
Abstract
Transcriptome analysis of complex tissues remains challenging due to assembly errors, isoform diversity, and annotation bias, necessitating optimized computational pipelines. Scorpion venoms are a treasure trove of bioactive peptides with significant biomedical potential, but their complexity complicates transcriptome profiling. We present ToxIR (Toxin Identification and Recognition), an RNA-seq pipeline optimized for accurate toxin transcriptome analysis, validated in Odontobuthus doriae venom glands. ToxIR combines deep sequencing, rnaSPAdes based de novo assembly, and a tailored annotation strategy to detect even low-abundance toxins and resolve isoforms with high accuracy. It incorporates rigorous quality control (FastQC, Trimmomatic), curated UniProt toxin homology searches, and integrated structural analyses (SignalP, TMHMM, Pfam, InterProScan) to prioritize candidates based on signal peptides, cysteine content, and toxin-specific domains. Unlike general-purpose or previous toxin pipelines, ToxIR minimizes misassemblies and annotation bias through its modular design, automated structural queries, and SQLite-backed data integration. The pipeline identified 378 putative toxin candidates, including 192 high-confidence candidates (Group A) and 23 novel, divergent toxins (Group C). These included 180 sodium channels, 111 potassium channels, and 69 chloride channel toxins. By enabling flexible cross-species use and enhancing annotation precision, ToxIR provides a robust framework that accelerates the discovery of therapeutic toxins.
Similar content being viewed by others


Introduction
Accurate transcriptome profiling is essential for understanding gene expression in complex biological systems. Venom glands of scorpions, enriched in diverse toxin peptides, represent a particularly challenging model that highlights the limitations of general-purpose RNA-seq pipelines1. Animal venoms are extraordinary biochemical arsenals, evolved to target physiological systems with exquisite precision. In scorpions, venom is a complex cocktail dominated by peptides and proteins, many of which act as potent neurotoxins modulating ion channels and receptors2,3. Beyond their ecological roles in prey capture and defense, these toxins have drawn increasing attention for their potential in biomedical applications, including the development of novel analgesics, anticancer agents, and immunomodulators4,5.
Odontobuthus doriae, a scorpion species distributed across parts of the Middle East, is of particular medical relevance due to the severe neurotoxic effects of its venom6. Despite its clinical importance and pharmacological promise, our molecular understanding of O. doriae toxins remains fragmented, largely constrained by the limitations of traditional proteomic studies and low-throughput molecular approaches7.
Venom gland transcriptome analyses have revolutionized toxin discovery by allowing researchers to directly investigate toxin gene expression at the mRNA level, capturing even low-abundance transcripts often missed by proteomics8. However, conventional RNA-seq studies face technical hurdles, including incomplete assembly of toxin precursors, challenges in distinguishing closely related toxin isoforms, and annotation biases that hinder comprehensive characterization of toxin diversity9,10.
In this study, we present an optimized RNA-seq framework specifically designed for the identification of toxin-encoding transcripts. By integrating deep sequencing coverage, an advanced de novo assembly strategy, and a refined annotation system focused on toxin gene families, this approach substantially enhances the sensitivity and accuracy of toxin transcript detection across diverse species. It enables reliable identification of low-abundance toxins, reconstruction of full-length precursors, and precise classification of isoforms with potential biomedical relevance.
This targeted transcriptomic strategy offers a detailed view of the molecular diversity and evolutionary innovations of toxin systems, while providing a flexible platform for the discovery of novel bioactive compounds. By focusing on the toxin gene repertoire rather than the entire transcriptome, the method generates a high-resolution map that captures both well-characterized and previously unrecognized toxin genes. Ultimately, this study highlights the potential of specialized RNA-seq approaches to bridge biodiversity research with applied toxicology and facilitate the development of nature-derived therapeutic agents.
Materials and methods
Biological material and RNA-seq experiment
Sample preparation
According to the World Health Organisation’s (WHO) ethical guidelines, the O.doriae scorpions were captured from the central desert regions of Iran in October 2024. The scorpion species was identified and confirmed by a taxonomist. The milking was carried out by electroshock to stimulate venom gland regeneration and induce the secretory phase of venom-producing cells. Three days after milking, the telsons of scorpion individuals were removed and preserved in RNAprotect Tissue Reagent® (Cat. No. /ID: 76,104) for immediate stabilization of RNA.
RNA isolation and cDNA library construction from O.doriae venom glands
Total RNA was isolated from venom gland tissues preserved in RNAprotect Tissue Reagent according to the manufacturer’s instructions of the RNeasy Mini Kit (Cat. 74,104). RNA integrity was assessed based on electrophoretic profiles, and samples corresponding to RIN-equivalent quality scores > 7 were selected. The library was constructed using TruSeq® Stranded mRNA Library Prep (cat. number 20020594) according to the manufacturer’s instructions.
RNA-seq method
The constructed cDNA library was sequenced by High-throughput RNA sequencing (Illumina Next-Generation Sequencing). The samples were subjected to paired-end sequencing (150 bp) on an Illumina HiSeq 2000 platform (Macrogen Inc., Seoul, South Korea).
ToxIR computational pipeline
Phase I: transcriptome assembly and annotation
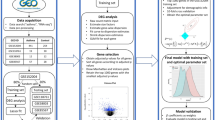
The ToxIR pipeline provides a Comprehensive ORF Annotation Database in this phase, offering a complete collection of data and annotations for researchers (Fig. 1).
Quality control, trimming, and de novo transcriptome assembly
Initial quality assessment of the raw paired-end reads was performed using FastQC v0.12.111. This step allowed for the identification of adapter contamination, low-quality bases, and sequence content biases before trimming.
Reads were subsequently trimmed using Trimmomatic v0.3912. Adapter sequences were removed (ILLUMINACLIP), and low-quality bases were trimmed from both ends (LEADING:2, TRAILING:2). Additional filtering was applied using a sliding window of 4 bases with a minimum.

Phase I diagram of the ToxIR pipeline, whose final output is a comprehensive ORF annotation database.
average quality of 8 (SLIDINGWINDOW:4:8), and only reads longer than 38 bp were retained (MINLEN:38). These settings were applied after multiple rounds of trimming optimization and were finalized based on quality control assessments conducted both before and after assembly to ensure optimal performance.
Trimmed high-quality reads were de novo assembled using rnaSPAdes v4.2.0 (RNA mode)13 with default parameters. rnaSPAdes was selected due to its demonstrated ability to reconstruct a higher number of ≥ 95%-assembled genes and isoforms, with fewer misassemblies and a lower duplication ratio compared to other commonly used assemblers such as Trinity.
Assembly completeness was assessed using BUSCO v5.8.314 in transcriptome mode (euk_tran), with two lineage datasets: arthropoda_odb10 and arachnida_odb10.
To evaluate read representation, trimmed reads were mapped back to the assembled transcriptome using Bowtie2 v2.5.415, and assembly contiguity metrics were computed using QUAST v5.2.016.
Prediction of coding sequences, protein translation, and redundancy reduction
Open Reading Frames (ORFs) were predicted from the assembled transcripts using ORFipy v0.0.417. The analysis was conducted with the following parameters: start codon set to ATG, a minimum ORF length of 90 nucleotides (corresponding to 30 amino acids), and extraction of both nucleotide and amino acid sequences. Only complete ORFs were retained, excluding partial sequences. To eliminate redundant protein sequences and improve computational efficiency for downstream analyses, clustering was performed using CD-HIT v4.8.118 with stringent parameters: 100% sequence identity, 100% alignment coverage for the shorter sequence, and greedy clustering mode. Descriptions were preserved in full.
Toxin reference database construction and BLAST homology search
To construct a comprehensive and up-to-date toxin reference database, protein sequences associated with the UniProt19 keyword KW-0800 (Toxin) were retrieved programmatically via the UniProt REST API (release 2025_02, retrieved on 20 April 2025). The query included both reviewed and unreviewed entries with isoforms enabled and no taxonomic restrictions. A total of 109,442 toxin-related protein sequences were collected in FASTA format for use as a custom BLAST database.
A homology-based search was performed using BLASTP v2.16.0 + 20 to compare the clustered ORFs against a curated UniProt toxin protein database. The search was conducted with an e-value cutoff of 1e-5, and the output was generated in tabular format. To ensure specificity, for each ORF, only the best-scoring hit was retained. Selection was based on a two-level criterion: lowest e-value, and in case of ties, highest bit score.
Signal peptide and transmembrane domain prediction
To predict N-terminal signal peptides and identify potentially secreted proteins, SignalP v6.021was employed in fast mode with the organism type set to eukarya. SignalP 6.0 supports detection of all known classes of signal peptides across diverse taxa. The analysis was conducted on the full set of clustered protein sequences. To assist in the interpretation of secretion potential and subcellular localization, transmembrane domain prediction was performed on the full set of clustered protein sequences using TMHMM v2.0c22. ToxIR does not distribute the proprietary tools SignalP v6.0 and TMHMM v2.0c. Users are required to run these programs independently under valid licenses and provide the resulting output files for downstream integration.
Protein domain annotation using Pfam
Protein domain annotation was performed on the complete set of clustered proteins using HMMER v3.3 (hmmscan)23 against the Pfam-A database (release 37.3, April 2025). The search was executed in domtblout format, and only domain matches with an e-value ≤ 1e-5 were retained to ensure high-confidence annotations. When multiple domains were detected for a single protein, all were included in the final entry.
Cysteine content analysis
To enrich protein annotation with structural features, a Python-based script was implemented to analyze cysteine content and positional distribution within each predicted protein sequence. For each protein, the total sequence length was calculated, cysteine residues were identified with their absolute positions (1-based indexing), and the number of cysteines as well as the theoretical maximum number of disulfide bonds (based on all pairwise combinations) were determined.
Integration of annotations into a unified database
All functional and structural annotations were systematically integrated into a unified SQLite database to facilitate downstream filtering, prioritization, and exploration. The integrated dataset comprised open reading frame (ORF) identifiers, corresponding full-length nucleotide sequences, and their in silico-translated protein sequences. Functional annotations included best-hit results from BLASTP searches against a curated UniProt-derived toxin database, with associated species information for taxonomic traceability. High-confidence Pfam domain predictions (E-value ≤ 1e-5), signal peptide predictions from SignalP v6.0, and transmembrane domain predictions from TMHMM v2.0 were incorporated. Cysteine-related features—including residue counts, theoretical disulfide bond potential, and positional motifs—were extracted using a custom Python-based annotation script. Each ORF was also linked to its source transcript via the original transcript identifier, enabling complete traceability across annotation layers. The SQLite database follows an ORF-centric relational schema, in which each predicted ORF is assigned a unique ORF identifier that serves as the primary key across all tables. All annotation layers are linked through this ORF identifier. This design enables flexible querying across annotation layers (e.g., filtering by secretion status, domain architecture, or homology strength) while preserving full traceability between transcripts and derived protein features. The database structure ensures reproducibility and allows re-analysis or re-classification without re-running upstream computational steps.
Physicochemical property annotation
To further enrich the ORF-level annotation, a custom Python pipeline was implemented using the Bio.SeqUtils.ProtParam module from Biopython24 to calculate key physicochemical features for each predicted protein. All calculations were performed on the full-length precursor sequences. For each ORF-derived protein sequence, the following properties were computed: molecular weight, aromaticity index, instability index, isoelectric point (pI), GRAVY (grand average of hydropathy), mean backbone flexibility (based on normalized residue-specific scales), and a binary protein stability label. The stability label was assigned as “STABLE” for sequences with an instability index below 40, and “UNSTABLE” otherwise. The resulting values were appended as additional columns to an extended version of the SQLite annotation database, enabling structured retrieval during downstream analyses. Sequences for which no physicochemical properties could be recorded were found to contain unresolved or non-standard amino acids, precluding calculation of their parameters.
Functional domain validation via InterProScan
To enhance the functional characterization of putative toxin proteins, InterProScan v5.65-97.025 was used to annotate conserved domains, protein families, and sequence motifs. Protein sequences that showed significant similarity to entries in a custom toxin-focused UniProt dataset based on prior BLASTP analysis were selected for this step. InterProScan was used to annotate conserved domains and functional signatures across multiple databases26. InterProScan was executed as a locally installed standalone instance due to its Java-based architecture and substantial external database requirements. ToxIR does not bundle InterProScan or its associated databases; instead, it expects user-generated InterProScan output files produced under standard configurations, which are subsequently parsed and integrated into the pipeline during downstream analysis.
Resulting annotation files were parsed and stored in a structured SQLite database. Only entries corresponding to the BLAST-validated open reading frames (ORFs) were retained. Domain predictions were considered valid if they either met an E-value threshold of ≤ 1e-5 or were labeled with a trusted (T) status by the InterProScan tool. For each accepted hit, associated metadata including the prediction method, domain signature, functional description, InterPro identifier, Gene Ontology (GO) terms, and pathway annotations were extracted. Each ORF was linked to its corresponding domain annotations and assigned summary-level features such as total domain count and sequence length.
Phase II: toxin classification strategy
To comprehensively identify and prioritize candidate toxin proteins, a three-tier classification framework was implemented based on an integrative assessment of sequence similarity (via BLASTP against the curated toxin dataset), domain annotations (Pfam or InterProScan), and toxin-like features such as signal peptide (SP), protein length, and cysteine content. Each predicted protein was assigned to one of the following three categories, designed to capture both high-confidence known toxins and novel or divergent toxin candidates (Fig. 2).

Phase II diagram of the ToxIR pipeline, which performs multi-criteria classification of predicted toxin candidates.
Group A contained high-confidence toxins, defined by BLASTP identity ≥ 80% to the custom toxin dataset, toxin-associated Pfam/InterProScan domains (Section “Identification of toxin-associated pfam and interpro domains”), and specific features (signal peptide, ≤ 300 aa, > 1 cysteine).
Group B included moderate-homology candidates (BLASTP 0–80%) with toxin-associated domains and the same features as Group A, representing divergent homologs or convergent proteins.
Group C represents putative novel or divergent toxin candidates, lacking BLASTP hits but showing toxin-associated Pfam/InterProScan domains and required features. ORFs were identified by filtering annotated sequences in the SQLite database against the curated toxin-related Pfam/InterPro lists (Section “Identification of toxin-associated pfam and interpro domains”). An overview of these classifications is shown in Table 1.
To uncover potential toxin candidates that lacked BLAST similarity but still exhibited hallmark toxin-like properties—such as the presence of a signal peptide and a short protein length—the following pipeline was implemented:
Step 1: Filtering for Unannotated Proteins.
All ORFs predicted to possess a signal peptide (SP), but lacking any BLAST hit were selected from the integrated annotation database. To prioritize compact, secretion-prone proteins, a maximum sequence length threshold of 300 amino acids was enforced. This selection was implemented using a custom Python script that filtered rows meeting all of the following criteria: (i) the prediction was labeled as ‘SP’; (ii) all BLAST-related columns were empty; (iii) the sequence length was less than or equal to 300 amino acids; (iv) and more than one cysteine residue. The resulting subset of sequences was exported in FASTA format for further domain analysis.
Step 2: Domain Scanning with InterProScan.
The filtered sequences were then scanned using InterProScan v5.65-97.0 to annotate conserved domains and functional signatures across multiple databases. Subsequently, InterPro IDs (see Section “Identification of toxin-associated pfam and interpro domains”) were matched against the InterPro annotations of the filtered candidates using a comparison script.
After selecting representatives from each group, BLAST searches were performed against the NCBI database to verify toxin prediction and assess the pipeline’s accuracy.
Identification of toxin-associated Pfam and interpro domains
A reproducible list of Pfam IDs related to toxin functions was generated via the InterPro REST API (https://www.ebi.ac.uk/interpro/api/)27 using the keyword “toxin.” A Python script automated the queries, extracted valid Pfam accessions (PFxxxxx), removed duplicates, and saved them as a text file, yielding 292 unique Pfam IDs. In parallel, 410 toxin-related InterPro IDs were retrieved via the InterPro API ( https://www.ebi.ac.uk/interpro/api/entry/InterPro/?search=toxin ) to assess potential functional relevance. A Python script automated the extraction of InterPro accession numbers and names, storing them in a plain text file. This approach ensured transparency, reproducibility, and allowed updates by re-running the queries.
All tools in the ToxIR pipeline were executed using explicitly defined command-line parameters and tool versions, summarized in Supplementary Table 1. For tools with substantial external dependencies (InterProScan, SignalP, TMHMM), ToxIR expects user-generated output files produced using the specified configurations, which are subsequently processed by the provided parsing and integration scripts. Execution can be performed using either Conda or Mamba environments, ensuring reproducibility and proper dependency management. The pipeline was implemented in a modular fashion, and detailed instructions for environment setup and execution are provided in the link available in the Data Availability section.
Results
After quality trimming, a total of 41,051,118 paired-end reads (20,525,559 read pairs) were retained for downstream analysis.
Transcriptome assembly and evaluation
The de novo transcriptome assembly yielded 96,058 transcripts, with an average transcript length of 620.7 bp, a total assembled length of 59.6 Mb, and an N50 value of 1009 bp. Among these, 26,492 transcripts were longer than 500 bp, and 14,036 exceeded 1000 bp in length.
BUSCO analysis showed high completeness of the assembly. Against the arthropoda_odb10 dataset, 90.7% of BUSCOs were identified as complete, comprising 72.9% single-copy and 17.9% duplicated BUSCOs, while 5.9% were fragmented and 3.4% were missing (n = 1013). Similarly, analysis against the arachnida_odb10 dataset revealed 87.5% complete BUSCOs (68.5% single-copy, 19.0% duplicated), with 4.1% fragmented and 8.4% missing BUSCOs (n = 2934). Read mapping with Bowtie2 confirmed high assembly representation, with an overall alignment rate of 98.84%, indicating that the majority of trimmed reads were successfully incorporated into the final assembly.
ORF prediction, protein extraction and redundancy filtering
A total of 201,849 complete ORFs were identified across the transcriptome. Corresponding nucleotide and amino acid sequences were successfully extracted. Clustering of the 201,849 predicted ORFs using CD-HIT resulted in 175,696 non-redundant protein sequences. This reduction highlights the presence of highly similar or identical ORFs in the transcriptome, and the filtering process ensured a high-confidence dataset for functional and comparative analyses.
Identification of putative toxins
In total, 2,965 high-confidence BLAST hits were retrieved, corresponding to the same number of unique ORFs, highlighting the effectiveness of the homology-based approach.
Signal peptide and transmembrane domain prediction
SignalP analysis revealed a subset of proteins containing N-terminal signal peptides, indicating their potential for secretion. A total of 2504 sequences were predicted to contain signal peptides, highlighting their likelihood of being secreted proteins. The TMHMM analysis showed that 139,162 sequences lacked any predicted transmembrane helices (TMHs = 0), 32,642 had a single TMH, 2645 had two TMHs, and 1247 sequences contained more than two TMHs.
Functional domain annotation
Pfam domain analysis enabled the identification of conserved protein domains across the entire set of clustered ORFs. By applying a strict e-value threshold of 1e-5, only high-confidence domain matches were retained. The inclusion of multiple domains per ORF where applicable, provided deeper insight into potential toxin-related activities. In total, 30,008 ORFs were found to contain at least one Pfam domain passing the defined threshold, highlighting the prevalence of conserved functional elements across a substantial portion of the dataset. Among the identified domains, several were notably frequent, including PF00537 (201 occurrences), PF05294 (74 occurrences), and PF00451 (51 occurrences), suggesting potential roles in functional specialization or toxin-related mechanisms.
Disulfide bond potential profiling
Based on cysteine content, 99,756 sequences contained only 0 or 1 cysteine residue, 71,094 had between 2 and 8 cysteines, and 4846 sequences contained 9 or more cysteines. This distribution provides a basis for identifying proteins with potential disulfide-rich architectures.
Physicochemical property annotation results
A total of 175,696 ORF-derived protein sequences were processed through the Biopython ProtParam pipeline. After computing the instability index for each sequence, 79,442 proteins (45.2%) were classified as STABLE (instability index < 40), while 96,113 proteins (54.8%) were classified as UNSTABLE. For 141 sequences (0.1%), no stability label could be assigned; these sequences contained non-standard or unresolved amino acids that precluded calculation of their physicochemical parameters.
Analysis of GRAVY scores across the dataset revealed that 103,703 proteins (59.1%) had a positive GRAVY value (indicating overall hydrophobic character), whereas 71,849 proteins (40.9%) exhibited negative GRAVY values (indicating overall hydrophilic character). The same 141 sequences lacking stability labels also had no GRAVY values recorded for the reasons noted above.
Functional domain validation via InterProScan
To refine the functional annotation of the BLAST-validated putative toxin proteins, domain-level analysis was performed using InterProScan. Out of the analyzed open reading frames (ORFs), several conserved functional domains were identified with high confidence, either by meeting the E-value threshold (≤ 1e-5) or being marked as trusted (T) by InterProScan. Three distinct InterPro domain identifiers were frequently observed among the toxin candidate sequences. The most prevalent domain was IPR036574, associated with 351 ORFs. This was followed by IPR003614 (144 ORFs) and IPR018218 (101 ORFs). All domain assignments were linked to their respective ORFs and stored in a structured SQLite database along with associated metadata, including functional descriptions, prediction methods, and GO annotations.
Multi-criteria classification of putative toxins
Qualitative expression of putative toxin types across all groups, including sodium, potassium, chloride channel toxins, and other categories, based on BLAST results or domain identification, is summarized in Fig. 3. As shown, the highest proportion of toxins corresponds to sodium channel toxins.

Total number of identified toxins categorized according to their putative functional roles.
Multi-criteria classification of putative toxins—Results for group A
Out of a total of 269 ORFs that exhibited ≥ 80% identity to known toxins, 192 ORFs fulfilled all predefined criteria for classification into Group A – High-Confidence Toxins. These candidates demonstrated strong sequence similarity, presence of toxin-associated domains, a predicted signal peptide, appropriate sequence length (≤ 300 amino acids), and more than one cysteine residue. The convergence of these features indicates a high likelihood of conserved toxin function. Figure 4 summarizes the toxin composition in Group A.

Distribution of predicted toxins across all groups based on toxin type.
The remaining 77 ORFs, while meeting the sequence similarity threshold, failed to satisfy one or more of the additional toxin-associated criteria. Although excluded from Group A, these ORFs remain potential toxin candidates and may represent non-canonical or divergent toxin variants that warrant further investigation. Three representative ORFs from Group A were selected for downstream functional analysis (Table 2).
Multi-criteria classification of putative Toxins – Results for group B
A total of 2,696 ORFs with BLASTP identity greater than 0% and less than 80% to known toxins were evaluated for classification into Group B – Moderate-Homology Candidates. Of these, 163 ORFs met all defined criteria, including the presence of toxin-associated domains (Pfam or InterProScan), a predicted signal peptide, suitable protein length (≤ 300 amino acids), and more than one cysteine residue. These sequences likely represent divergent homologs of known toxins or functionally convergent proteins with similar biochemical properties. Figure 4 shows the toxin composition in Group B. The remaining 2533 ORFs, although falling within the same BLAST identity range, lacked one or more of the additional required features. While excluded from Group B, these sequences remain potential toxin candidates and may be prioritized for further investigation as part of exploratory or hypothesis-generating studies. Three representative ORFs from Group B were selected for downstream analysis (Table 3).
Multi-criteria classification of putative Toxins – Results for group C
Group C encompassed toxin candidates that did not exhibit significant BLASTP similarity to known toxins but were retained due to the presence of toxin-related Pfam and InterPro domains, as well as structural features associated with toxic functions. From the full dataset, 1,060 ORFs passed the initial filtering criteria and were subjected to domain analysis using InterProScan. Of these, 211 sequences returned domain annotation results with InterPro ID. Comparison of these results with a curated list of toxin-related InterPro and Pfam IDs—retrieved via automated API queries—revealed that 23 sequences contained domains with known toxin associations. These sequences were thus considered putative toxins. The distribution of these putative toxins in Group C is shown in Fig. 4. Three representative ORFs from Group C were selected for downstream analysis (Table 4). The identification of Group C highlights the pipeline’s capability to uncover novel or under-characterized toxins that deviate from canonical sequence patterns. Such sequences are particularly valuable for evolutionary studies and for broadening our understanding of structurally diverse toxin architectures.
Comprehensive information for all nine selected candidates, including their full annotation details from the internal database, is provided in the Supplementary Table 2.
Discussion
This study presents a modular bioinformatics pipeline for de novo toxin discovery in non-model organisms, addressing key limitations of existing tools such as DeTox27 and ToxCodAn28, with improved accuracy, flexibility, and interpretability across diverse species.
We used rnaSPAdes for de novo transcriptome assembly due to its superior recovery of full-length and low-abundance transcripts compared to alternatives like Trinity, which is crucial for toxin mining, where such transcripts are often rare29. Unlike deterministic pipelines such as DeTox, we emphasize data-specific optimization, with read quality assessment, trimming, and post-assembly validation (BUSCO, read mapping)14,15. As these steps are data-driven and require researcher oversight, we deliberately avoided full automation.
Instead of incomplete manually compiled toxin lists, we programmatically retrieved UniProt KW-0800 entries (reviewed and unreviewed), enabling updatable, name-independent reference datasets. Homology searches (BLAST, e-value ≤ 1e-5) retained only the top hit per ORF to reduce redundancy, while integrating species metadata in a SQLite database to trace taxonomic origin and exclude non-target matches19,20.
Our pipeline incorporates SignalP 6.0, which detected all known signal peptide classes across taxa, aiding the identification of secreted toxin candidates30,31. TMHMM analysis complemented SignalP by detecting membrane-associated proteins, helping to distinguish truly secreted toxins from those retained in the membrane. Rather than serving as a strict filter, these predictions provided valuable biological context, informing likely localization and mode of action. Integrating signal peptide and transmembrane domain data significantly improved toxin identification accuracy and—depending on species context—facilitated the separation of toxins from non-toxic proteins32.
Unlike methods such as Venomix33, we applied functional domain annotation using InterProScan to provide more precise biological context for interpreting toxin candidates. Functional domain annotation via InterProScan enriched BLAST-validated candidates with structural context, retaining multiple domain matches for classification. Sequences lacking BLAST hits but positive for signal peptides and short ORFs were also scanned, balancing computational efficiency with depth. This guided approach produced curated, traceable toxin profiles, unlike uniform scans in DeTox.
Cysteine content analysis enables rapid screening and prioritization of cysteine-rich proteins for structural and functional studies. While it does not predict actual disulfide bond patterns, it highlights proteins—especially those with high cysteine density—as strong candidates for extracellular or structurally stable roles34, suitable for structural modeling or experimental validation.
Only complete ATG-initiated ORFs predicted by ORFipy v0.0.4 were retained to avoid ambiguous partial sequences lacking functional motifs. Focusing exclusively on full-length ORFs enhanced accuracy in domain prediction, toxin classification, and experimental validation, whereas less curated pipelines such as DeTox often retain partial sequences, introducing annotation noise, false positives, and prioritization errors. The optimal start codon, however, may vary depending on the organism or research context35.
Strict CD-HIT clustering at 100% identity and coverage was applied to remove redundant isoforms and assembly artifacts. The -aS 1.0 parameter enforced full-length alignment coverage for the shorter sequence in each pairwise comparison, preventing partial alignments from being incorrectly clustered and avoiding inappropriate collapsing of distinct protein variants—particularly important in toxin discovery, where minor sequence differences may correspond to distinct functional profiles. Without this parameter, sequences with high identity over only partial regions could cluster together, potentially masking biologically relevant diversity.
All annotations—sequence, structure, and physicochemical properties—were merged in a relational database for direct exploration, avoiding fragmented outputs seen in older pipelines27,33. Unlike pipelines such as DeTox and Venomix, the present approach integrates physicochemical properties (including stability, hydropathy, and charge) to deliver functional insights beyond structural motifs and sequence similarity.
The pipeline concludes with a three-tier classification framework (Groups A–C) that integrates sequence homology, domain architecture, signal peptide presence, and cysteine content, eliminating the need for extensive manual inspection. Thresholds and criteria can be adjusted depending on organism-specific biology or research goals (e.g., disabling signal peptide filtering for membrane toxin studies). By combining homology, structural domains, and functional signals, this strategy maximizes sensitivity and novelty detection, enabling toxin identification across broad evolutionary distances. The 80% sequence-identity threshold was selected based on evidence that proteins exceeding this similarity have a 95–97% probability of sharing Gene Ontology (GO) Biological Process annotations36.
Group A contains high-confidence toxins that meet strict criteria, including BLASTP identity ≥ 80% to known toxins, presence of toxin-related domains, signal peptides, appropriate length (≤ 300 amino acids, chosen because nearly all characterized scorpion toxins fall below this length)2, and cysteine content. Most members are well-characterized and conserved, representing reliably annotated toxins suitable for benchmarking.
Group B represents functionally relevant toxin-like proteins with moderate sequence similarity, including divergent homologs or convergent analogs not easily detected by homology alone. Integration of structural and physicochemical features enhances detection. Optional filters for Groups A and B (as described in the ToxIR README) include:
-
no_sp (ignore signal peptide requirement).
-
max_len N (maximum sequence length, default: 300).
-
cys_gt N (minimum cysteine count).
-
cys_lt N (maximum cysteine count).
Group C highlights the domain-centric strategy of the pipeline, enabling the detection of distant homologs or poorly characterized proteins that lack detectable sequence-level similarity. This group comprises candidate toxins that are likely novel or highly divergent, identified based on conserved domain architecture and hallmark sequence features. It captures cryptic, lineage-specific, or rapidly evolving toxins, including taxon-specific variants and previously uncharacterized secreted peptides. Supported by comprehensive domain resources such as Pfam and InterPro, this approach reveals hidden toxin candidates by leveraging domain-based and sequence-informed signatures beyond conventional sequence similarity–based methods.
Automated scripts retrieved toxin-associated domain IDs from Pfam and InterPro APIs, ensuring reproducible and up-to-date annotations without manual bias—unlike DeTox, where manual annotation introduced subjectivity, errors, and limited reusability. This automation improves accuracy, reproducibility, and scalability in toxin identification, addressing a major limitation of earlier methods in which researchers faced large, ambiguous domain lists without clear guidance on toxin-related IDs. By generating targeted, current domain annotations, the pipeline establishes a standardized and biologically meaningful workflow for toxin discovery. While Kunitz-type proteins were included in the unified database and correctly detected by BLAST, they were excluded from domain extraction and classification since Kunitz domains are not strictly defined as toxins. Nevertheless, the pipeline can be extended to include inhibitors such as Kunitz by adding their domain IDs during grouping; these proteins remain available in the comprehensive database for manual review if needed.
In this study, unlike methods such as Venomix and DeTox, expression-based filters (e.g., TPM or FPKM) were excluded from toxin candidate selection due to their limited accuracy in de novo transcriptome assemblies without reference genomes, isoform ambiguity, and assembly fragmentation37, as well as the risk of discarding low-abundance but biologically important toxins38. Since some venom peptides with low expression have critical roles, strict cutoffs could eliminate them. Instead, the discovery strategy was sequence- and feature-based to maintain sensitivity for novel toxin detection.
A limitation of this framework is its reliance on the completeness and annotation depth of databases such as UniProt, Pfam, and InterProScan. Although multi-layer filtering (sequence similarity, domain architecture, toxin-associated features) reduces this dependency, identification remains influenced by database coverage and accuracy. As these resources expand, particularly for under-characterized taxa and protein families, classification sensitivity and precision will improve. While this dependency is inherent to functional annotation pipelines, the present design mitigates its impact by integrating multiple annotation perspectives, reducing the chance of missing biologically meaningful toxins.
Although toxin candidates were classified into defined groups, some sequences—whether with high or moderate BLAST similarity (≥ 80% or < 80%) or toxin-like features such as signal peptides and cysteine richness—may lack recognizable domains in Pfam or InterProScan. These sequences, while outside strict group definitions, remain biologically significant. To avoid missing them, all annotated ORFs were stored in a comprehensive pre-classification SQLite database, enabling researchers to manually revisit sequences excluded from final tiers that may represent novel or lineage-specific toxins. Such ORFs warrant further study via experimental validation, structural modeling, or comparative analyses. This database extends toxin discovery beyond automated classification and supports hypothesis-driven exploration of atypical or borderline candidates.
The pipeline features a modular architecture, enabling automation and adaptation to diverse toxin discovery projects. While optimized for rnaSPAdes output, it can be readily applied to other transcriptome assemblies. Core components (ORF prediction, domain annotation, database integration) are reconfigurable to suit various organisms, datasets, and research goals. Critical steps such as quality control, assembly, and trimming should be dataset-specific, benefiting from manual oversight and parameter tuning. This flexibility supports both standardized and fine-tuned workflows, as well as future partial automation of repetitive tasks. Predictions require experimental validation (in vitro/in vivo) to confirm toxin activity, with computational analyses serving mainly for hypothesis generation and prioritization.
Designed for interpretability, the pipeline centralizes, links, and organizes annotations into an integrated system—ready for analysis, visualization, and publication—avoiding the fragmented outputs typical of some existing methods.
Supplementary data statement
Supplementary Table 1 summarizes the command-line parameters and tool versions used in the ToxIR pipeline. Supplementary Table 2 provides an overview of the ToxIR pipeline outputs for the selected sequences.
Data availability
All raw RNA-seq data generated in this study have been deposited in the National Centre for Biotechnology Information (NCBI) under BioProject accession number PRJNA1293699 and BioSample accession number SAMN50032453. The corresponding Sequence Read Archive (SRA) dataset has been submitted but remains in private status; it will be released to qualified researchers upon reasonable request accompanied by a justified rationale. In addition, nine representative sequences are available in GenBank under the following accession numbers: PV928892, PV926128, PV928893, PV928894, PV928895, PV928896, PV928897, PV928898, and PV928899. All scripts required to run the ToxIR pipeline, along with detailed step-by-step usage instructions, are provided in the README file available at the following DOI: [https://doi.org/10.5281/zenodo.17931954].
References
Gonçalves, C., Cabral, M., Alves de Matos, I., Grosso, A. P., Costa, P. M. & A. R. & Transcriptome profiling of the posterior salivary glands of the cuttlefish sepia officinalis from the Portuguese West Coast. Front. Mar. Sci. 11, 1362824 (2024).
Xia, Z., He, D., Wu, Y., Kwok, H. F. & Cao, Z. Scorpion venom peptides: Molecular diversity, structural characteristics, and therapeutic use from channelopathies to viral infections and cancers. Pharmacol. Res. 197, 106978 (2023).
Ortiz, E., Gurrola, G. B., Schwartz, E. F. & Possani, L. D. Scorpion venom components as potential candidates for drug development. Toxicon 93, 125–135 (2015).
King, G. Venoms To Drugs: Venom as a Source for the Development of Human Therapeutics (Royal Society of Chemistry, 2015).
Petricevich, V. L., Navarro, L. B. & Possani, L. D. Therapeutic use of Scorpion venom. Mol. Asp Inflamm. 9, 209–231 (2013).
Dehghani, R. & Kassiri, H. Geographical distribution of Scorpion odontobuthus doriae in Isfahan Province, central Iran. J. Arthropod-Borne Dis. 11, 433 (2017).
Modahl, C. M., Brahma, R. K., Koh, C. Y., Shioi, N. & Kini, R. M. Omics technologies for profiling toxin diversity and evolution in snake venom: Impacts on the discovery of therapeutic and diagnostic agents. Annu. Rev. Anim. Biosci. 8, 91–116 (2020).
Roman-Ramos, H. & Ho, P. L. Current technologies in snake venom analysis and applications. Toxins 16, 458 (2024).
Von Reumont, B. M. Studying smaller and neglected organisms in modern evolutionary venomics implementing RNASeq (transcriptomics)—a critical guide. Toxins 10, 292 (2018).
von Reumont, B. M. et al. Modern venomics—Current insights, novel methods, and future perspectives in biological and applied animal venom research. GigaScience 11, giac048 (2022).
Andrews, S. & FastQC A Quality Control Tool for High Throughput Sequence Data. (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 4 May 2023).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Bushmanova, E., Antipov, D., Lapidus, A. & Prjibelski, A. D. rnaSPAdes: A de novo transcriptome assembler and its application to RNA-Seq data. GigaScience 8, giz100 (2019).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: Novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol. Biol. Evol. 38, 4647–4654 (2021).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with bowtie 2. Nat. Methods. 9, 357–359 (2012).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075 (2013).
Singh, U. & Wurtele, E. S. Orfipy: A fast and flexible tool for extracting ORFs. Bioinformatics 37, 3019–3020 (2021).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
UniProt. The universal protein knowledgebase in 2025. Nucleic Acids Res. 53, D609–D617 (2025).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Teufel, F. et al. SignalP 6.0 predicts all five types of signal peptides using protein Language models. Nat. Biotechnol. 40, 1023–1025 (2022).
Krogh, A., Larsson, B., Von Heijne, G. & Sonnhammer, E. L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 305, 567–580 (2001).
Eddy, S. R. Accelerated profile HMM searches. PLoS Comput. Biol. 7, e1002195 (2011).
Cock, P. J. et al. Biopython: Freely available python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422 (2009).
Jones, P. et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014).
Blum, M. et al. The interpro protein families and domains database: 20 years on. Nucleic Acids Res. 49, D344–D354 (2021).
Ringeval, A. et al. DeTox: A pipeline for the detection of toxins in venomous organisms. Brief. Bioinform. 25, bbae094 (2024).
Nachtigall, P. G. et al. ToxCodAn: A new toxin annotator and guide to venom gland transcriptomics. Brief. Bioinform. 22, bbab095 (2021).
Espín-Angulo, J. & Vela, D. Computational modeling of Low-Abundance proteins in venom gland transcriptomes: Bothrops Asper and Bothrops Jararaca. Toxins 17, 262 (2025).
Popoff, M. R. Overview of bacterial protein toxins from pathogenic bacteria: Mode of action and insights into evolution. Toxins 16, 182 (2024).
Jenner, R. A., Casewell, N. R. & Undheim, E. A. What is animal venom? Rethinking a manipulative weapon. Trends Ecol. Evol. (2025).
Nonin-Lecomte, S., Fermon, L. & Felden, B. Pinel-Marie, M.-L. Bacterial type I toxins: Folding and membrane interactions. Toxins 13, 490 (2021).
Macrander, J., Panda, J., Janies, D., Daly, M. & Reitzel, A. M. Venomix: A simple bioinformatic pipeline for identifying and characterizing toxin gene candidates from transcriptomic data. PeerJ 6, e5361 (2018).
Cheek, S., Krishna, S. S. & Grishin, N. V. Structural classification of small, disulfide-rich protein domains. J. Mol. Biol. 359, 215–237 (2006).
Asano, K. Why is start codon selection so precise in eukaryotes? Translation 2, e28387 (2014).
Joshi, T. & Xu, D. Quantitative assessment of relationship between sequence similarity and function similarity. BMC Genom. 8, 222 (2007).
Hsieh, P., Oyang, Y. & Chen, C. Effect of de Novo transcriptome assembly on transcript quantification. Sci. Rep. 9 (1), 8304 (2019).
Tan, C. H., Tan, K. Y., Fung, S. Y. & Tan, N. H. Venom-gland transcriptome and venom proteome of the Malaysian King Cobra (Ophiophagus hannah). BMC Genom. 16, 687 (2015).
Acknowledgements
The authors gratefully acknowledge the laboratory and financial support provided by Shahid Chamran University of Ahvaz. (Grant No: SCU.SB1403.47678).
Funding
This research received a specific grant from the Shahid Chamran University of Ahvaz (Grant No: SCU.SB1403.47678).
Author information
Authors and Affiliations
Contributions
M.E. and M.N.S. contributed equally to the conception and design of the study, performed the experiments, analyzed the data, and co-wrote the manuscript. Both authors have read and approved the final version of the manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This study involved only Odontobuthus doriae scorpions and did not require institutional ethical approval for human participation. All procedures for scorpion collection, electroshock milking, and tissue preservation were conducted in accordance with the World Health Organization (WHO) guidelines for the ethical handling of animals and were supervised by Shahid Chamran University of Ahvaz, Iran. The source data were generated under the ethical approval code IRAJUMS.REC.1396.916.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ebadi, M., Soorki, M.N. ToxIR: an accurate RNA-seq pipeline for high-precision toxin transcriptome profiling, validated in odontobuthus doriae venom glands. Sci Rep 16, 3529 (2026). https://doi.org/10.1038/s41598-025-33632-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-33632-0
