
Abstract
The emergence and expansion of methicillin-resistant Staphylococcus aureus (MRSA) clones in healthcare facilities pose a significant public health concern due to their adaptability and resistance to commonly used antibiotics. In this study, the genomic and epidemiological characteristics of 81 MRSA isolates collected between February and June 2022 were analyzed. ST5 (25.9%) and ST6 (19.7%) emerged as the dominant sequence types, collectively accounting for 45.6% of infections. Whole-genome sequencing and phenotypic analyses revealed that ST5 clone exhibited a broader multidrug-resistant profile compared to ST6 clone, with higher prevalence of β-lactam, tetracycline, and trimethoprim resistance. ST6 clone showed more variable resistance, including aminoglycoside and macrolide genes, suggesting a possible community origin before adaptation to hospital settings. Phylogenetic analysis demonstrated ongoing microevolution within Clonal Complex 5 (CC5), including the identification of novel single-locus variants such as ST8111. Correlation analyses highlighted significant associations between key resistance genes and their phenotypic profiles, reflecting complex co-resistance mechanisms. Additionally, virulence profiling revealed that ST5 uniquely carried tsst-1, whereas PVL genes were absent in both ST5 and ST6 and appeared only in a small subset of other MRSA clones. The emergence of highly resistant clones like ST672 underscores the potential concern for sustained genomic surveillance. The observed clonal shift from ST239 to ST5 and ST6 signals a dynamic MRSA landscape in Saudi Arabia, emphasizing the need for integrated molecular epidemiology and targeted infection control strategies.
Similar content being viewed by others


Introduction
Methicillin-resistant Staphylococcus aureus (MRSA) is a highly adaptable, pathogenic bacterium that poses a serious public health risk1. It has the potential to cause severe infections and is resistant to common antibiotics, making treatment more challenging2. The emergence and expansion of MRSA clones in healthcare facilities underscores the urgent need for comprehensive epidemiological surveillance and intervention strategies3. In Saudi Arabia, studies indicate a recent concerning increase in MRSA diversity, with 18 distinct clonal complexes (CC) identified among MRSA isolates in Riyadh4. These include notable new strains such as CC152 and CC3614. Previous analyses further elucidated the prevalence of established clonal complexes like CC80, CC6, and CC5, which were predominant from 2009 to 2015, alongside the emergence of novel sequence types (STs) such as ST8-MRSA-IV and ST72-MRSA-IV5.
The prevalence of MRSA in healthcare settings remains critical, with a recent study reporting a 45.4% MRSA prevalence among S. aureus cases in a maternity and children’s hospital6. Meanwhile another tertiary hospital reported a stable MRSA prevalence of approximately 41.2% during 2020, despite disruptions caused by the COVID-19 pandemic7. Such statistics emphasize the importance of continuous monitoring of MRSA within healthcare facilities to enhance patient safety and reduce infection rates. Moreover, the growing prevalence of community-associated MRSA ST5 and ST6 in Riyadh’s healthcare facilities raises significant concerns about patient-to-patient transmission and infection control8. Epidemiological data have revealed a high incidence of healthcare-associated infections (HAIs) involving MRSA, especially in intensive care units, prompting the need for targeted infection control measures9. Additionally, studies have highlighted the alarming prevalence of multidrug-resistant strains, particularly those harboring the Panton-Valentine leukocidin (PVL) gene, which is linked to severe clinical outcomes10,11.
Notably, the decline of the pandemic clone CC8/ST239-III, alongside the emergence of new clones, highlights a shifting resistance landscape that continues to pose a significant threat to public health12,13. Despite these developments, the genomic features, resistance mechanisms, and clonal relationships of the increasingly dominant ST5 and ST6 clones in Saudi Arabia remain poorly understood4,5,8. A clearer understanding of their evolutionary dynamics is essential for developing tailored infection control practices and antimicrobial stewardship efforts.
Previous investigations of MRSA in Saudi Arabia have reported a shifting epidemiology, where older clones such as ST239 and ST80 were historically dominant, while more recent studies have noted increasing detection of community associated lineages including ST5, ST6, ST22, and ST88 across different regions. However, national genomic surveillance remains limited, and most published datasets include small numbers of isolates, underscoring the need for broader genomic characterization to understand current MRSA population structure.
Considering the ongoing challenges posed by MRSA, this study aimed to investigate the emergence and expansion of MRSA ST5 and ST6 within hospitals in Riyadh. By elucidating the genetic characteristics and transmission dynamics of these prevalent strains, we aimed to contribute valuable insights that can effectively inform public health strategies and enhance patient outcomes in the ongoing fight against antibiotic-resistant pathogens.
Results
Epidemiological characteristics of MRSA isolates
All 81 MRSA isolates included in this study underwent phenotypic confirmation. Among these isolates, 55 were confirmed by VITEK2, with 54 testing positive on the cefoxitin screen and having an oxacillin MIC ≥ 4 mg/L, while one isolate was cefoxitin-negative with an oxacillin MIC of 0.5 mg/L. Additionally, 26 MRSA isolates were confirmed using BD Phoenix, all of which had cefoxitin MIC ≥ 8 mg/L and oxacillin MIC > 2 mg/L. The distribution of MRSA clones, stratified by age group and infection type, showed significant variations in sequence type prevalence (Table 1). The 31–50 age group had the highest number of MRSA infections, with ST5 and ST6 predominating in respiratory and wound infections. In the younger age group (0–12 years), ST5 and ST6 were notably prevalent in wound, systemic, and respiratory tract infections. The (0–1 years) and (31–50 years) age groups exhibited a diverse range of clones across various infection types, with notable representations of ST88 in the younger age group and ST22 in the older age group, along with prominent ST5, ST6 and other clone representations in different infection types. Statistical analysis indicated significant differences in the prevalence of MRSA clones across age groups (p < 0.05), highlighting the importance of demographic factors in understanding MRSA transmission dynamics.
Distribution of MRSA clones (MLST)
Multi locus sequence typing (MLST) analysis identified 19 distinct STs among the 81 MRSA isolates. The analysis showed that ST5 and ST6 were highly prevalent among MRSA isolates obtained during the study. ST5 accounted for 21/81 (25.9%), while ST6 contributed 16/81 (19.7%). The distribution of ST5 and ST6 among MRSA sequence types (p < 0.001) was significantly different from that of other MRSA clones. ST88 and ST22 accounted for 9/81 (11.1%) and 7/81 (8.6%), respectively. Other sequence types had a prevalence of 5/81 (6.1%), including ST672 and ST97, while ST30 and ST8 each represented 3/81 (3.7%). ST152 was identified in 2/81 isolates (2.4%), and the remaining sequence types appeared as singletons (Fig. 1).

Distribution of MRSA isolates by sequence type based on MLST analysis.
SCCmec and spa typing
The analysis of MRSA isolates revealed a diverse distribution of Staphylococcal cassette chromosome mec (SCCmec) types and Staphylococcal protein A (spa) types (Table 2). The most common SCCmec type was IVa, representing 51.2% (41/80). Within SCCmec IVa, the predominant STs were ST6, ST88, and ST22, which were mainly associated with spa types t304 and t690, indicating limited spa diversity within this SCCmec background. SCCmec type V was the second most frequent, accounting for 21.2% (17/80). This group showed greater spa variability, with ST672 commonly linked to t3841 and ST5 associated with t311. SCCmec type VI, which comprised 7.5% (6/80), included ST5 and ST30 and was characterized by spa types t688 and t018.
Less common SCCmec types included Vc (11/80, 13.7%) and IVc (4/80, 5%). Most SCCmec Vc isolates were ST5 and carried spa type t311, although one isolate lacked a resolvable spa type. One ST152 isolate remained unclassified for SCCmec. Overall, spa typing revealed moderate diversity across the MRSA population, with t304, t690, t311, and t3841 being the most common spa types. These spa types were distributed across wound, respiratory, and systemic specimens without a clear specimen-specific pattern, reflecting their circulation across multiple clinical sources.
Molecular relationships among MRSA clones (MST analysis)
The minimum spanning tree (MST) analysis of S. aureus STs based on MLST data, revealed the genetic relationships among 19 distinct S. aureus STs identified in 81 MRSA isolates (Fig. 2). The MST analysis identified ST5 and ST6 (CC5) as the most central and highly connected sequence types in the ST network, comprising 45.6% (37/81) of all MRSA isolates. ST149 differs from ST5 by a single allele (tpi), whereas ST6 differs from ST5 by two alleles (arcC and yqiL); therefore, ST149 is more closely related to ST5 than ST6 within the MRSA/CC5 STs. Meanwhile, ST8111, a newly emerging ST, differs from ST6 by a single allele (yqiL), indicating its close evolutionary relationship with ST6.

Minimum spanning tree (MST) showing clonal relationships among S. aureus sequence types (STs) based on MLST. Node sizes reflect isolate counts, colors indicate CCs, and numbers on lines represent allelic differences. The inset pie chart shows the distribution of isolates by CC.
The genomic relationships among MRSA isolates revealed distinct clonal lineages. ST88 (CC88) and ST22 (CC22) are the most common after ST5, differing by six alleles, indicating genetic divergence. Other notable lineages include ST97 (CC97), a livestock-associated clone, which clusters with ST8 (CC8) and shows moderate clustering with ST672 (CC361), differing by four alleles. Additionally, ST30 (CC30) was identified as a separate lineage, while ST8110, a newly emerged ST, and ST1930 are positioned outside major clonal complexes. These findings highlight the genetic diversity within the MRSA population.
Genotypic antimicrobial resistance and virulence profiles
Both ST5 and ST6 demonstrated high resistance to methicillin, with ST5 exhibiting a broader antimicrobial resistance profile (Table 3). The presence of resistance genes such as mecA and blaZ was more frequent in ST5, contributing to its increased resistance compared to that of ST6. Statistical analysis revealed significant differences between these STs in resistance to non-beta-lactam antibiotics (p < 0.01).
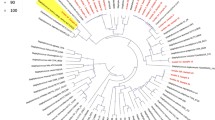
The phylogenetic analysis of 81 MRSA isolates reveals a diverse distribution of antibiotic resistance genes (ARGs) and virulence factors (Fig. 3). Key resistance determinants include mecA (conferring methicillin resistance), blaZ (β-lactam resistance), ermC (macrolide resistance), and tetK (tetracycline resistance), among others like aph(3’)-IIIa, aph(2’’)-Ih, ant(4’)-Ia, mphC, msrA, tetM, tet38, dfrG, fusC, fosB-saur, fexA, and lnuA. Virulence genes such as tsst-1(toxic shock syndrome gene) and lukS-PV/lukF-PV show variable presence among the STs. The PVL genes (lukS-PV/lukF-PV) were detected in only three isolates belonging to non-CC5 lineages such as ST88 and ST30. The tsst-1 gene was identified in five isolates, predominantly within ST5. In contrast, adhesion-related genes such as fnbA were present in nearly all isolates, while enterotoxin genes (sea and seb) and hemolysin genes (hla and hlb) showed moderate to low frequencies across different STs. Overall, virulence gene carriage was limited compared to antibiotic resistance determinants.

Phylogenetic tree of 81 MRSA isolates showing the relationship between sequence types (STs), clonal complexes (CCs), and the distribution of antibiotic resistance and virulence genes. Isolates are labeled (C1–C84). The presence of resistance genes and virulence factors is indicated alongside each isolate.
The analysis also revealed that certain ARGs were associated with specific STs. ST5 isolates consistently carried mecA and blaZ, along with frequent occurrences of ermC, tetM, and aminoglycoside resistance genes (aph(3’)-IIIa, aph(2’’)-Ih, ant(4’)-Ia). This suggests that ST5 represents a multidrug-resistant lineage with strong resistance to β-lactams, macrolides, and aminoglycosides. In contrast, ST6 isolates also carried mecA and blaZ but exhibited a lower prevalence of tetM. Despite this, they maintained resistance genes against β-lactams and aminoglycosides. Unlike ST5, ST6 displayed greater heterogeneity in the presence of ARGs; certain isolates, such as those belonging to ST22 and ST30, lacked some resistance genes, and the frequency of fusC and fosB-saur was reduced in these clones. Notably, the ST672 clone appeared to have a particularly high prevalence of resistance genes, harboring multiple ARGs, including mecA, blaZ, ermC, tetK, fusC, and fosB-saur. This suggests that ST672 may represent an epidemiologically relevant clone with extensive antibiotic resistance.
Correlation between ARGs and drug resistance
The correlation analysis between antimicrobial resistance genes and their corresponding antibiotics in MRSA isolates exhibited varying degrees of association (Fig. 4). For example, strong positive correlations were observed between mecA and penicillin (PEN), cefoxitin (FOX), and oxacillin (OXA) (R = 1.0), consistent with its well-established role in β-lactam resistance. Additionally, ermC demonstrated a moderate correlation with erythromycin (ERY) (R = 0.52) and clindamycin (CLI) (R = 0.49), reinforcing its involvement in macrolide-lincosamide resistance. A high correlation was also observed between dfrG and trimethoprim-sulfamethoxazole (SXT) (R = 0.59), consistent with its role in trimethoprim resistance. Moreover, aph exhibited a strong correlation with gentamicin (GEN) (R = 0.75), indicating aminoglycoside resistance. Another notable correlation was observed between blaZ and SXT (R = 0.45), suggesting potential co-resistance mechanisms. The correlation matrix further revealed possible co-resistance patterns, such as the association between mecA and multiple β-lactam antibiotics, indicating that the presence of mecA may contribute towards a broader resistance profile. Overall, this study indicates that the genetic determinants of antibiotic resistance in multidrug-resistant MRSA isolates are suggested by statistically significant associations (p < 0.05) between the identified genetic markers and resistance phenotypes.

Correlation matrix illustrating the relationships between antimicrobial resistance genes (ARGs) and corresponding antibiotics in MRSA isolates. The color gradient and numerical values represent the strength and direction of correlations, providing insights into co-resistance patterns and the genetic basis of antimicrobial resistance.
Discussion
This study provides a comprehensive genomic and phenotypic characterization of MRSA isolates circulating in Riyadh healthcare facilities, revealing important insights into the evolving epidemiology of antimicrobial resistance in Saudi Arabia. The rise of ST5 and ST6 highlights their strong adaptation to hospital environments under antibiotic pressure and improved adaptation mechanisms. This represents a notable transition from previous reports of ST239-III dominance in Saudi hospitals14 and mirrors global trends where traditional pandemic lineages are being replaced by genetically distinct clones15.
MRSA clones ST5 and ST6 are globally significant due to their multidrug resistance and virulence. Originating from the acquisition of SCCmec elements (types I–III) carrying the mecA gene (conferring β-lactam resistance), these clones exhibited broad antibiotic resistance via genes such as ermB (macrolide-lincosamide resistance), msrA (macrolide efflux), blaZ (penicillin resistance), and tetM (tetracycline resistance), often acquired through horizontal gene transfer16,17,18. Their dissemination is achieved through key virulence factors, including PVL (a cytolytic toxin that causes necrotizing infections), TSST-1 (induction of systemic shock), fibronectin-binding protein (FnBP; mediates host cell adhesion), and enterotoxins (SEA/SEB; triggers food poisoning and immune hyperactivation)19. ST5 is predominantly healthcare-associated, spreading via hospital outbreaks due to its strong biofilm formation and resistance to disinfectants, while ST6 is linked to community transmission in crowded settings20. Molecular typing methods (MLST, PFGE, spa typing, WGS) revealed their clonal evolution and zoonotic potential, emphasizing the need for integrated One Health strategies21.
The genomic architecture of these predominant clones in this study reveals distinct evolutionary pathways that likely reflect different adaptation strategies to hospital environments. ST5’s multidrug resistance suggests high selective pressure, reinforcing its role as a dominant healthcare clone. This multidrug-resistant phenotype aligns with ST-5’s well-established characterization as a pandemic HA-MRSA clone22. The significant link between ST5 and hospital-acquired infections (OR: 3.2 for isolates with ≥ 4 resistance genes) reflects its evolutionary adaptation to hospital settings, where antibiotic pressure and vulnerable patients favor multidrug-resistant strains23. This finding is consistent with reports from other Middle Eastern countries documenting the increasing prevalence of ST5 in critical care settings24.
In contrast to ST5 clone, ST6 demonstrated a more variable resistance pattern that suggests a different evolutionary history. While like ST5 clone, universal mecA presence (100%) is maintained, it showed significantly lower prevalence of blaZ (18.7% compared to 90.2% in ST5) but a higher prevalence of aminoglycoside resistance genes. This resistance profile, combined with its frequent detection in pediatric and general-ward patients, suggests that ST6 may have originated in community settings before adapting to hospital environments; a phenomenon increasingly recognized worldwide25. Higher fosB-Saur in ST6 may reflect local fosfomycin use and clone-specific adaptation26.
The microevolution of these clones within CC5 was highlighted by the MST analysis. ST149 and ST8111 indicate ongoing microevolution, signaling potential future emerging variants of interest. These findings are supported by similar reports from other Gulf Cooperation Council (GCC) countries documenting the emergence of region-specific variants through point mutations and genetic recombination5,27,28. The detection of these variants is particularly important as they may represent early stages in the development of new epidemic clones with potentially enhanced virulence or resistance properties.
Many genotype-phenotype associations with clinical and epidemiological consequences were identified via the correlation matrix analysis. The correlations between mecA and resistance β-lactams (R = 1.0 for all) confirms this genetic determinant’s pivotal involvement in MRSA development and supports its ongoing application as a major diagnostic marker29. However, some discordance was observed between resistance patterns that warrants further investigation. Most notably, while tet 38 was present in all the MRSA isolates, its correlation with tetracycline resistance was weaker than expected, suggesting the involvement of additional regulatory mechanisms or alternative resistance pathways that may modulate phenotypic expression.
The resistance gene profiles showed several clone-specific patterns with important clinical implications. ST5 isolates demonstrated a high prevalence of ermC (65.8%), which showed moderate correlation with resistance to erythromycin (R = 0.52) and clindamycin (R = 0.49). This was particularly relevant for antimicrobial stewardship programs, as these antibiotics are commonly used for MRSA treatment30. The co-occurrence of ermC and msrA in certain ST6 isolates may have contributed to the considerably lower correlation values compared to those reported from European populations (typically R > 0.7), which can make resistance predictions more difficult31,32. Another finding was the strong association between aph genes and gentamicin resistance (R = 0.75), which exceeded correlation values typically observed in Western MRSA populations (R ~ 0.6) and likely reflects region-specific aminoglycoside usage patterns33,34.
One of the most important results of this study was the moderate correlation (R = 0.45) between blaZ and trimethoprim-sulfamethoxazole resistance. While these resistance mechanisms are not biochemically linked, this association may indicate co-localization of resistance determinants on mobile genetic elements, as previously observed in Middle Eastern EMRSA-15 isolates5,35. Such arrangements can facilitate the co-selection of resistance traits under antibiotic pressure, potentially accelerating the development of multidrug resistance36. This phenomenon needs attention in antimicrobial stewardship programs, as it suggests that use of one antibiotic class could simultaneously select for resistance to unrelated agents.
The virulence gene distribution patterns revealed important differences between the predominant clones. While ST5 exclusively carried tsst-1, both ST5 and ST6 lacked the PVL genes that are characteristic of many community-associated MRSA strains. This contrasts sharply with the PVL positivity rates observed in ST88 (20%) and ST30 (33%) isolates in our study and aligns with previous reports from Saudi pediatric populations6. The presence of tsst-1 gene in ST5 is particularly concerning given this clone’s association with critical infections, as the toxin can cause life-threatening toxic shock syndrome in vulnerable patients. These findings suggest that different MRSA clones may occupy different ecological niches within healthcare facilities, with varying pathogenic potentials that could influence patient health outcomes.
On the other hand, the emergence of recent clones, such as ST672, as clones of potential concerns warrant special attention. For example, MRSA ST672 isolates carried an exceptionally high number of resistance genes (seven or more per isolate), including blaZ, ermC, and tet(K), demonstrating a resemblance to multidrug-resistant strains recently reported from India37. While currently representing a small proportion of isolates (6.1%), the genetic flexibility demonstrated by this clone suggests it may have significant potential for future expansion, particularly if selective antibiotic pressures continue.
Overall, our findings highlight key implications for MRSA control in Saudi healthcare settings. The dominance of multidrug-resistant ST5 in hospitals calls for enhanced surveillance and targeted precautions, while ST6’s presence in both hospital and community settings suggests that integrated transmission tracking is required. High antibiotic resistance genes such as, blaZ, tet, and fosB-Saur prevalence are examples of region-specific resistance patterns that suggest the necessity of local antibiotic management. Emerging clones (ST672) and novel variations (ST8111) highlight the importance of continuous genomic monitoring. One limitation was the study’s focus on a single region (Riyadh city hospitals, and regional laboratories) and potential undetected resistance mechanisms. Additionally, the study design was cross-sectional, and longitudinal genomic surveillance would be needed to confirm clonal dynamics over time. Further research is needed to clarify transmission routes and guide targeted interventions against clinically significant clones such as ST5 and ST6.
Conclusion
This study provides comprehensive genomic and epidemiological insights into the emergence and dominance of MRSA ST5 and ST6 in Riyadh’s healthcare facilities. Key findings include the high prevalence of ST5 in ICUs and its multidrug-resistant phenotype, the broader dissemination of ST6 in general hospital wards, and the active clonal diversification within CC5. Strong gene-to-phenotype correlations highlight the complexity of co-resistance patterns, complicating treatment and containment efforts. The evolutionary replacement of older clones like ST239 with more adaptive ones such as ST5 and ST6 highlights the evolving epidemiological landscape of MRSA. Future directions should focus on long term genomic surveillance, clone-specific infection control interventions, and the identification of environmental reservoirs to reduce transmission. Integrated strategies leveraging molecular epidemiology, clinical data, and public health measures are essential for mitigating the burden of MRSA in Saudi Arabia and beyond.
Methods
Sample collection and isolation
Between February and June 2022, 81 non-duplicate MRSA isolates were collected from human specimens at the Riyadh Regional Laboratory and Blood Bank.
Each isolate originated from a unique clinical specimen obtained from a different patient; no repeat samples were included. The specimens included wound swabs, blood cultures, respiratory tract samples, and sterile body fluids. Patients included both males and females across pediatric and adult age groups who presented for routine diagnostic testing. The identification of MRSA was performed using the VITEK2 system (bioMérieux, Marcy-L’Etoile, France) and BD Phoenix (BD Diagnostics, Franklin Lakes, NJ, USA). All samples were subjected to microbiological analyses within 24 h of collection.
Identification and confirmation of S. aureus and MRSA
The S. aureus isolates were cultured on self-prepared sheep blood agar plates using Columbia blood agar powder (OXOID, Basingstoke, UK), and the clinically isolated samples showing β-hemolysis were compared with the ATCC 25,923 reference strain (ATCC, Manassas, VA, USA). Confirmatory tests were conducted on isolated colonies using mannitol salt agar (MSA; Neogen, Lansing, MI, USA), Gram staining, catalase, and coagulase tests (PROLEX™, Pro-Lab Diagnostics, Richmond hill, ON, Canada). MRSA detection was performed using Harlequin MRSA Chromogenic Agar supplemented with cefoxitin (Neogen). Positive identification was indicated by the presence of blue colonies, which are characteristic of MRSA, following 24 h of incubation. Following initial confirmation of S. aureus and MRSA identity through culture-based methods, isolates underwent molecular testing to detect the mecA gene, thereby ensuring both phenotypic and genotypic validation of resistance.
PCR-based mecA detection
DNA was extracted from pure bacterial colonies utilizing the Qiagen DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany), according to the manufacturer’s instructions. DNA purity was assessed using a QIAxpert Spectrophotometer (Qiagen), and DNA concentration was quantified with a Qubit™ Flex Fluorometer (Thermofisher Scientific, Waltham, MA, USA). The mecA gene, which encodes penicillin-binding protein 2a (PBP2a), was targeted to confirm methicillin resistance. A 310-bp fragment was amplified using specific primers: forward primer 5′-GTAGAAATGACTGAACGTCCGATAA-3′ and reverse primer 5′-CCAATTCCACATTGTTTCCGTAA-3′ both synthesized by Macrogen (Seoul, South Korea)38,39. The PCR reaction included DreamTaq™ 2X Green PCR Master Mix (Thermofisher Scientific), primers at a concentration of 10 pmol, and 2 µL of DNA template, achieving a total volume of 25 µL. The positive MRSA control used was S. aureus ATCC 43,300 (ATCC). The amplification process involved an initial denaturation step at 95 °C for 4 min, followed by 30 cycles of denaturation at 95 °C for 45 s, annealing at 56 °C for 1 min, and extension at 72 °C for 1 min, concluding with a final extension at 72 °C for 4 min. The annealing temperature of 56 °C was optimized to ensure specific binding of the primers to the mecA gene, thereby minimizing non-specific amplification. The resulting PCR product was analyzed through electrophoresis on 1.6% agarose gel stained with ethidium bromide, and visualized using Syngene image analysis software (Syngene, Bangalore, India).
WGS and bioinformatics analysis
WGS libraries were prepared using a QIAseqFX DNA library preparation kit (Qiagen), with 100 ng of DNA as input, in accordance with the manufacturer’s protocols. Libraries were selected for a 300–350 bp insert size. Sequencing was conducted on a NovaSeq 6000 platform (Illumina, San Diego, CA, USA) across two SP lanes. Data quality control was executed, applying a Phred score cutoff of Q30 to ensure high-quality sequencing data with a base call accuracy of 99.9%. Bioinformatics analyses were performed using the Bactopia V2 pipeline, tailored specifically to S. aureus workflows (https://github.com/bactopia/bactopia)39. The CARD and ResFinder databases were employed for the detection of resistance markers, while screening for PVL was conducted using the local BLAST database (accessed in December 2023).
Statistical analysis
Data analysis was conducted using GraphPad Prism version 10.0.0 software (GraphPad Software, Boston, MA, USA). The significance of differences was evaluated using the chi-square (χ²) test in a two-tailed format, with p < 0.05 considered statistically significant. A correlation matrix comprising Pearson’s correlation coefficients was calculated using the “corrplot” package in R version 4.2.140.
Data availability
The datasets generated and analyzed during the current study are available in the European Nucleotide Archive (ENA) under the accession number PRJEB64197 and can be accessed at https://www.ebi.ac.uk/ena/browser/view/PRJEB64197.
References
Turner, N. A. et al. Methicillin-resistant Staphylococcus aureus: an overview of basic and clinical research. Nat. Rev. Microbiol. 17 (4), 203–218. https://doi.org/10.1038/s41579-018-0147-4 (2019).
Ventola, C. L. The antibiotic resistance crisis: part 1: causes and threats. Pharm. Ther. 40 (4), 277 (2015).
Udo, E. E. Community-acquired methicillin-resistant Staphylococcus aureus: The new face of an old foe? Med. Principles Pract. 22(Suppl 1), 20 (2013).
Senok, A. et al. Emergence of novel methicillin-resistant Staphylococcus aureus strains in a tertiary care facility in Riyadh, Saudi Arabia. Infect. Drug Resist. 12, 2739–2746 (2019).
Senok, A., Ehricht, R., Monecke, S., Al-Saedan, R. & Somily, A. Molecular characterization of methicillin-resistant Staphylococcus aureus in nosocomial infections in a tertiary-care facility: emergence of new clonal complexes in Saudi Arabia. New. Microbes New. Infect. 14, 13–18 (2016).
Almutairi, H. et al. Prevalence and antimicrobial susceptibility pattern of methicillin-resistant Staphylococcus aureus (MRSA) at a maternity and children hospital in Saudi arabia: A cross-sectional study. Saudi Pharm. J. : SPJ. 32 (4), 102001 (2024).
Ahmed, O. B. et al. The prevalence and antimicrobial susceptibility of Methicillin-Resistant Staphylococcus aureus before and after the COVID-19 pandemic in a tertiary Saudi hospital. Cureus 16 (2), 1 (2024).
Al-Saleh, A., Shahid, M., Farid, E. & Bindayna, K. Trends in methicillin-resistant Staphylococcus aureus in the Gulf Cooperation Council countries: antibiotic resistance, virulence factors and emerging strains. East. Mediterr. Health J. 28 (6), 434–443 (2022).
Alamer, A. et al. Healthcare-Associated infections (HAIs): challenges and measures taken by the radiology department to control infection transmission. Vaccines (Basel) [Internet]. 10 (12), 1 (2022).
Thabit, A. K., Alabbasi, A. Y., Alnezary, F. S. & Almasoudi, I. A. An overview of antimicrobial resistance in Saudi Arabia (2013–2023) and the need for National surveillance. Microorganisms 11 (8), 1 (2023).
Alkuraythi, D. M. et al. Comparative genomic analysis of antibiotic resistance and virulence genes in Staphylococcus aureus isolates from patients and retail meat. Front. Cell. Infect. Microbiol. 13, 1339339 (2023).
Al Yousef, S. & Taha, E. Methicillin-Resistant Staphylococcus aureus in Saudi arabia: genotypes distribution review. Saudi J. Med. Med. Sci. 4 (1), 2 (2016).
El-Mahdy, T. S., Al-Agamy, M. H., Emara, M., Barakat, A. & Goering, R. V. Complex clonal diversity of Staphylococcus aureus nasal colonization among community Personnel, healthcare Workers, and clinical students in the Eastern Province, Saudi Arabia. Biomed. Res. Int. 2018, 1–9 (2018).
Ward, M. J. et al. Identification of source and sink populations for the emergence and global spread of the East-Asia clone of community-associated MRSA. Genome Biol. [Internet]. 17 (1), 1. https://doi.org/10.1186/s13059-016-1022-0 (2016).
Chen, H. et al. Drivers of methicillin-resistant Staphylococcus aureus (MRSA) lineage replacement in China. Genome Med. 13 (1), 1 (2021).
Lakhundi, S. & Zhang, K. Methicillin-Resistant Staphylococcus aureus: molecular Characterization, Evolution, and epidemiology. Clin. Microbiol. Rev. 31 (4), 1–103 (2018).
Shoaib, M. et al. MRSA compendium of epidemiology, transmission, pathophysiology, treatment, and prevention within one health framework. Front. Microbiol. 13, 1 (2023).
Tenover, F. C. et al. Characterization of Staphylococcus aureus isolates from nasal cultures collected from individuals in the united States in 2001 to 2004. J. Clin. Microbiol. 46 (9), 2837–2841 (2008).
File, T. M. Methicillin-resistant Staphylococcus aureus (MRSA): focus on community-associated MRSA. South. Afr. J. Epidemiol. Infect. 23 (2), 13–15 (2008).
Tacconelli, E. & De Angelis, G. The control of MRSA. In: Antibiotic Policies. New York, NY: Springer New York; 63–79. (2012).
Zaghen, F. et al. Epidemiology of antimicrobial resistance genes in Staphyloccocus aureus isolates from a public database in a one health Perspective—Sample characteristics and isolates’ sources. Antibiotics 12 (7), 1225 (2023).
Maruri-Aransolo, A. et al. Genomic characterization of MRSA recovered from people with cystic fibrosis during two Spanish multicentre studies (2013 and 2021). JAC Antimicrob. Resist. 6 (5), dlae160 (2024).
Monegro, A. F., Muppidi, V., Regunath, H., Hospital-Acquired & Infections Patient Safety: A Case-based Innovative Playbook for Safer Care: Second Edititon [Internet]. 2023 Feb 12 [cited 2025 Apr 12];183–98. Available from: https://www.ncbi.nlm.nih.gov/books/NBK441857/.
Tabaja, H., Hindy, J. R. & Kanj, S. S. Epidemiology of Methicillin-Resistant Staphylococcus aureus in Arab countries of the middle East and North African (MENA) region. Mediterr. J. Hematol. Infect. Dis. 13 (1), e2021050 (2021).
Coll, F. et al. Longitudinal genomic surveillance of MRSA in the UK reveals transmission patterns in hospitals and the community. Sci. Transl Med. 9, 413 (2017).
Lee, Y. C., Chen, P. Y., Wang, J. T. & Chang, S. C. Prevalence of fosfomycin resistance and gene mutations in clinical isolates of methicillin-resistant Staphylococcus aureus. Antimicrob. Resist. Infect. Control [Internet]. 9 (1), 135 (2020).
Hadyeh, E., Azmi, K., Seir, R. A., Abdellatief, I. & Abdeen, Z. Molecular characterization of methicillin resistant Staphylococcus aureus in West Bank-Palestine. Front. Public. Health. 7 (May), 450080 (2019).
Soliman, M. S. et al. Genomic characterization of methicillin-resistant Staphylococcus aureus (Mrsa) by high-throughput sequencing in a tertiary care hospital. Genes (Basel). 11 (10), 1–17 (2020).
Tluanpuii, V. & Mahajan, R. K. Detection of the mecA Gene and Its Association With Antimicrobial Resistance Among Coagulase-Negative Staphylococci Isolated From Clinical Samples in a Tertiary Care Hospital: A Cross-Sectional Study. Cureus [Internet]. 2025 Apr 3 [cited 2025 Apr 13]; Available from: https://www.cureus.com/articles/294836-detection-of-the-meca-gene-and-its-association-with-antimicrobial-resistance-among-coagulase-negative-staphylococci-isolated-from-clinical-samples-in-a-tertiary-care-hospital-a-cross-sectional-study.
Ali Alghamdi, B. et al. Antimicrobial resistance in methicillin-resistant Staphylococcus aureus. Saudi J. Biol. Sci. 30 (4), 103604 (2023).
Bishr, A. S. et al. Association of macrolide resistance genotypes and synergistic antibiotic combinations for combating macrolide-resistant Mrsa recovered from hospitalized patients. Biology (Basel). 10 (7), 624 (2021).
Monecke, S. et al. Diversity of Staphylococcus aureus isolates in European wildlife. PLoS One. 11 (12), e0168433 (2016).
García-Álvarez, L. et al. Meticillin-resistant Staphylococcus aureus with a novel MecA homologue in human and bovine populations in the UK and denmark: A descriptive study. Lancet Infect. Dis. 11 (8), 595–603 (2011).
Klein, E. Y. et al. Global increase and geographic convergence in antibiotic consumption between 2000 and 2015. Proc. Natl. Acad. Sci. U S [Internet]. 115 (15), E3463–E3470. https://doi.org/10.1073/pnas.1717295115 (2018).
Monecke, S. et al. Characterisation of MRSA strains isolated from patients in a hospital in Riyadh, Kingdom of Saudi Arabia. BMC Microbiol. 12 (1), 1–9. https://doi.org/10.1186/1471-2180-12-146 (2012).
Abebe, A. A. & Birhanu, A. G. Methicillin resistant Staphylococcus aureus: molecular mechanisms underlying drug resistance development and novel strategies to combat. Infect. Drug Resist. 16, 7641 (2023).
Mohamad Farook, N. A. et al. Diversity and dissemination of Methicillin-Resistant Staphylococcus aureus (MRSA) genotypes in Southeast Asia. Trop. Med. Infect. Dis. 7 (12), 438 (2022).
Parvin, M. S. et al. Prevalence and multidrug resistance pattern of methicillin resistant S. aureus isolated from frozen chicken meat in Bangladesh. Microorganisms 9 (3), 636 (2021).
Petit, R. A. & Read, T. D. Bactopia: a flexible pipeline for complete analysis of bacterial genomes. mSystems 5 (4), 1 (2020).
R Core Team. R: A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2022).
Acknowledgements
The authors gratefully acknowledge the University of Jeddah and King Saud University for their academic support and collaboration. We also thank the Saudi Food and Drug Authority (SFDA) for facilitating sample collection and analysis. Special thanks to King Abdullah University of Science and Technology (KAUST) for providing technical resources and bioinformatics support essential to the success of this study.
Funding
This research did not receive external funding but was supported by institutional resources from the Saudi Food and Drug Authority (SFDA) and King Abdullah University of Science and Technology (KAUST).
Author information
Authors and Affiliations
Contributions
Conceptualization: DA., MMA., and SA. Methodology, DA., and MA. software, DA., MA.; validation, MMA., SA.; formal analysis, DA., MA.; investigation, DA.; resources, TG., MM., SAA. Data curation, D. A.; writing—original draft preparation, D. A.; writing—review and editing, DA.DT.MMA., MM., MA., and SA.; visualization, DA.MMA., MM., MA., SA.; visualization, DA.MMA.; supervision, MMA., SA.; project administration, MMA., SA.; funding acquisition, TG., SA. All the authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical statement
The samples used in this study were collected during routine clinical diagnostics and fully anonymized prior to analysis. The study protocol was reviewed and approved by the Institutional Review Board of King Saud Medical City (approval number: H-01-R-053) and the Institutional Biosafety and Bioethics Committee of King Abdullah University of Science and Technology (approval number: 22IBEC051). The requirement for informed consent was waived by both committees in accordance with national regulations and institutional policies.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Alkuraythi, D.M., Alkhulaifi, M.M., Altwiley, D. et al. Genomic and epidemiological insights into the emergence and dominance of MRSA clones in Riyadh’s healthcare facilities. Sci Rep 16, 3835 (2026). https://doi.org/10.1038/s41598-025-34001-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-34001-7
