
Abstract
Photobiomodulation (PBM) is a non-invasive therapeutic strategy that uses specific light wavelengths to stimulate cellular processes that promote tissue repair, reduce inflammation, and alleviate pain. In this study, we evaluated the efficacy of a high-intensity 980 nm near-infrared (NIR) pulsed laser in a murine model of ovalbumin (OVA)-induced allergic asthma. In OVA-sensitized mice, typical allergic features were noted, including airway wall thickening, significant peribronchial and perivascular inflammatory cell infiltration, and compressed alveolar spaces. NIR PBM treatment markedly attenuated these histological changes. At the molecular level, PBM significantly downregulated type 2 cytokine gene expression, including IL-4, IL-5 and IL-13, in the lung tissue. Flow cytometry analyses further revealed that PBM reduced the infiltration of pulmonary eosinophils and inflammatory monocytes. Transcriptomic profiling coupled with gene set enrichment analysis (GSEA) indicated that PBM suppressed NF-κB-mediated inflammatory signaling while modulating Th2-related responses. Collectively, our findings demonstrate that high-intensity 980 nm NIR PBM effectively mitigates key features of allergic asthma in a murine model, supporting its potential as a non-pharmacological, non-invasive adjunct therapy for asthma management.
Similar content being viewed by others

Introduction
Photobiomodulation (PBM) is a non-invasive therapy that uses specific wavelengths of light to stimulate cellular functions, and it has evolved significantly since its inception. In recent years, PBM has been widely applied in numerous fields, including the treatment of hair loss, dermatology, rehabilitation medicine, inflammation regulation, and wound healing1.
Early research focused on red light (600–700 nm), demonstrating efficacy in wound healing and anti-inflammatory applications through mechanisms such as cytochrome c oxidase activation, which enhances mitochondrial ATP production2,3.In addition to its impact on ATP synthesis, red light has been shown to increase the production of nitric oxide and reactive oxygen species. This modulation further influences downstream signaling pathways, promoting cellular proliferation, migration, and differentiation4,5.
More recent advancements have highlighted near-infrared (NIR, 700–1100 nm) wavelengths for their deeper tissue penetration (up to 5 cm), attributed to reduced scattering and absorption by chromophores like hemoglobin and melanin6,7. This capability positions NIR PBM as a promising tool for targeting subdermal tissues, expanding its therapeutic potential beyond superficial conditions8. Moreover, through the development of semi-conductor based high intensity laser coupled with pulsed laser, nowadays the High-Intensity Laser Therapy (HILT) (power > 500 mW) has been used for therapeutic protocol for musculoskeletal pain management9. Compared to Low-intensity laser therapy (power ≤ 500 mW), HILT could irradiated more intense laser beam which could penetrate deeper under the skin and the pulsed laser could reduce the thermal effect.
Asthma, a chronic respiratory disorder affecting over 262 million people globally (WHO, 2021), is characterized by airway inflammation, bronchoconstriction, and hyper-responsiveness. Both environmental factors and genetic predispositions contribute to its development10. Exposure to viruses, bacteria, dust mites, and certain foods can trigger asthma, while tobacco smoke and airborne particulates may exacerbate the condition11,12. Typically, the pathogenesis of asthma involves the uptake of allergens by dendritic cells, which then present these antigens to naïve T cells, prompting their differentiation into TH2 cells. TH2 cells release cytokines such as IL-4, IL-5, and IL-13 that stimulate B cells-derived IgE production, which in turn activates mast cells, leading to the release of mediators including leukotrienes, histamine, and prostaglandins. These mediators increase mucus secretion from epithelial cells, induce goblet cell hyperplasia, and exacerbate airway constriction, while also recruiting eosinophils as the predominant inflammatory cell type13,14,15. Moreover, allergens can directly stimulate epithelial cells to produce IL-25, IL-33, and TSLP, further promoting the recruitment of eosinophils via the activation of ILC2 cells that secrete IL-5 and IL-1316. Although asthma phenotypes can be heterogeneous, they are generally classified into TH2 high and non-TH2 variants, with the TH2 high phenotype marked by elevated eosinophils in lung tissue and bronchoalveolar lavage fluid, along with increased expression of type 2 cytokines such as IL-4, IL-5, and IL-1317,18,19,20,21. In murine models induced by allergens such as ovalbumin (OVA)22,23,24 house dust mite25, Alternaria26, or papain27, increased eosinophil counts have been consistently observed in lung tissues or bronchoalveolar lavage fluid. Furthermore, IL-4, IL-5, and IL-13 are well-established contributors to the progression of asthma28,29,30. IL-4 stimulates B cells to produce IgE31, IL-5 promotes tissue eosinophilia32, and IL-13 is responsible for airway hyper-responsiveness and goblet cell differentiation33.
Current treatments, including inhaled corticosteroids, β2-agonists, and leukotriene modifiers, often fail to achieve sustained control in 5–10% of severe cases34. Limitations such as corticosteroid resistance35 and systemic side effects (e.g., osteoporosis, immunosuppression) underscore the need for adjunctive therapies35,36. Nonsteroidal anti-inflammatory drugs (NSAIDs) further pose risks of gastrointestinal and cardiovascular complications, highlighting the demand for safer alternatives37.
PBM has emerged as an alternative asthma intervention owing to its ability to modulate inflammation with minimum adverse effects. Previous reports have demonstrated that PBM can attenuate bronchial inflammation in murine models, in part by suppressing the release of proinflammatory cytokines and reducing levels of inflammatory mediators such as leukotriene B4 (LTB4) and thromboxane B2 (TXB2)38. Moreover, clinical trials in chronic obstructive pulmonary disease show improved spirometry metrics39. Prior studies investigating PBM in murine models of allergic lung inflammation have shown that treatment with 660 nm red light can reduce eosinophil counts and lower levels of TH2 cytokines, including IL-4, IL-5, and IL-13, in bronchoalveolar lavage fluid40. In another study, researchers using 660 nm PBM in a chronic asthma murine model observed a decrease in TH2 cytokine levels in bronchoalveolar lavage fluid alongside an increase in IL-10 secreting regulatory T cells, suggesting that the downregulation of TH2 inflammation may be mediated by enhanced IL-10 production and Treg cell expansion41.
By leveraging advantage of deeper tissue penetration potential of NIR, NIR PBM might serve as alternative therapeutic approach to target airway smooth muscle and submucosal glands, potentially mitigating asthmatic related symptoms. In this study, we evaluated the efficacy of 980 nm NIR PBM in a murine model of OVA-induced allergic asthma. Our findings demonstrate that high-intensity 980 nm laser treatment significantly attenuates OVA-induced allergic responses by suppressing the recruitment of eosinophils and inflammatory monocytes, as well as reducing TH2-mediated responses. Collectively, these results support the mechanistic rationale and functional efficacy of NIR PBM, positioning it as a compelling, non-pharmacological, and non-invasive candidate for translational asthma research.
Results
NIR PBM attenuates OVA-induced allergy symptom
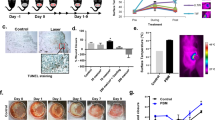
Initially, we evaluated the safety profile of the high-energy pulsed 980 nm laser. Mice received direct exposure to the upper chest area for five consecutive days (Fig. 1A). Detailed examination of the treated lung histology revealed no structural abnormalities or pathological changes (Fig. 1B). This confirms that the applied NIR PBM are safe and do not induce local tissue damage.

980 nm PBM alleviates OVA-induced allergic responses. (A) Schematic diagram of the OVA-allergy model and the PBM treatment regimen. (B) Representative H&E-stained lung sections from control and PBM-treated mice. (C) H&E-stained lung sections from control, OVA-allergic, and PBM-treated OVA-allergic mice following five consecutive days of aerosolized OVA sensitization. (D) Quantitative analysis of lung inflammation scores. Group sizes: control (n = 2), OVA (n = 4), and OVA-PBM (n = 4). (E) Periodic Acid-Schiff stain lung sections from control, OVA-allergic, and PBM-treated OVA-allergic mice following five consecutive days of aerosolized OVA sensitization. (F) Quantitative analysis of pathology scores from PAS staining. Group sizes: control (n = 2), OVA (n = 4), and OVA-PBM (n = 4). Statistical significance was determined using an unpaired, two-tailed Student’s t-test (*P < 0.05).
In the OVA-induced allergic asthma model, untreated mice exhibited classic histopathological features of allergic inflammation, including thickened airway walls, partial compression of alveolar spaces, and marked peribronchial as well as perivascular inflammatory cell infiltration (Fig. 1C). In contrast, mice treated with NIR PBM displayed attenuated histopathological changes (Fig. 1C). The airway architecture in the PBM-treated group was closer to normal, with reduced immune cell infiltration and thinner airway walls. The overall inflammation score was also lower in the PBM group (Fig. 1D). PAS staining revealed that the PBM group exhibited a significant reduction in goblet cell hyperplasia compared to the OVA group (Fig. 1E), along with a marked decrease in the overall pathology score (Fig. 1F). These improvements suggest that NIR PBM can effectively counteract the structural lung alterations induced by allergen exposure.
NIR PBM dampens type 2 cytokine expression
Th2 cytokines, such as IL-4, IL-5, and IL-13, are central mediators in the pathogenesis of allergic asthma. Our qPCR analysis demonstrated that OVA-allergic mice had a significant upregulation of these cytokine genes compared to controls. In animals receiving NIR PBM, we observed a marked downregulation of IL-4, IL-5, and IL-13 expression in the lung tissue (Fig. 2A–C). This reduction is critical, as IL-5 is known to drive eosinophil recruitment and survival, while IL-4 and IL-13 promote IgE class switching and mucus hypersecretion. Serum was collected from mice and assessed for total IgE levels (Fig. 2D). The results demonstrated a significant reduction in IgE concentration in the PBM group, which aligns with the concurrent downregulation of type 2 cytokines. The downregulation of these cytokines and IgE by NIR PBM indicates a direct modulation of the type 2 inflammatory response.

980 nm PBM reduces expression of classical type 2 cytokine genes. Gene expression levels of (A) IL-4, (B) IL-5, and (C) IL-13 were measured by real-time PCR in lung tissues from control, OVA-allergic, and PBM-treated OVA-allergic mice. Group sizes: control (n = 2), OVA (n = 3), and OVA-PBM (n = 3). (D) Serum total IgE level. Group sizes: control (n = 3), OVA (n = 3), and OVA-PBM (n = 3). Statistical significance was determined using an unpaired, two-tailed Student’s t-test (*P < 0.05).
Reduction of eosinophils and inflammatory monocytes infiltration
Flow cytometry analysis was performed at both baseline on Day 20 and Day 25 post-OVA aerosol sensitization to assess the pulmonary immune cell composition. Gating strategy was shown as in (Fig. 3A). Despite the fact that pulmonary eosionphils, neutrophils were increased in OVA allergy group at day 25, suggesting successful induction of acute allergic response, there is no difference between the OVA group and the PBM group, PBM treatment did not alter cellular composition (Fig. 3B). Similar pattern was seen in B, CD4 and CD8 T cells (Fig. 3C). The immune cell composition seems not reflective of the observed attenuation of histology and Th2 cytokine expression. We speculate that this might be due to the fact that at Day25, via the consecutive 5 days of OVA-aerosol sensitization, the immune cell infiltration is reaching the pletaeu and we might need to examine the immune cell composition in an early timepoint.

Characterization of pulmonary immune cell infiltration in the OVA-allergy model at day 25. (A) Flow cytometry gating strategy for identifying immune cell subsets in lung tissue. (B) Quantification of pulmonary eosinophils, alveolar macrophages, neutrophils, and Ly6hi monocytes. (C) Quantification of pulmonary B cells, CD4 T cells, and CD8 T cells. Group sizes: PBS day 25 (n = 3), OVA day 25 (n = 4), and OVA-PBM day 25 (n = 4). Statistical significance was determined using an unpaired, two-tailed Student’s t-test (*P < 0.05).
To this end, further flow cytometry analysis was performed at Days 23 post-OVA aerosol sensitization. We observed a significant increase of eosinophils, alveolar macrophages, neutrophils and inflammatory Ly6hi monocytes infiltration after OVA sensitization, and PBM treatment damped the infiltrated eosinophils and inflammatory Ly6Chi monocytes (Fig. 4A). The increase of B, CD4 and CD8 cells were less pronounced as that at Day25, with PBM seems to suppress lymphocytes infiltration (Fig. 4B). OVA sensitization resulted in a significant expansion of the eosinophil population. In contrast, PBM treatment led to a reduction in the proportion of inflammatory Ly6Chi monocytes (Figure S1). Together, these results suggest a time-dependent increase in pulmonary eosinophils—a hallmark of the acute allergic response. In NIR PBM-treated mice. In parallel, inflammatory monocytes, which contribute to the propagation of the inflammatory milieu, were also diminished in the PBM group. And the reduced infiltration of B and CD4 T cells at Day 23, together suggesting that PBM could induce beneficial effects in OVA allergy, in part, by the suppression of key inflammatory cell recruitment into the lung.

Pulmonary immune cell characterization in the OVA-allergy model at basal and day 23. (A) Quantification of pulmonary eosinophils, alveolar macrophages, neutrophils, and Ly6hi monocytes. (B) Quantification of pulmonary B cells, CD4 T cells, and CD8 T cells. Group sizes: Basal PBS (n = 4), Basal OVA (n = 4), PBS day 23 (n = 3), OVA day 23 (n = 4), and OVA-PBM day 23 (n = 4). Data were analyzed using an unpaired, two-tailed Student’s t-test (*P < 0.05).
NIR PBM attenuates OVA-induced allergy via suppressing Th2 response and NF-kb pathway
To gain insights into the molecular mechanisms underlying the anti-allergic effects of NIR PBM, we performed RNA sequencing on lung tissue collected at baseline and 3 days after the OVA aerosol challenge (Day 23). Principal component analysis (PCA) clearly separated the OVA-treated groups from the baseline samples (Fig. 5A), confirming a robust transcriptomic shift induced by OVA allergy.

Transcriptomic analysis of the PBM effect in the OVA-allergy model. (A) Principal component analysis (PCA) of RNA-seq data from lung tissues obtained from Basal PBS, Basal OVA, PBS day 23, OVA day 23, and OVA-PBM day 23 groups. (B) Volcano plot of differentially expressed genes between the OVA day 23 and Basal PBS groups. (C) Volcano plot of differentially expressed genes between the OVA-PBM day 23 and Basal PBS groups. (D) Gene set enrichment analysis (GSEA) showing pathways upregulated in the OVA-PBM group versus the OVA group. Representative GSEA plots for (E) HALLMARK_INFLAMMATORY_RESPONSE, (F) HALLMARK_TNF_SIGNALING_VIA_NFKB, (G) Eosinophil_vs_Mac_IL25_Treatment, and (H) a heatmap of Th2-related gene sets are also presented. (I) Western blot analysis was performed to determine the levels of p-P65 and P65.Data were analyzed using an unpaired, two-tailed Student’s t-test (*P < 0.05).
Comparing differentially expressed genes (DEGs) between the OVA_Day23 and Basal_Ova groups, the volcano plot demonstrates the successful induction of an acute allergic response (Fig. 5B). In particular, type 2 response-related genes such as Il33, Chil3, and the inflammatory mediator Saa3 were significantly upregulated, along with the prominently differentially expressed Clca1, a gene critical for goblet cell hyperplasia and mucus secretion. Similar DEG patterns were observed in the OVA_PBM_Day23 group (Fig. 5C). However, using the same stringent analysis criteria, there were no obvious DEGs or associated pathway differences between the PBM-treated and untreated groups. This suggests that the transcriptomic differences induced by PBM are modest and may not be captured by strict DEG analysis—a conclusion further supported by the overlapping distribution of the OVA_Day23 and OVA_PBM_Day23 groups in the PCA plot.
Given that NIR PBM attenuated both histological pathology and the infiltration of eosinophils and inflammatory monocytes, we adopted Gene Set Enrichment Analysis (GSEA). Unlike strict DEG filtering, GSEA ranks the expression of a set of genes between conditions, thereby highlighting pathway-level differences. GSEA revealed that hallmark pathways associated with the inflammatory response—specifically TNF signaling via NF-κB, apical surface, and other inflammatory gene sets—were downregulated in the PBM-treated group compared to the untreated OVA group (Fig. 5D–F). Furthermore, when we performed GSEA against the C7 inflammatory gene set from msigdbr, the eosinophil_versus_Mac_IL25_treated_lung subset was negatively enriched, suggesting that PBM treatment leads to downregulation of eosinophil-specific signatures compared to the non-treated group (Fig. 5G). Additionally, a focused heatmap of Th2 signature genes (including Il4, Il5, Il13, and Tslp) confirmed that NIR PBM significantly reduced the expression of these key mediators (Fig. 5H). To verify that PBM downregulates the NF-κB signaling pathway, we collected mouse lung tissues three hours after the last aerosol challenge and performed Western blot analysis. The results showed a significant expression of p-P65 protein in the OVA group, while the expression level of p-P65 in the OVA PBM group was significantly lower than that in the OVA group (original gels are presented in Supplementary Material) . This finding is consistent with our RNA sequencing results. (Fig. 5I).
Together, these transcriptomic findings indicate that although overall gene expression changes are modest, pathway-level modulation—particularly of NF-κB signaling and Th2-mediated responses—plays a critical role in the therapeutic effects of NIR PBM.
Discussion
Our study demonstrates that high-intensity 980 nm NIR photobiomodulation attenuates allergic airway inflammation in a murine model of OVA-induced asthma. We show that NIR PBM improves lung histology by reducing airway wall thickening and inflammatory cell infiltration, decreases the expression of key Th2 cytokines (IL-4, IL-5, IL-13), and limits the recruitment of eosinophils and inflammatory monocytes. Additionally, RNAseq analysis coupled with GSEA indicates that NIR PBM modulates inflammatory pathways—most notably by suppressing TNF/NF-κB signaling—and downregulates a Th2 gene signature.
These findings provide support for the mechanistic rationale of using NIR PBM as a non-pharmacological intervention in allergic asthma. The reduction in type 2 cytokines is particularly important, as these mediators are directly linked to eosinophilic inflammation, IgE production, and airway hyperresponsiveness. By dampening these responses, NIR PBM may offer a novel therapeutic approach for patients who are resistant or intolerant to conventional corticosteroid-based therapies42,43.
The transcriptomic analysis further suggests that the beneficial effects of NIR PBM may extend to modulating broader inflammatory cascades via the NF-κB pathway44,45,46. Although the overall transcriptomic differences between PBM-treated and untreated OVA groups were subtle, pathway-level analyses uncovered significant differences that align with the observed histological and cellular changes. Downregulation of NF-κB–driven pathways is consistent with the observed decrease in inflammatory cell recruitment and cytokine expression47,48,49, and the upregulation of DNA repair pathways may reflect a cellular protective response that contributes to tissue recovery following allergen-induced injury50.
One important observation from our study is that the effects of NIR PBM appear to be mediated primarily through the modulation of immune cell recruitment and cytokine expression rather than through large-scale changes in gene expression detectable by stringent differential gene analysis. This suggests that NIR PBM may act as a fine-tuner of immune responses, exerting its effects by subtly altering key regulatory pathways that govern inflammation. Such a mechanism would be advantageous in clinical settings, where complete suppression of immune function is undesirable.
While these results are promising, several limitations must be acknowledged. First, the study was conducted in an acute model of OVA-induced allergic asthma, which may not capture all aspects of chronic human asthma. Second, although our RNAseq and GSEA approaches provided insights into the molecular pathways modulated by PBM, further studies are needed to dissect the precise intracellular signaling mechanisms and to identify potential direct targets of NIR light. Finally, while we observed clear reductions in inflammatory markers and improvements in lung histology, additional studies assessing lung function parameters (such as airway hyperresponsiveness) would provide further evidence of the clinical relevance of NIR PBM. To investigate whether NIR PBM treatment can suppress the OVA-induced activation of the NF-κB signaling pathway in asthmatic mice, lung tissues were collected three hours post-challenge for Western blot analysis. The results demonstrated a significant reduction in the ratio of p-P65 to total P65 in the PBM-treated group compared to the OVA group. This finding indicates that the therapeutic effect of NIR PBM on the asthmatic phenotype is mediated, at least in part, through the modulation of the NF-κB signaling pathway.
In conclusion, our findings support the use of high-intensity 980 nm NIR photobiomodulation as a promising adjunctive therapy for allergic asthma. By suppressing Th2-mediated inflammation and modulating key inflammatory signaling pathways, NIR PBM may provide a safe, non-invasive strategy to improve outcomes in patients with asthma. Future work should aim to translate these findings into clinical studies and further elucidate the molecular underpinnings of PBM’s therapeutic effects.
Materials and methods
Animals
Wild-type C57BL/6 mice were obtained from the Xiamen University Laboratory Animal Center and maintained under specific pathogen-free conditions. All experimental procedures involving animals were approved by the Institutional Animal Care and Use Committee at Xiamen University, in accordance with the center’s guidelines for animal welfare.
Sensitization and antigen challenge
Mice were sensitized with an intraperitoneal injection of 100 μL phosphate-buffered saline (PBS) containing 100 μg of ovalbumin (OVA; Sigma, A5253) and 5 mg of aluminum hydroxide (Sigma, A6435). Two weeks later, a booster injection with the same dose was administered intraperitoneally. Three weeks post initial sensitization, mice were exposed daily for 30 min to aerosolized 5% OVA (dissolved in PBS) over a five-day period using an ultrasonic nebulizer (Medisana, SD-8063CS) connected to a plastic inhalation chamber. Twenty-four hours following the final challenge, the animals were euthanized via intraperitoneal injection of 200 μL of 1% sodium pentobarbital, and lung tissues were harvested for further analysis.
Photobiomodulation
Mice were irradiated by a laser device (LaserMedix1500, Fotonmedix Mecal Laser, China) set at an output power of 0.48 W with a 980 nm wavelength, which covered a spot size of 12.56 cm2. Two distinct irradiation protocols were implemented:
Following each nebulized challenge, mice were irradiated on the skin over the trachea and both lung regions immediately for three to five consecutive days. Lung tissues were collected at 3 h for early evaluation or at 24 h after the final antigen exposure to assess pulmonary inflammation.
Histology
The left lungs were fixed in 4% paraformaldehyde for 24 h, followed by dehydration and clearing using an automatic tissue processor (Leica, TP1020), and were embedded in paraffin. Sections (8 μm thick) were then stained with Hematoxylin and Eosin (H&E) to assess inflammatory cell infiltration. For each mouse, at least six images were captured and analyzed. Inflammatory cell infiltration was scored on a scale of 1 to 10: a score of 1 indicated no detectable inflammation; scores of 2–4 were assigned for increasing infiltration of inflammatory cells encircling the bronchi with a thin layer; scores of 5–7 indicated thicker layers of inflammatory cells surrounding the bronchi; while scores of 8 and 9 denoted severe to extreme inflammation spreading into the interstitial areas. The final histologic inflammation score for each mouse was calculated as the mean score.
For the PAS staining, tissue sections were sequentially subjected to dewaxing and hydration, followed by staining using an Alcian Blue Periodic Acid Schiff (AB-PAS) Stain Kit (Acmec, AC11577). Subsequently, the sections were dehydrated through a graded alcohol series, cleared, and mounted with neutral resin. A semi-quantitative scoring system was employed for the assessment.
Flow cytometry
Lung tissues were homogenized and passed through a 70-μm cell strainer. To lyse red blood cells, samples were incubated with ACK buffer for 3 min. The remaining cells were then centrifuged and resuspended in FACS buffer (1% FBS and 2 mM EDTA in PBS).
Cell suspensions were labeled for flow cytometric analysis using the following dye and antibodies: Live/Dead marker (Cat. 65-0865-14, Invitrogen), CD45 (Cat. 103147, Clone 30-F11, BioLegend), CD4 (Cat. 100451, Clone RM4-5, BioLegend), CD8 (Cat. 11–0081-85, Clone 53–6.7, eBioscience), CD11b (Cat. 48-0112-82, Clone M1/70, eBioscience), Ly6C (Cat. 128044, Clone HK1.4, BioLegend), Ly6G (Cat. 127628, Clone 1A8, BioLegend), CD19 (Cat. 152408, Clone 1D3/CD19, BioLegend), CD11c (Cat. 117338, Clone N418, BioLegend), SiglecF (Cat. 562680, Clone E50-2440, BD).
Cells were incubated on ice in FACS buffer with the antibody cocktail for 45 min. Fixed and stained cells were analyzed using an ID7000™ Spectral Cell Analyzer (Sony Biotechnology) and subsequently processed using FlowJo software.
RNA extraction and quantitative real-time PCR
Total RNA was extracted from lung tissues using TRIzol Reagent (Sangon Biotech, B511311-0500). RNA was then reverse transcribed into cDNA using a mix of dNTPs (Beyotime, D7366), oligo-dT primers, RNA transcriptase (Accurate Biology, AG11605), and RNase inhibitor (Accurate Biology, AG11608). Quantitative PCR (qPCR) was conducted using the SYBR Green method (Accurate Biology, AG11701). Gene expression levels were normalized to the housekeeping gene β₂-microglobulin (β2M) using the ΔCT method. The primer sequences used were as follows:
mIL4 Forward: 5'-TCACAGCAACGAAGAACACCA-3'.
mIL4 Reverse: 5'-CAGGCATCGAAAAGCCCGAA-3'.
mIL5 Forward: 5'-TGACAAGCAATGAGACGATGAGG-3'.
mIL5 Reverse 5’-TACCCCCACGGACAGTTTGATTC-3'.
mIL13 Forward: 5'-GTGTCTCTCCCTCTGACCCT-3'.
mIL13 Reverse: 5'-CAGGGCTACACAGAACCC-3'.
β2M Forward: 5'-TTCTGGTGCTTGTCTCACTGA-3'.
β2M Reverse: 5'-CAGTATGTTCGGCTTCCCATTC-3'.
ELISA
Level of total serum IgE was measured by a mouse IgE uncoated enzyme-linked immunosorbent assay (ELISA) kit (Invitrogen,88-50460-22); according to the manufacturer’s instruction. Briefly, 96-well plates were coated with anti-mouse IgE . After blocking, the diluted serum samples were added into the wells and incubated for 2 h at room temperature. After incubation, the plates were washed with phosphate buffered solution with 0.1% tween-20 (PBST), and the anti-mouse IgE-conjugated biotin was added and incubated for 1 h at room temperature. The plate was then washed and incubated with streptavidin–horseradish peroxidase (HRP) for 30 min at room temperature. Finally, tetramethylbenzidine substrate was added, and the reaction was stopped by Phosphoric acid. The absorbance at 450 nm was measured using a plate reader.
Western blot
Lungs were lysed with RIPA buffer (1 mM Tris–HCl, 0.3 M NaCl, 0.01% SDS, 1.5% NP40, 120 mM deoxycholate, 1 M MgCl2) containing protease inhibitors and Phosphatase Inhibitors. The proteins were resolved by SDS-PAGE and transferred to PVDF membranes (catalog number Roche, # 3010040001). The membranes were then blocked for 1 h in blocking buffer (5% BSA and 0.1% tween-20 in TBS) at room temperature, and incubated with respective primary antibodies in 5% BSA containing TBST at 4 °C overnight. The membranes were then washed and subjected to HRP-coupled secondary antibodies in TBST at room temperature for 1 h. Antibody against β-Actin (catalog number 21338) was purchased from ABclonal. Antibodies against NF-κB p65 (catalog number 8242S) and antibodies against Phospho-NF-κB p65 (catalog number 3033S) were purchased from CST. The Western blot images were captured by ChampChemi 910plus (SINSAGE).
RNA sequencing
The isolated RNA was eluted with the 20 μL reverse transcription mix consisting of 100 unit Hiscript III Reverse Transcriptase (Vazyme, R303), 40 unit RNase Inhibitor (Vazyme, R301),1 μL 25 mM dNTP mix, 1 μL 100 mM DTT, 1 μL 1 val Betaine (Sigma, B0300-1VL), 0.5 μL 5% Digitonin, 0.5 μL TSO-LNA and 0.5 μL 5 μM Probe. The reaction was incubated at 42℃ for 90 min. Following cDNA synthesis, the sample was pooled, cleaned and concentrated with cleanup beads at a 1:0.8 of DNA to beads ratio and eluted in 30 μL of ddH2O. Second-strand synthesis and pre-amplification were performed in a 50 μL reaction, consisting of 2 × KAPA HiFi Ready mix and 0.5 μM TSO-PCR primer, with the following PCR setup initial denaturation at 98 °C for 3 min, (denaturation at 98 °C for 15 s, annealing at 65 °C for 30 s, elongation at 72 °C for 6 min) repeated for 8 cycles, and a final elongation at 72 °C for 10 min. The DNA was cleanup using DNA cleanup beads at a ratio of 1:0.8 of DNA to beads and eluted with 30 μL of ddH2O. The library was prepared with the TruePrep DNA Library Prep Kit V2 for Illumina according to the manufacturer. Probe was designed by TSO, sample barcode(7-mer),UMI(8-mer) and Oligo(dT).
RNA-seq data analysis
The raw data was quality-controlled using fastqc and then trimmed of the Nextera adaptor. Following trimming, the Dropseq Tools pipeline was used to filter the data with a Phred quality score threshold of 30 for sample barcode and UMI identify and poly(A) trimming. The filtered data was mapped to the mouse genome (mm10) using STAR 2.5.1a,and the reads counted using Drop-seq-Tools.
Paired-end reads obtained from RNA sequencing were aligned to the reference human genome using HISAT2 with default parameters. Read counts for each gene were generated using feature Counts, and expression levels were calculated as RPKM based on gene length and raw counts. Differential gene expression was analyzed using DESeq2, with differentially expressed genes selected based on fold-change thresholds and statistical significance. Functional pathway analysis was then conducted using ClusterProfiler in R with a p-value cutoff of 0.05.
In addition, gene set enrichment analysis (GSEA) was performed to further explore the biological processes and pathways impacted by the treatment. GSEA was conducted using the Molecular Signatures Database (MSigDB) with both the HALLMARK and C7 (immunologic signature) gene sets. Enrichment plots were generated to visualize the normalized enrichment scores for significantly enriched pathways, with significant pathways defined as those with a nominal p-value < 0.05 and a false discovery rate (FDR) < 0.25. Heatmaps for the genes within these pathways were generated using the ComplexHeatmap package in R. The processed RNA-seq data have been deposited in the GEO database (accession number GSE294668).
Quantification and statistical analysis
Comparisons among experimental groups were performed using the t-test. Data are presented as means ± standard deviation (SD), and a p-value of less than 0.05 was considered statistically significant, denoted with a star symbol ( ∗). All statistical analyses were conducted using GraphPad Prism software.
All methods were done in accordance with ARRIVE guidelines.
Data availability
The RNA sequencing data generated in this study have been deposited in the NCBI Gene Expression Omnibus (GEO) under accession number GSE294668 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE294668). Theoriginal, uncropped Western blot images generated in this study have been deposited in Zenodo and are accessible via the following link: https://doi.org/10.5281/zenodo.17645268.
References
Liebert, A., Bicknell, B., Markman, W. & Kiat, H. A potential role for photobiomodulation therapy in disease treatment and prevention in the era of COVID-19. Aging Dis. 11, 1352–1362. https://doi.org/10.14336/ad.2020.0901 (2020).
Hamblin, M. R. Mechanisms and applications of the anti-inflammatory effects of photobiomodulation. AIMS Biophys. 4, 337–361. https://doi.org/10.3934/biophy.2017.3.337 (2017).
Poyton, R. O. & Ball, K. A. Therapeutic photobiomodulation: nitric oxide and a novel function of mitochondrial cytochrome c oxidase. Discov. Med. 11, 154–159 (2011).
Maghfour, J. et al. Photobiomodulation CME part I: Overview and mechanism of action. J. Am. Acad. Dermatol. 91, 793–802. https://doi.org/10.1016/j.jaad.2023.10.073 (2024).
Hamblin, M. R. & Liebert, A. Photobiomodulation therapy mechanisms beyond cytochrome c oxidase. Photobiomodul. Photomed. Laser Surg. 40, 75–77. https://doi.org/10.1089/photob.2021.0119 (2022).
de Freitas, L. F. & Hamblin, M. R. Proposed mechanisms of photobiomodulation or low-level light therapy. IEEE J. Sel. Top. Quant. Electron https://doi.org/10.1109/jstqe.2016.2561201 (2016).
Hudson, D. E., Hudson, D. O., Wininger, J. M. & Richardson, B. D. Penetration of laser light at 808 and 980 nm in bovine tissue samples. Photomed. Laser Surg. 31, 163–168. https://doi.org/10.1089/pho.2012.3284 (2013).
Chu, Y., Xu, X. Q. & Wang, Y. Ultradeep photothermal therapy strategies. J. Phys. Chem. Lett. 13, 9564–9572. https://doi.org/10.1021/acs.jpclett.2c02642 (2022).
Nazari, A., Moezy, A., Nejati, P. & Mazaherinezhad, A. Efficacy of high-intensity laser therapy in comparison with conventional physiotherapy and exercise therapy on pain and function of patients with knee osteoarthritis: a randomized controlled trial with 12-week follow up. Lasers Med. Sci. 34, 505–516. https://doi.org/10.1007/s10103-018-2624-4 (2019).
Papi, A., Brightling, C., Pedersen, S. E. & Reddel, H. K. Asthma. Lancet 391, 783–800. https://doi.org/10.1016/s0140-6736(17)33311-1 (2018).
Castillo, J. R., Peters, S. P. & Busse, W. W. Asthma exacerbations: Pathogenesis, prevention, and treatment. J. Allergy Clin. Immunol. Pract. 5, 918–927. https://doi.org/10.1016/j.jaip.2017.05.001 (2017).
Wills, T. A., Soneji, S. S., Choi, K., Jaspers, I. & Tam, E. K. E-cigarette use and respiratory disorders: an integrative review of converging evidence from epidemiological and laboratory studies. Eur. Respir. J. https://doi.org/10.1183/13993003.01815-2019 (2021).
Suraya, R. et al. Molecular mechanism of asthma and its novel molecular target therapeutic agent. Respir. Investig. 59, 291–301. https://doi.org/10.1016/j.resinv.2020.12.007 (2021).
Miller, R. L., Grayson, M. H. & Strothman, K. Advances in asthma: New understandings of asthma’s natural history, risk factors, underlying mechanisms, and clinical management. J. Allergy Clin. Immunol. 148, 1430–1441. https://doi.org/10.1016/j.jaci.2021.10.001 (2021).
Hussain, M. & Liu, G. Eosinophilic asthma: Pathophysiology and therapeutic horizons. Cells https://doi.org/10.3390/cells13050384 (2024).
Lloyd, C. M. & Snelgrove, R. J. Type 2 immunity: Expanding our view. Sci. Immunol. https://doi.org/10.1126/sciimmunol.aat1604 (2018).
D’Aiuto, V. et al. Eosinophil-driven vs. eosinophil-associated severe asthma: Practical implications for target treatment. Int. J. Mol. Sci. https://doi.org/10.3390/ijms26041729 (2025).
Nakagome, K. & Nagata, M. The possible roles of IL-4/IL-13 in the development of eosinophil-predominant severe asthma. Biomolecules https://doi.org/10.3390/biom14050546 (2024).
Pritam, P. et al. Eosinophil: a central player in modulating pathological complexity in asthma. Allergol. Immunopathol. (Madr) 49, 191–207. https://doi.org/10.15586/aei.v49i2.50 (2021).
Steffan, B. N., Townsend, E. A., Denlinger, L. C. & Johansson, M. W. Eosinophil-epithelial cell interactions in asthma. Int. Arch. Allergy Immunol 185, 1033–1047. https://doi.org/10.1159/000539309 (2024).
Van Hulst, G. et al. Eosinophil diversity in asthma. Biochem. Pharmacol. 179, 113963. https://doi.org/10.1016/j.bcp.2020.113963 (2020).
Kianmeher, M., Ghorani, V. & Boskabady, M. H. Animal model of asthma, various methods and measured parameters: A methodological review. Iran J. Allergy Asthma Immunol. 15, 445–465 (2016).
Hirota, J. A., Hackett, T. L., Inman, M. D. & Knight, D. A. Modeling asthma in mice: what have we learned about the airway epithelium?. Am. J. Respir. Cell Mol. Biol. 44, 431–438. https://doi.org/10.1165/rcmb.2010-0146TR (2011).
Kumar, R. K., Herbert, C. & Foster, P. S. The “classical” ovalbumin challenge model of asthma in mice. Curr. Drug Targets 9, 485–494. https://doi.org/10.2174/138945008784533561 (2008).
Huang, H. J., Sarzsinszky, E. & Vrtala, S. House dust mite allergy: The importance of house dust mite allergens for diagnosis and immunotherapy. Mol. Immunol. 158, 54–67. https://doi.org/10.1016/j.molimm.2023.04.008 (2023).
Zhou, W. et al. COX inhibition increases alternaria-induced pulmonary group 2 innate lymphoid cell responses and IL-33 release in mice. J. Immunol. 205, 1157–1166. https://doi.org/10.4049/jimmunol.1901544 (2020).
Kabata, H. et al. Targeted deletion of the TSLP receptor reveals cellular mechanisms that promote type 2 airway inflammation. Mucosal. Immunol. 13, 626–636. https://doi.org/10.1038/s41385-020-0266-x (2020).
Lambrecht, B. N., Hammad, H. & Fahy, J. V. The cytokines of asthma. Immunity 50, 975–991. https://doi.org/10.1016/j.immuni.2019.03.018 (2019).
Habib, N., Pasha, M. A. & Tang, D. D. Current understanding of asthma pathogenesis and biomarkers. Cells https://doi.org/10.3390/cells11172764 (2022).
Komlósi, Z. I. et al. Cellular and molecular mechanisms of allergic asthma. Mol. Aspects Med. 85, 100995. https://doi.org/10.1016/j.mam.2021.100995 (2022).
Gandhi, N. A. et al. Targeting key proximal drivers of type 2 inflammation in disease. Nat. Rev. Drug Discov. 15, 35–50. https://doi.org/10.1038/nrd4624 (2016).
Dougan, M., Dranoff, G. & Dougan, S. K. GM-CSF, IL-3, and IL-5 family of cytokines: Regulators of inflammation. Immunity 50, 796–811. https://doi.org/10.1016/j.immuni.2019.03.022 (2019).
Ingram, J. L. & Kraft, M. IL-13 in asthma and allergic disease: asthma phenotypes and targeted therapies. J. Allergy Clin. Immunol. 130, 829–842. https://doi.org/10.1016/j.jaci.2012.06.034 (2012).
Chung, K. F. et al. International ERS/ATS guidelines on definition, evaluation and treatment of severe asthma. Eur. Respir. J. 43, 343–373. https://doi.org/10.1183/09031936.00202013 (2014).
Barnes, P. J. Corticosteroid resistance in patients with asthma and chronic obstructive pulmonary disease. J. Allergy Clin. Immunol. 131, 636–645. https://doi.org/10.1016/j.jaci.2012.12.1564 (2013).
Jha, S. S., Kumar, M., Agrawal, P. K. & Thakur, D. K. Osteoporosis in asthma and COPD. Indian J. Orthop. 57, 200–208. https://doi.org/10.1007/s43465-023-01048-5 (2023).
Imam, K. H. & Woessner, K. M. Nonsteroidal anti-inflammatory drug-exacerbated respiratory disease: diagnosis and current management. Pol. Arch. Intern. Med. https://doi.org/10.20452/pamw.16544 (2023).
Brochetti, R. A. et al. Beneficial effects of infrared light-emitting diode in corticosteroid-resistant asthma. Lasers Med. Sci. 37, 1963–1971. https://doi.org/10.1007/s10103-021-03457-0 (2022).
Barrett, D. W. & Gonzalez-Lima, F. Transcranial infrared laser stimulation produces beneficial cognitive and emotional effects in humans. Neuroscience 230, 13–23. https://doi.org/10.1016/j.neuroscience.2012.11.016 (2013).
Silva, V. R. et al. Low-level laser therapy inhibits bronchoconstriction, Th2 inflammation and airway remodeling in allergic asthma. Respir. Physiol. Neurobiol. 194, 37–48. https://doi.org/10.1016/j.resp.2014.01.008 (2014).
de Brito, A. A. et al. Photobiomodulation therapy restores IL-10 secretion in a murine model of chronic asthma: Relevance to the population of CD4(+)CD25(+)Foxp3(+) cells in lung. Front. Immunol. 12, 789426. https://doi.org/10.3389/fimmu.2021.789426 (2021).
Barnes, P. J. Inflammatory endotypes in COPD. Allergy 74, 1249–1256. https://doi.org/10.1111/all.13760 (2019).
Barnes, P. J. et al. Chronic obstructive pulmonary disease. Nat. Rev. Dis. Primers 1, 15076. https://doi.org/10.1038/nrdp.2015.76 (2015).
Curra, M. et al. Photobiomodulation reduces oral mucositis by modulating NF-kB. J. Biomed. Opt. 20, 125008. https://doi.org/10.1117/1.Jbo.20.12.125008 (2015).
Ma, Y. et al. Photobiomodulation Attenuates neurotoxic polarization of macrophages by inhibiting the Notch1-HIF-1α/NF-κB signalling pathway in mice with spinal cord injury. Front. Immunol. 13, 816952. https://doi.org/10.3389/fimmu.2022.816952 (2022).
Ryu, J. H. et al. Photobiomodulation ameliorates inflammatory parameters in fibroblast-like synoviocytes and experimental animal models of rheumatoid arthritis. Front. Immunol. 14, 1122581. https://doi.org/10.3389/fimmu.2023.1122581 (2023).
Capece, D. et al. NF-κB: blending metabolism, immunity, and inflammation. Trends Immunol. 43, 757–775. https://doi.org/10.1016/j.it.2022.07.004 (2022).
Sun, S. C. The non-canonical NF-κB pathway in immunity and inflammation. Nat. Rev. Immunol. 17, 545–558. https://doi.org/10.1038/nri.2017.52 (2017).
Yu, H., Lin, L., Zhang, Z., Zhang, H. & Hu, H. Targeting NF-κB pathway for the therapy of diseases: mechanism and clinical study. Signal Transduct. Target. Ther. 5, 209. https://doi.org/10.1038/s41392-020-00312-6 (2020).
da Silva Sergio, L. P., Mencalha, A. L., de Souza da Fonseca, A. & de Paoli, F. DNA repair and genomic stability in lungs affected by acute injury. Biomed. Pharmacother. 119, 109412. https://doi.org/10.1016/j.biopha.2019.109412 (2019).
Acknowledgements
This study was supported by the Natural Science Foundation of Xiamen, China (Grant No. 3502Z20227003, awarded to Zhang Jia). SC.C. gratefully acknowledges financial support from the National Natural Science Foundation of China (grants 32161133020), the Fundamental Research Funds for the Central Universities (grant 20720220003), and the Startup Fund of Xiamen University.
Funding
This study was supported by the Natural Science Foundation of Xiamen, China (Grant No. 3502Z20227003, awarded to Zhang Jia). S-C.C. gratefully acknowledges financial support from the National Natural Science Foundation of China (grants 32161133020), the Fundamental Research Funds for the Central Universities (grant 20720220003), and the Start-up Fund of Xiamen University.
Author information
Authors and Affiliations
Contributions
S-C.C. conceptualized and supervised the study and secured funding, while also contributing to both the original draft and the subsequent review and editing. Methodology was established by Z.W., J.Z., D.W., Y.Z., and H.Y., with Z.W., J.Z., and D.W. carrying out the investigation; the manuscript was originally drafted by Z.W. and S-C.C. and further refined through collaborative review and editing by all authors, including Z.W., J.Z., D.W., Y.Z., H.Y., and S-C.C.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, Z., Zhang, J., Gao, Q. et al. Pulsed 980 nm near-infrared photobiomodulation attenuates OVA-induced allergic asthma by modulating Th2 cytokine responses and inflammatory cell infiltration. Sci Rep 16, 3214 (2026). https://doi.org/10.1038/s41598-025-33195-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-33195-0
