1524-4741.2010.00908.x (2010).
Marini, C. et al. Myocardial metabolic response predicts chemotherapy curative potential on hodgkin lymphoma: A Proof-of-Concept study. Biomedicines https://doi.org/10.3390/biomedicines9080971 (2021).
Yin, J. et al. Doxorubicin-induced mitophagy and mitochondrial damage is associated with dysregulation of the PINK1/parkin pathway. Toxicol. Vitro. https://doi.org/10.1016/j.tiv.2018.05.001 (2018).
Christensen, I. B. et al. Peripheral blood mononuclear cells exhibit increased mitochondrial respiration after adjuvant chemo- and radiotherapy for early breast cancer. Cancer Med. https://doi.org/10.1002/cam4.6333 (2023).
Qu, F. et al. Leukocyte mitochondrial DNA content: a novel biomarker associated with prognosis and therapeutic outcome in colorectal cancer. Carcinogenesis https://doi.org/10.1093/carcin/bgv042 (2015).
Acknowledgements
This research was funded by the grant AIRC (IG 23201) for the project “Chemotherapy Effect on Cell Energy Metabolism and Endoplasmic Reticulum Redox Control”, by “Ricerca Corrente” program of the Italian Ministry of Health, and by H2UB project (code T4-AN-10) of the Italian Ministry of Health. The funding sources had no role in gathering, analyzing, or interpreting the data.
Funding
This research was funded by the grant AIRC (IG 23201) for the project “Chemotherapy Effect on Cell Energy Metabolism and Endoplasmic Reticulum Redox Control”, by “Ricerca Corrente” program of the Italian Ministry of Health, and by H2UB project (code T4-AN-10) of the Italian Ministry of Health. The funding sources had no role in gathering, analyzing, or interpreting the data.
Author information
Authors and Affiliations
Contributions
Designing the study: GS, MB and CM. Clinical identification of patients and their accrual: CG and EA; analysis of PET images for patient classification GS, FL, MB and CM; selection and analysis of metabolism of mononucleated cells populating the peripheral blood: VC, S Carta, KP, MG, SL, FV, S Chiesa and AMO; analysis of mitochondrial DNA copy number and its sequencing: S Coco, DC, MC and SM. All authors participated to the interpretation of the data and the preparation of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study was conducted in accordance with the Declaration of Helsinki, and the protocol was approved by the Ethical Committee of Regione Liguria (50/20-DB id 10306). All subjects gave their informed consent for inclusion before they participated in the study.
Consent for publication
All subjects gave their informed consent for data publication.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Sambuceti, G., Coco, S., Cossu, V. et al. Anthracyclines effect on mitochondrial function and biogenesis in normal blood cells of hodgkin lymphoma patients. Sci Rep 16, 2246 (2026). https://doi.org/10.1038/s41598-025-31980-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-31980-5
