
Abstract
The electrochemical conversion of biomass-derived 5-hydroxymethylfurfural (HMF) represents a promising, economically viable, and environmentally sustainable approach for producing value-added chemicals using renewable energy and in situ hydrogen generated through water electrolysis. However, the electrochemical hydrogenation (ECH) of HMF remains challenging due to the inherently low catalytic activity and selectivity of the electrodes, compounded by competition with the kinetically favored hydrogen evolution reaction (HER) in aqueous electrolytes. In this work, we demonstrate that CuxNi100−x heteroatomic thin films, fabricated via direct current (DC) magnetron co-sputtering, achieve a more than one order of magnitude increase in the HMF to 2,5-Bis-hydroxymethylfuran (BHMF) conversion rate, with nearly 50% faradic efficiency (FE) for BHMF, when compared to pure Cu and Ni electrodes (~ 10% BHMF FE). Our results suggest that the synergistic interaction between Cu and Ni creates an optimal catalytic environment for both HMF and adsorbed hydrogen (Hads) species, thereby enhancing BHMF formation through the ECH pathway.
Similar content being viewed by others
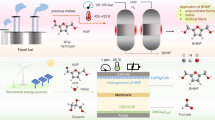

Introduction
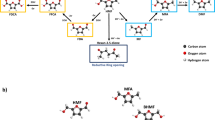
The integration of biomass into the energy and chemical sectors presents a promising strategy to mitigate the depletion of fossil resources while advancing toward a more sustainable and resilient future1,2,3,4,5,6. A key step in realizing this transition lies in identifying versatile platform molecules that can efficiently convert biomass-derived carbohydrates into value-added chemicals and fuels. In the biorefinery roadmap, 5-hydroxymethylfurfural (HMF, C6H6O3) obtained through the dehydration of fructose or glucose acts as a crucial intermediary between biomass raw materials and the production of valuable commodities, including alternative fuels, chemicals, and monomers2,7,8. The bio-based polymer building blocks and fuel additives such as 2,5-Bis-hydroxymethylfuran (BHMF), 2,5-dimethylfuran (DMF), 2,5-dimethyltetrahydrofuran (DMTHF), and others can further be synthesized via selective reduction of HMF molecules8,9,10,11,12,13,14,15,16,17. The HMF reduction reactions can be performed via chemical catalytic, thermocatalytic, enzymatic catalytic, hydrogen transfer routes, electro/photoreduction, and other processes7,18,19,20,21. Among these, the electrochemical hydrogenation (ECH) process is an attractive approach due to operational conditions (e.g., ambient temperature and pressure), eliminating H2 requirements from external resources, tunability based on intrinsic properties of used electrodes and electrolytes, and renewable energy resources and water can also be utilized to access required electrons and H+ for HMFRR respectively20,21,22. The electrochemical reduction of HMF can proceed through hydrogenation, where hydrogen atoms are selectively added to form alcohols without breaking the C−O bonds; through hydrogenolysis, which involves hydrogen-induced cleavage of carbonyl and ether bonds to produce CHx fragments; or through a combination of both pathways, leading to various ECH products (Fig. 1)23,24,25. For example, HMF can be reduced to BHMF and 5-methylfurfural (5MF) via the hydrogenation and hydrogenolysis pathways respectively, while compounds like 5-methylfurfural alcohol (MFA ), DMF are typically formed through the combination of electrochemical hydrogenation and hydrogenolysis pathways. (Fig. 1). However, the selectivity of the product and the competition with the more favourable hydrogen evolution reaction (HER), due to its low kinetic barriers and thermodynamic potentials, make the ECH process more challenging26. Therefore, designing an effective electrocatalytic system capable of selectively converting HMF into the desired products is essential.
Many electrocatalysts such as metal catalysts (e.g., Fe, Ni, Ag, Co, Zn, Sn, Sb, Au, and others)12,25,27, single atoms alloy28, transition-metal nanostructures29,30, oxide derived metal catalysts9, and bimetallic/heteroatomic catalysts have been studied for HMF electrochemical hydrogenation reaction in search of higher selectivity and production rate. Among these, heteroatomic catalysts, distinguished by the intimate co-localization of two distinct atomic species within a shared matrix, have attracted considerable scientific interest. Heteroatomic catalysts demonstrate a distinct bimetallic synergistic effect, which has been proven to greatly enhance the efficiency of HER31,32,33, oxygen evolution reaction (OER)34, hydrogenolysis35, and other vital electrochemical processes, including CO2 electrochemical reduction, glucose oxidation, and nitrate reduction reactions36,37,38. For example, it has been reported that introducing single-atom Ru into Cu significantly promotes HMF electrochemical reduction by accelerating the dissociation rate of water molecules to generate H* species, thereby enhancing the hydrogen-mediated HMF reduction reaction28. In general, the intrinsic properties of individual atoms play a pivotal role in defining the overall catalytic behaviour of the catalyst in the targeted reaction, and an efficient catalytic system for HMF can be designed by preparing a heteroatomic structure where atoms can create favourable binding sites for adsorbed hydrogen (Hads) and HMF molecules respectively5.

Electrochemical reduction pathways of 5-hydroxymethylfurfural (HMF): hydrogenation, hydrogenolysis, and corresponding products.
Herein, we present CuxNi100−x heteroatomic thin films catalysts prepared via DC magnetron sputtering technique by co-sputtering Cu and Ni atoms on degenerately doped p-type Si < 100 > substrates. We hypothesis that Cu and Ni can provide desired catalytic active sites for HMF and Hads molecules and promote HMF hydrogenation reaction via synergistic effect. The novelty of the present work lies in elucidating the role of CuxNi100–x heteroatomic nanometer-scale films as efficient electrocatalysts for the electrochemical hydrogenation of HMF. Interestingly, with minimal content of Ni (1.7%at), Cu98.3Ni1.7 catalyst significantly higher (~ 10 times) HMF conversion efficiency with ~ 40% Faradic Efficiency (FE) in comparison to the single atomic Cu and Ni catalysts (< 10% BHMF FE). The results indicate that Ni atoms enhance the surface coverage of Hads atoms which can migrate or directly participate in the reduction of the carbonyl group of HMF preferably interacting with nearby Cu atoms to produce BHMF via an ECH mechanism. In brief, HMF-derived compounds contribute to sustainable and circular chemical processes by enabling the valorization of lignocellulosic biomass, offering an eco-friendly alternative to petroleum-based feedstocks8,39,40,41. Similarly, CuxNi100−x-based catalysts benefit from the natural abundance and low cost of copper and nickel compared to noble metals, resulting in a reduced environmental footprint in both material sourcing and synthesis42. These factors highlight the potential of CuxNi100−x systems for scalable and sustainable biomass valorization applications.
Results
Catalysts morphological and structural analysis
Thin film (50–150 nm) of single atomic Ni and CuxNi100−x heteroatomic thin films were prepared via DC magnetron sputtering on cleaned Si substrate (degenerately doped p-type Si substrate) at ambient temperature and low pressure (see the method section) (Fig. 2, Fig. S1)38. Pure copper (99.99% Cu) foil with micrometer thickness was used to compare the results with CuxNi100−x and Ni catalysts. Here, it should be noted that pure Cu has featureless morphology except for some physical scratches due to physical polishing to remove oxide-based impurities followed by chemical cleaning to eliminate physically adsorbed impurities prior to use as catalysts (Fig. S2). The SEM image of sputtered Ni thin film evidenced continuous film formation of densely packed grain (< 50 nm) (Fig. 2B). The co-sputtered CuxNi100−x heteroatomic thin films consists similar size of grains but forming islands separated via random line defects (Fig. 2C,D). The EDS analysis suggested that by tuning of power of Cu and Ni sputtering targets, we were able to obtain different Cu and Ni atomic compositions in the films (Table S1).

Physical characterization of the grown film: (A) Thickness profile of sputter-deposited thin films. SEM images of sputter deposited (B) Ni, (C) Cu64Ni36, (D) Cu88Ni12, and (E) Cu98.3Ni1.7 thin films. Scale bar on SEM images is 250 nm. (F) XRD patterns of Cu foil and sputter deposited thin films.
The XRD measurements of the as-prepared electrodes were performed to analyze the crystal structure of fabricated electrodes (Fig. 2F). The XRD characterization peaks observed at 44.7 and 50.48 degrees for sputtered Ni and pristine Cu samples confirmed the presence of Ni (111) and Cu (200) crystals respectively. In the case of Cu, one more diffraction pattern at 43.4 associated to Cu (111) crystal structures was also observed. We have not observed Ni diffraction peak for the CuxNi100−x heteroatomic samples. This might be due to alloy formation and low amount of crystallized Ni structures in CuxNi100−x heteroatomic thin film. However, CuxNi100−x sputtered samples have Cu (111) phase dominated over Cu (200) in the case of Cu64Ni36, and Cu88Ni12. The Cu-rich CuxNi100−x exhibited a similar XRD pattern alike pure Cu except for a broader peak probably due to Cu and Ni interatomic interaction (Fig. 2F, Table S2).
The prepared electrodes were further characterized by using X-ray photoelectron spectroscopy (XPS) for understanding the elemental states of Cu and Ni atoms on the surface of the thin film (Fig. 3, Figs. S3–S7). In case of pristine Cu film, the Cu 2p spectra consist of two specific peaks associated to Cu 2p½ and Cu 2p3/2 located at 952.2 eV and 932.5 eV with peak split spin–orbit components of 19.7 eV confirming presence of metallic Cu atoms (Fig. 3A)43,44. For Cu 2p3/2, a shift from 932.3 eV for pristine Cu film to 932.5 eV, 932.6 eV, and 932.5 eV has been observed for Cu98.3Ni1.7, Cu88Ni12, and Cu64Ni36 respectively (Fig. 3A,D). We have also noticed that intensity of Cu2+ peak increases in presence of Ni atoms. Similarly, Ni 2p spectra for pristine Ni film evidence the presence of typical Ni 2p½ (at 869.9 eV) and Ni 2p3/2 peaks (at 852.5 eV) with peak split spin–orbit components of 17.5 eV, confirming the formation of metallic Ni (Fig. 3E)43,45,46. Alike for Cu 2p3/2 peaks, we also observed a small shift from 852.5 eV for pristine Ni film to 852.6 eV, 852.7 eV, and 852.7 eV for Cu98.3Ni1.7, Cu88Ni12, and Cu64Ni36 respectively (Fig. 3E,H). The pristine Ni has also higher Ni0 peak and, which decreases gradually for Cu98.3Ni1.7, Cu88Ni12, and Cu64Ni36 respectively. Here it should be noted that the positive shift in the Cu and Ni peaks indicate the existence of strong electronic effect due to interaction of Cu and Ni atoms in all CuxNi100−x heteroatomic thin films44,47. The modified electronic structure has been found in accelerating CO2 reduction reaction in comparison to the competing HER44. Thus, we hypothesis that it will enhance the HMF reduction reaction positively.

High-resolution XPS spectra of CuxNi100−x catalysts: Deconvoluted Cu 2p spectra of (A) pure Cu, (B) Cu98.3Ni1.7, (C) Cu88Ni12, and (D) Cu64Ni36; and deconvoluted Ni 2p spectra of (E) pure Ni, (F) Cu98.3Ni1.7, (D) Cu88Ni12, and (H) Cu64Ni36. The spectra reveal the chemical states of Cu and Ni in the heteroatomic catalysts, indicating compositional and electronic structure changes with varying Ni content.
HMF electrochemical hydrogenation study
The electrochemical hydrogenation characteristics of Cu, Ni, and different compositions of Cu100 − xNix were initially investigated by performing linear sweep voltammetry (LSV) with 20 mM HMF and without HMF buffer solution (borate buffer solution, pH 9.2) using two compartments separated by frits three electrodes electrochemical cell (Fig. S8). Initially, N2 was purged into the electrolyte (without HMF) and potential was scanned from 0 to − 0.8 V vs. RHE followed by similar experiments in 20 mM HMF buffer solution used as electrolyte.
Pristine Ni exhibits very low HER and HMF ECH rates as 4 and 5 mA cm− 2 current densities were recorded at − 0.8 V vs. RHE applied potential (Fig. 4). Surprisingly Cu exhibited relatively higher HER activity under-examined experimental conditions, and a 16 mA cm− 2 current density was recorded at a similar operating potential (i.e., − 0.8 V vs. RHE). The Cu100 − xNix film consisting of the lowest Ni amount has shown the highest current density for both HER and ECH reactions. Further increase in Ni content results in a decrease in the electrochemical current densities as shown in Fig. 4. Figure 4F indicated the ~ 20 and 1.5 times higher reaction rates of Cu98.3Ni1.7, catalysts in comparison to the pure Ni and Cu film respectively. The results indicate that the insertion of Ni atoms with densely packed Cu film boosts the ECH activity for the HMF electrochemical conversion process.

Linear sweep voltammetry (LSV) curves of prepared catalysts: (A) Ni, (B) Cu64Ni36, (C) Cu88Ni12, (D) Cu98.3Ni1.7 thin films, and (E) Cu foil with (solid) and without (dotted) HMF. (F) Current density at − 0.8 V vs. RHE with and without HMF of all samples.
To understand HMF hydrogenation’s selectivity, we performed a chronoamperometry (CA) experiment at different potentials (− 0.6 V vs. RHE, − 0.7 V vs. RHE, − 0.8 V vs. RHE, and − 0.9 V vs. RHE) for Cu, Ni, Cu64Ni36, Cu88Ni12, and Cu98.3Ni1.7, in 20 mM HMF solution for 30 min (Fig. 5A, Fig. S9–S13, Table S3). The collected catholyte solutions were analyzed using 1 H NMR spectroscopy minutes (Fig. 5B, Figs. S9–S13). Figure 5A represent the CA graphs showing steady performance over 30 min and higher current at higher applied negative potentials. The 1 H NMR spectra of HMF and BHMF are distinct due to their chemical structure. The NMR spectra of 20 mM HMF/solution indicate the presence of three HMF characteristics peaks at 6.3 (labelled as C),7.4 (labelled as B) and 9.3 ppm (labelled as A), respectively. Similarly, two peaks at ~ 4.0 (labelled as B2) and 6.0 ppm (labelled as A2) were observed for the BHMF solution. In the case of ECH of HMF on all studied samples we have observed a minimal BHMF characteristics A2 peak (at 6 ppm) for the solution collected after performing an electrochemical reaction at − 0.6 V vs. RHE potential. The results indicate the formation of BHMF via HMF electrochemical hydrogenation at this negative potential. The intensity of the BHMF characteristic peak increased with respect to the negatively increased applied potential (Fig. 5B). This is the case for all studied samples.

Electrochemical performance of Cu98.3Ni1.7 catalysts. (A) Choronoamperometry (CA) experiments at − 0.6, − 0.7, − 0.8 and − 0.9 V vs. RHE. All CA experimental were conducted in a 20 mM HMF solution at different potentials for 30 min in presence of continueous N2 bubbling as carrier gas for in-situ gasseus products analysis. (B) corresponding NMR analysis of the catholyte collected at different potentials, and (C) Ratio of A2 (associated to BHMF) to A (associated to HMF) NMR peaks for Cu98.3Ni1.7 sample.
We also plotted the ratio of A and A2 peaks associated to HMF and BHMF respectively. In the case of Cu98.3Ni1.7, we observed a linear relationship between applied potential and intensity of the A2/A ratio (Fig. 5C and Fig. S15). The A2/A ratio for collected catholyte after 30 min CA experiments using pristine Cu and Ni remains very low at − 0.6 and − 0.7 V vs. RHE potentials indicating low conversion rates due to consumption of low charges (− 0.84 C cm− 2, − 1.71 C cm− 2 for Cu, respectivly) or higher side hydrogen evolution reaction (HER) (Table S3). At higher potentials, Cu and Ni show improved BHMF formation rate as A2/A peak ratio reached 0.1 (at − 0.8 V vs. RHE) and 0.08 (at − 0.9 V vs. RHE) for Cu and Ni catalysts respectively. Interestingly, all prepared CuxNi100−x heteroatomic catalysts exhibit up to four times higher A2/A ratio indicating a higher HMF conversion rate (SI file).
Considering hydrogen evolution reaction (HER) as a competitive reaction, we also performed gaseous products analysis during CA experiments using online gas chromatography (GC) (see method section) and calculated the hydrogen faradic efficiency (FE) for examined catalysts (SI file)9,33. First, alike other pristine noble metal catalysts (e.g., Pt and Au)23, hydrogen was identified as a single gaseous product indicating absence of any gaseous side products (Fig. 6A). We found that the Ni and Cu catalysts produced mainly hydrogen (> 90% FE) and only ~ 10% of charges consumed for BHMF production (Fig. 6A). Interestinlgy, a significant increase in the BHMF FEs for Cu64Ni36, Cu88Ni12, and Cu98.3Ni1.7 catalysts have been recorded, and it confirms that CuxNi100−x heteroatomic structure preferably suppress the HER and promote HMF ECH reaction (Fig. 6). Moreover, the BHMF partial current density is almost 10 times higher in comparison to the pristine Cu and Ni based catalysts (Fig. 6B,C). When the BHMF partial current density is normalized with respect to the pristine Ni, Cu64Ni36, Cu88Ni12, and Cu98.3Ni1.7, show approximately ~ 9, ~10 and ~ 11 folds higher BHMF partial current densities respectively (Fig. 6C). Here it should be noted that CuxNi100−x heteroatomic catalysts exhibit relatively higher BHMF FE with ~ 100% BHMF selectivity as HMF liquid phase product when compared with pristine noble metal catalysts (e.g., Pt and Au)23 AgPb heteroatomic film48, nanostructured Cu catalysts24, and Ga doped Ga-doped Ag-Cu nanomaterials catalysts49 (Fig. 6D).

Product ananlysis of CuxNi100−x catalysts: Product analysis of CuxNi100−x catalysts: (A) FE for BHMF and H2 production at − 0.9 V vs. RHE for all CuxNi100−x catalysts. (B) Absolute partial current density for BHMF and H2 formation at − 0.9 V vs. RHE. (C) BHMF and H2 partial current density normalized to that of the pure Ni catalyst at − 0.9 V vs. RHE. (D) Comparison of BHMF FE for CuxNi100−x catalysts with values reported in the literature (Refs.23,24,48,49).
Discussion
The HMF hydrogenation reaction depends on the adsorption of the reactant and electron transfer from the surface of the catalyst. Initially, HMF molecules adsorb on the surface of the catalyst based on the available active sites and build organic adsorbed intermediate on the catalyst’s surface. In the second step, the hydrogenation step may proceed either via proton transfer from the solution through proton-coupled electron transfer (PCET) or through the direct addition of adsorbed hydrogen (Hads) to the intermediate by hydrogen atom transfer (HAT). In alkaline/neutral environments, the limited proton availability constrains direct PCET, which usually results in HMF dimerization. The Hads formed during the Volmer step of the hydrogen evolution reaction support the hydrogenation of the aldehyde group. Finally, the desired BHMF is released via a HAT mechanism (Fig. 7). In the case of Cu and Ni-based catalysts, the higher Tafel slope for HER represents that the Volmer step is more dominant for Ni in comparison to Cu under similar conditions indicating favourable sites for hydrogen adsorption (Fig. S17–S21 and Table S4). Here it should be noted that in CuNi alloys, the incorporation of Ni, which binds hydrogen strongly in comparison to Cu, modifies the intrinsic hydrogen-adsorption landscape of the surface. Despite the higher adsorption energy of H on Ni (2.89 eV) compared with Cu (2.39 eV), previous studies have shown that H adatoms nonetheless can migrate from Ni to neighboring Cu sites. This confirms that spillover occurs even against the expected thermodynamic preference for Ni–H bonding50,51,52. Based on these observation similar HMF hydrogenation mechanism with other Cu-based heteroatomic Ru1Cu catalysts, where water molecules were activated over single-atom Ru with concomitant electron transfer and dissociation into Hads species, then two Hads species react with the carbonyl of HMF to produce BHMF via an ECH mechanism has been proposed28.

Proposed electrochemical reduction mechanism of HMF over the CuxNi100−x heteroatomic catalyst.
Conclusion
The electroreduction of 5-hydroxymethylfurfural (HMF) to 2,5-bis(hydroxymethyl)furan (BHMF) was effectively realized using CuxNi100−x co-sputtered thin films. The CuxNi100−x catalysts demonstrated excellent electrochemical performance, including high HMF conversion rate, and superior BHMF Faradaic efficiency. Based on earlier study, we predict that the enhanced catalytic activity of the CuxNi100−x electrodes can be attributed to the synergistic interplay between Cu and Ni: Cu exhibits a strong adsorption affinity for HMF, facilitating its activation, while Ni plays a crucial role in driving the selective reduction of HMF to BHMF by facilitating hydrogen adsorption. However further study is highly recommended to validate the proposed mechanism. This cooperative interaction between Cu and Ni within the alloy matrix is pivotal in optimizing the electroreduction process, resulting in a highly efficient and selective transformation of HMF to BHMF.
Materials and methods
Chemicals
Acetone, ethanol, and buffer oxide etchant (BOE) were purchased from VWR chemicals. Cu, Ni metal targets, with purity of 99.95% and a diameter of 2 inches, were obtained from Kurt J. Leskar company. Sodium hydroxide (NaOH, pellets), Boric acid (H3BO3, ≥ 99.5%), 5-(Hydroxymethyl)furfural, (C6H6O3, ≥ 99%) were purchased from Millipore Sigma.
Preparation of samples
The Cu-Ni samples were prepared via DC magnetron sputtering technique as discussed in our previous report38. In brief, Cu–Ni alloy thin films with compositions of Cu₆₄Ni₃₆, Cu₈₈Ni₁₂, and Cu₉₈.₃Ni₁.₇ were deposited on degenerately doped p-type Si(0.001–0.005Ohm-cm ) substrates using DC magnetron sputtering at room temperature. The substrate holder was rotated at a constant speed of 40 rpm during deposition to ensure uniform film growth. The sputtering targets were arranged in a confocal configuration, with the substrates positioned approximately 25 cm from the target surface. Prior to deposition, a pre-sputtering step was performed to stabilize the sputtering power and plasma conditions, thereby ensuring reproducibility and minimizing contamination. The deposition system is equipped with a radiant substrate heating capability of up to 850 °C and a 50 W RF bias, enabling controlled film adhesion, density, and crystallinity. The p-type Si substrates were cleaned by sequential sonication for 5 min in acetone, ethanol, and deionized water respectively. Afterword, the substrates were immersed in buffered oxide etchant (BOE) for 2 min, rinsed with DI water, and dried under nitrogen. Deposition was performed using commercially available 2-inch diameter, 99.95% pure Cu and Ni targets. The chamber was pumped down to ~ 10−8 Torr before sputtering. Films were grown for 1 h at 100 W DC power in a 10 mTorr ultrahigh-purity Ar (99.999%) atmosphere. During deposition, rotating degenerately doped p-type Si (0.001–0.005 Ohm-cm ) substrate was placed 25 cm away from the target to achieve uniform deposition.
Characterisation
The samples were characterized using XRD, SEM, and XPS to assess their physical structure and surface chemistry9,33,38. Intially the thickness and composition of the Cu–Ni films were determined from EDS spectra. Structural characterization was performed using X-ray diffraction (XRD), while surface morphology was examined by scanning electron microscopy (SEM, JEOL JSM-6060LV) equipped with a Thermo Scientific UltraDry EDS detector for chemical analysis. XRD measurements were carried out with a Rigaku Miniflex diffractometer. Surface chemical composition was further analyzed by X-ray photoelectron spectroscopy (XPS, SPECS) using a 300 W Mg Kα source (hν = 1253.6 eV). Wide-scan spectra (0–1100 eV, step size 0.5 eV) were recorded to identify elemental constituents, followed by high-resolution scans of C 1s, O 1s, Cu 2p, and Ni 2p regions at 0.1 eV step size. Spectral deconvolution was performed using CasaXPS software, with all binding energies calibrated against the adventitious carbon C 1s peak at 285.0 eV.
Electrochemical characterisation
All electrochemical measurements were carried out in a conventional two-compartment, three-electrode H-type cell connected to a Biologic SP-300 potentiostat. The electrolyte consisted of a borate buffer solution (pH 9.2), prepared by dissolving boric acid and adjusting the pH with sodium hydroxide in deionized water. The working electrodes used in this study included the prepared Cu, Cu64Ni36, Cu88Ni12, Cu98.3Ni1.7, and Ni samples. A platinum mesh served as the counter electrode, while an Ag/AgCl electrode (saturated in KCl) was employed as the reference. The cathodic and anodic chambers were separated by a glass frit with a pore size of 20 μm. The hydrogenation kinetics of HMF were examined usingLSV, where the potential was scanned from 0.0 to − 0.8 V versus RHE at a rate of 20 mV s−1. All potentials were converted to the RHE using the Nernst equation, Potential vs. RHE = Applied potential versus Ag/AgCl (saturated KCl) + 0.197 V + 0.0592*pH. The CA experiments were conducted in a 20 mM HMF solution (catholyte solution volume − 10 ml, anolyte solution volume − 10 ml, magnetic steering at 500 rpm) prepared in borate buffer (pH 9.2) at constant potentials ranging from 0.6 to 0.9 V vs. RHE for 30 min. During CA experiments, continuous N2 bubbling was used as carrier gas for online gaseous products analysis. The gaseous products were analyzed using an online gas chromatograph (GC), while the collected catholyte solutions were examined by nuclear magnetic resonance (NMR) spectroscopy as discussed in our previous reports9,53.
Gas chromatography and NMR characterization
The gas-phase products were quantified using a gas chromatograph (GC, SRI Instruments, Model 8610 C) equipped with both a thermal conductivity detector (TCD) and a flame ionization detector (FID). For each measurement, 1 mL of headspace gas was sampled and injected into the GC at an exhaust flow rate of 10 sccm, following 5 and 20 min of constant potential electrolysis. Faradaic efficiencies were calculated by comparing the charge corresponding to the formation of each detected product—derived from its measured concentration—with the total charge recorded by the potentiostat during electrolysis32,37. Liquid-phase products were analyzed by nuclear magnetic resonance (NMR) spectroscopy using a Bruker 400 MHz solution-state instrument equipped with z-gradients and a broadband indirect detection probe. All NMR measurements were performed in the COSMIC Laboratory at Old Dominion University with 10% D₂O as the solvent. The data were collected at room temperature, with a relaxation delay of 1.5 s, and with 256 scans. One-dimensional 1 H spectra were acquired using a water suppression pulse sequence, Pew5shapepr (a modified Perfect Echo WATERGATE W5 sequence) with a train of water selective shaped pulses applied during relaxation delay.
Data availability
The data sets used and/or analyzed during the current study available from the corresponding author on reasonable request.
References
Biddy, M. J., Scarlata, C. & Kinchin, C. in Chemicals from Biomass: A Market Assessment of Bioproducts with Near-Term Potential. 131 (eds Size) (ED, United States, 2016).
Hou, Q. et al. Biorefinery roadmap based on catalytic production and upgrading 5-hydroxymethylfurfural. Green Chem. 23, 119–231. https://doi.org/10.1039/D0GC02770G (2021).
Klein, B. C. et al. Economics and global warming potential of a commercial-scale delignifying biorefinery based on co-solvent enhanced lignocellulosic fractionation to produce alcohols, sustainable aviation fuels, and co-products from biomass. Energy Environ. Sci. 17, 1202–1215. https://doi.org/10.1039/D3EE02532B (2024).
Corma, A., Iborra, S. & Velty, A. Chemical routes for the transformation of biomass into chemicals. Chem. Rev. 107, 2411–2502. https://doi.org/10.1021/cr050989d (2007).
Liu, C., Chen, F., Zhao, B. H., Wu, Y. & Zhang, B. Electrochemical hydrogenation and oxidation of organic species involving water. Nat. Rev. Chem. 8, 277–293. https://doi.org/10.1038/s41570-024-00589-z (2024).
Lam, C. H. et al. Minireview on bio-oil upgrading via electrocatalytic hydrogenation: connecting biofuel production with renewable power. Energy Fuels. 34, 7915–7928. https://doi.org/10.1021/acs.energyfuels.0c01380 (2020).
Xu, C., Paone, E., Rodríguez-Padrón, D., Luque, R. & Mauriello, F. Recent catalytic routes for the preparation and the upgrading of biomass derived furfural and 5-hydroxymethylfurfural. Chem. Soc. Rev. 49, 4273–4306. https://doi.org/10.1039/D0CS00041H (2020).
van Putten, R. J. et al. Hydroxymethylfurfural, A versatile platform chemical made from renewable resources. Chem. Rev. 113, 1499–1597. https://doi.org/10.1021/cr300182k (2013).
Muchharla, B. et al. Reduced metal nanocatalysts for selective electrochemical hydrogenation of biomass-derived 5-(hydroxymethyl)furfural to 2,5-bis(hydroxymethyl)furan in ambient conditions. 11, https://doi.org/10.3389/fchem.2023.1200469 (2023).
Luo, J. et al. Mechanisms for high selectivity in the hydrodeoxygenation of 5-Hydroxymethylfurfural over PtCo nanocrystals. ACS Catal. 6, 4095–4104. https://doi.org/10.1021/acscatal.6b00750 (2016).
Thananatthanachon, T. & Rauchfuss, T. B. Efficient production of the liquid fuel 2,5-Dimethylfuran from Fructose using formic acid as a reagent. 49, 6616–6618. https://doi.org/10.1002/anie.201002267 (2010).
Nilges, P. & Schröder, U. Electrochemistry for biofuel generation: production of furans by electrocatalytic hydrogenation of furfurals. Energy Environ. Sci. 6, 2925–2931. https://doi.org/10.1039/C3EE41857J (2013).
Hauke, P., Merzdorf, T., Klingenhof, M. & Strasser, P. Hydrogenation versus hydrogenolysis during alkaline electrochemical valorization of 5-hydroxymethylfurfural over oxide-derived Cu-bimetallics. Nat. Commun. 14, 4708. https://doi.org/10.1038/s41467-023-40463-y (2023).
Morales, M. V., Conesa, J. M., Galvin, A. J., Guerrero-Ruiz, A. & Rodríguez-Ramos, I. Selective hydrogenation reactions of 5-hydroxymethylfurfural over Cu and Ni catalysts in water: effect of Cu and Ni combination and the reagent purity. Catal. Today. 423, 114021. https://doi.org/10.1016/j.cattod.2023.01.028 (2023).
Ye, F. et al. The role of oxygen-vacancy in bifunctional indium oxyhydroxide catalysts for electrochemical coupling of biomass valorization with CO2 conversion. Nat. Commun. 14, 2040. https://doi.org/10.1038/s41467-023-37679-3 (2023).
Das, S., Cibin, G. & Walton, R. I. Selective oxidation of biomass-derived 5-hydroxymethylfurfural catalyzed by an iron-grafted metal–organic framework with a sustainably sourced ligand. ACS Sustain. Chem. Eng. 12, 5575–5585. https://doi.org/10.1021/acssuschemeng.3c08564 (2024).
Bender, M. T., Yuan, X., Goetz, M. K. & Choi, K. S. Electrochemical hydrogenation, hydrogenolysis, and dehydrogenation for reductive and oxidative biomass upgrading using 5-hydroxymethylfurfural as a model system. ACS Catal. 12, 12349–12368. https://doi.org/10.1021/acscatal.2c03606 (2022).
Chen, S., Wojcieszak, R., Dumeignil, F., Marceau, E. & Royer, S. How catalysts and experimental conditions determine the selective hydroconversion of furfural and 5-Hydroxymethylfurfural. Chem. Rev. 118, 11023–11117. https://doi.org/10.1021/acs.chemrev.8b00134 (2018).
Qin, Y. Z., Li, Y. M., Zong, M. H., Wu, H. & Li, N. Enzyme-catalyzed selective oxidation of 5-hydroxymethylfurfural (HMF) and separation of HMF and 2,5-diformylfuran using deep eutectic solvents. Green Chem. 17, 3718–3722. https://doi.org/10.1039/C5GC00788G (2015).
Cha, H. G. & Choi, K. S. Combined biomass valorization and hydrogen production in a photoelectrochemical cell. Nat. Chem. 7, 328–333. https://doi.org/10.1038/nchem.2194 (2015).
Obata, K. et al. Solar-driven upgrading of biomass by coupled hydrogenation using in situ (photo)electrochemically generated H2. Nat. Commun. 14, 6017. https://doi.org/10.1038/s41467-023-41742-4 (2023).
de Luna, G. S. et al. Insights into the electrochemical reduction of 5-Hydroxymethylfurfural at high current densities. ChemSusChem 15, e202102504. https://doi.org/10.1002/cssc.202102504 (2022).
Zhang, D. et al. Selective electrocatalytic hydrogenation of lignocellulose-derived 5-hydroxymethylfurfural with superior productivities. iScience 26 https://doi.org/10.1016/j.isci.2023.108003 (2023).
Yuan, X., Lee, K., Bender, M. T., Schmidt, J. R. & Choi, K. S. Mechanistic differences between electrochemical hydrogenation and hydrogenolysis of 5-Hydroxymethylfurfural and their pH dependence. ChemSusChem 15, e202200952. https://doi.org/10.1002/cssc.202200952 (2022).
Lee, D. et al. (ed, K.) The impact of 5-Hydroxymethylfurfural (HMF)-Metal interactions on the electrochemical reduction pathways of HMF on various metal electrodes. ChemSusChem 14 4563–4572 https://doi.org/10.1002/cssc.202101037 (2021).
May, A. S. & Biddinger, E. J. Strategies to control electrochemical hydrogenation and hydrogenolysis of furfural and minimize undesired side reactions. ACS Catal. 10, 3212–3221. https://doi.org/10.1021/acscatal.9b05531 (2020).
Kwon, Y., de Jong, E., Raoufmoghaddam, S. & Koper, M. T. M. Electrocatalytic hydrogenation of 5-Hydroxymethylfurfural in the absence and presence of glucose. 6, 1659–1667. https://doi.org/10.1002/cssc.201300443 (2013).
Ji, K. et al. Electrocatalytic hydrogenation of 5-Hydroxymethylfurfural promoted by a Ru1Cu Single-Atom alloy catalyst. 61, e202209849. https://doi.org/10.1002/anie.202209849 (2022).
Carroll, K. J. et al. Electrocatalytic hydrogenation of oxygenates using earth-abundant transition-metal nanoparticles under mild conditions. 9, 1904–1910. https://doi.org/10.1002/cssc.201600290 (2016).
Zhang, Z. et al. Operando generated copper-based catalyst enabling efficient electrosynthesis of 2,5‐bis(hydroxymethyl)furan. Fundam. Res. 3, 763–769. https://doi.org/10.1016/j.fmre.2022.01.016 (2023).
Chen, C. H. et al. Ruthenium-Based Single-Atom alloy with high electrocatalytic activity for hydrogen evolution. 9, 1803913. https://doi.org/10.1002/aenm.201803913 (2019).
Kumar, B. et al. Electrochemical CO2 conversion commercialization pathways: A concise review on experimental frontiers and technoeconomic analysis. Environ. Sci. Technol. Lett. 11, 1161–1174. https://doi.org/10.1021/acs.estlett.4c00564 (2024).
Malali, P. et al. Low platinum-loaded molybdenum co-catalyst for the hydrogen evolution reaction in alkaline and acidic media. Langmuir 38, 9526–9531. https://doi.org/10.1021/acs.langmuir.2c00902 (2022).
Hu, Y. et al. Single Ru atoms stabilized by hybrid amorphous/crystalline feconi layered double hydroxide for ultraefficient oxygen evolution. 11, 2002816. https://doi.org/10.1002/aenm.202002816 (2021).
Zhang, X. et al. Platinum–copper single atom alloy catalysts with high performance towards glycerol hydrogenolysis. Nat. Commun. 10, 5812. https://doi.org/10.1038/s41467-019-13685-2 (2019).
Zheng, T. et al. Copper-catalysed exclusive CO2 to pure formic acid conversion via single-atom alloying. Nat. Nanotechnol. 16, 1386–1393. https://doi.org/10.1038/s41565-021-00974-5 (2021).
Kumar, B. et al. New trends in the development of heterogeneous catalysts for electrochemical CO2 reduction. Catal. Today. 270, 19–30. https://doi.org/10.1016/j.cattod.2016.02.006 (2016).
Barbee, B. et al. Cu and Ni Co-sputtered heteroatomic thin film for enhanced nonenzymatic glucose detection. Sci. Rep. 12, 7507. https://doi.org/10.1038/s41598-022-11563-4 (2022).
He, Y., Deng, L., Lee, Y., Li, K. & Lee, J. M. A review on the critical role of H2 donor in the selective hydrogenation of 5-Hydroxymethylfurfural. ChemSusChem 15, e202200232. https://doi.org/10.1002/cssc.202200232 (2022).
Chen, C. et al. 5-Hydroxymethylfurfural and its downstream chemicals: A review of catalytic routes. Adv. Mater. 36, 2311464. https://doi.org/10.1002/adma.202311464 (2024).
Jiang, Z. et al. Chemical transformations of 5-hydroxymethylfurfural into highly added value products: present and future. Green Chem. 25, 871–892. https://doi.org/10.1039/D2GC03444A (2023).
Henckens, M. L. C. M. & Worrell, E. Reviewing the availability of copper and nickel for future generations. The balance between production growth, sustainability and recycling rates. J. Clean. Prod. 264, 121460. https://doi.org/10.1016/j.jclepro.2020.121460 (2020).
Li, Y. et al. Implanting Ni-O-VOx sites into Cu-doped Ni for low-overpotential alkaline hydrogen evolution. Nat. Commun. 11, 2720. https://doi.org/10.1038/s41467-020-16554-5 (2020).
Pan, W. et al. Cu–Ni alloy decorating N-doped carbon nanosheets toward high-performance electrocatalysis of mildly acidic CO2 reduction. Inorg. Chem. Front. 10, 2276–2284. https://doi.org/10.1039/D3QI00207A (2023).
Ratcliff, E. L. et al. Evidence for near-Surface NiOOH species in Solution-Processed NiOx selective interlayer materials: impact on energetics and the performance of polymer bulk heterojunction photovoltaics. Chem. Mater. 23, 4988–5000. https://doi.org/10.1021/cm202296p (2011).
Anastaziak, B. et al. Magnetic patterning of Co/Ni layered systems by plasma oxidation. Sci. Rep. 12, 22060. https://doi.org/10.1038/s41598-022-26604-1 (2022).
Naghash, A. R., Etsell, T. H. & Xu, S. XRD and XPS study of Cu – Ni interactions on reduced Copper –Nickel – Aluminum oxide solid solution catalysts. Chem. Mater. 18, 2480–2488. https://doi.org/10.1021/cm051910o (2006).
Luo, X., Xie, C., Zhao, Z., Shi, M. & Zheng, H. Optimization of electrochemical reduction of biomass derived 5-Hydroxymethylfurfural (HMF): A volcano plot and bimetallic catalysts. ChemSusChem 17, e202400723. https://doi.org/10.1002/cssc.202400723 (2024).
Tian, C. et al. Reduction of 5-Hydroxymethylfurfural to 2,5-Bis(hydroxymethyl)Furan at high current density using a Ga-Doped agcu:cationomer hybrid electrocatalyst. Adv. Mater. 36, 2312778. https://doi.org/10.1002/adma.202312778 (2024).
Yao, Y. & Goodman, D. W. Direct evidence of hydrogen spillover from Ni to Cu on Ni–Cu bimetallic catalysts. J. Mol. Catal. A: Chem. 383–384, 239–242. https://doi.org/10.1016/j.molcata.2013.12.013 (2014).
Wang, X. et al. Tuning the apparent hydrogen binding energy to achieve high-performance Ni-based hydrogen oxidation reaction catalyst. Nat. Commun. 15, 1137. https://doi.org/10.1038/s41467-024-45370-4 (2024).
Xiong, M., Gao, Z. & Qin, Y. Spillover in heterogeneous catalysis: new insights and opportunities. ACS Catal. 11, 3159–3172. https://doi.org/10.1021/acscatal.0c05567 (2021).
Muchharla, B. et al. Underlying substrate effect on electrochemical activity for hydrogen evolution reaction with Low-Platinum-Loaded catalysts. Small Struct. 5, 2300265. https://doi.org/10.1002/sstr.202300265 (2024).
Funding
This work was primarly supported by the National Science Foundation Eager program (558016), and in part by U.S. Department of Energy, National Nuclear Security Administration Minority Serving Institution Partnership Program (NNSA- MSIPP) (559063).
Author information
Authors and Affiliations
Contributions
M.D.: Data curation, formal analysis, methodology, validation, writing-original draft, writing-review and editing. B.M.: Data curation, formal analysis, methodology, validation, visualization, writing-original draft. L.V.R.: Data curation, methodology, K.K.: Formal analysis, data curation, review and editing, supervision. S.S.: Formal analysis, data curation, writing-review and editing, K.K.S.: Formal analysis, data curation, writing-review and editing. A.K.: Formal analysis, data curation, writing-review and editing. S.K.: Conceptualization, formal analysis, data curation, writing-review and editing, A.A.: Formal analysis, data curation, methodology, writing-review and editing. B.K.: Conceptualization, data curation, funding acquisition, investigation, methodology, resources, supervision, writing-review and editing.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Dikshit, M., Muchharla, B., Rivera, L.V. et al. Co-sputtered CuNi heteroatomic electrocatalyst for enhanced 5-hydroxymethylfurfural selective electrochemical conversion. Sci Rep 16, 2780 (2026). https://doi.org/10.1038/s41598-025-32621-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-32621-7
