
Abstract
Yes-associated protein 1 (YAP1), a core effector of the Hippo signaling pathway, has garnered increasing attention for its role in ferroptosis signaling in breast cancer. This study investigates the antitumor properties of Lycium barbarum polysaccharides (LBPs) and develops LBPs-loaded poly(lactic-co-glycolic acid) (PLGA) nanoparticles (LBPs NPs), further exploring the mechanisms of free LBPs and LBPs NPs in normal and malignant breast cells. Results demonstrated that YAP1 overexpression drives malignant transformation in normal mammary cells, including aberrant proliferation, apoptosis resistance, cytoskeletal remodeling, and enhanced invasiveness, alongside mitochondrial hyperfunction and altered energy metabolism. However, LBPs effectively suppressed these malignant phenotypes and counteracted the associated mitochondrial and metabolic enhancements by inhibiting YAP1 upregulation. In breast cancer cells, YAP1 overexpression similarly promoted proliferation, invasion, cytoskeletal reorganization, and apoptosis resistance, accompanied by enhanced mitochondrial dysfunction and energy, and acquired ferroptosis resistance. Both LBPs and LBPs NPs significantly inhibited cell proliferation, invasion, and cytoskeletal reorganization, disrupted energy metabolism, and ultimately induced ferroptosis via downregulation of YAP1 expression. Notably, both free LBPs and LBPs NPs effectively suppressed YAP1 expression, with LBPs NPs exhibiting significantly superior efficacy compared to free LBPs. This enhancement is attributed to improved drug delivery efficiency facilitated by the PLGA-based nanocarrier system. Mechanistically, LBPs NPs target the Hippo-YAP1 signaling axis, leading to altered phosphorylation status of YAP1 (p-YAP1), and synergistically induce changes in energy metabolism and ferroptosis to inhibit breast cancer metastasis. These findings provide a proof-of-concept at the in vitro level and a potential nanoparticle-based formulation platform for the treatment of breast cancer.
Similar content being viewed by others


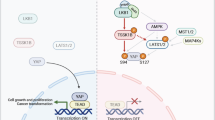
Introduction
Breast cancer is the most common malignant tumor in women and one of the leading causes of cancer-related deaths1. Despite continuous advancements in early detection and treatment strategies, the complex pathogenesis, diverse clinical manifestations, and high heterogeneity of breast cancer contribute to its persistently rising incidence and mortality rates in the coming years2. Therefore, in-depth investigation into the underlying molecular mechanisms of breast cancer development and the development of effective therapeutic agents are crucial for the prevention and treatment of this disease.
Ferroptosis is an iron-dependent, non-apoptotic form of cell death driven by lipid peroxidation. Its triggering mechanisms include glutathione depletion, reduced activity of Glutathione Peroxidase 4 (GPX4), and ferrous iron (Fe2+)-mediated lipid peroxidation. These factors collectively lead to massive accumulation of reactive oxygen species (ROS), ultimately inducing ferroptosis3. Studies indicate that ferroptosis can inhibit tumor growth, enhance responses to immunotherapy, and overcome chemoresistance4. Recent research has revealed the regulatory role of the Hippo signaling pathway in ferroptosis5. The Hippo pathway (also known as the Hippo-Yes-associated protein 1 (YAP1) signaling pathway) is an evolutionarily highly conserved signal transduction cascade involved in regulating cell proliferation, apoptosis, and tissue homeostasis6. Dysregulation of the Hippo-YAP1 pathway has been demonstrated to enhance cancer hallmarks, such as aberrant cell proliferation, chemoresistance, and metastasis7. Gu et al. found that cylindromatosis functions as a tumor suppressor gene in prostate cancer and promotes ferroptosis through Hippo/YAP signaling8. Furthermore, another study indicates that the glycolytic enzyme enolase 2 influences ferroptosis, glycolysis, and mitochondrial function in clear cell renal cell carcinoma by regulating Hippo-YAP1 signaling9. Therefore, targeting the Hippo-YAP1 signaling pathway to induce ferroptosis may represent a novel therapeutic strategy for cancer.
Lycium barbarum polysaccharides (LBPs), extracted from the fruit of Lycium barbarum, represent a promising anticancer bioactive compound. Their antitumor activity has been extensively documented in numerous studies10. Extensive in vitro and in vivo research has demonstrated that LBPs effectively inhibit cancer cell proliferation, migration, and invasion. These effects are mediated through mechanisms including the downregulation of matrix metalloproteinases (MMPs), inhibition of epithelial-mesenchymal transition, and suppression of the PI3K/AKT signaling pathway11,12. Despite exhibiting significant anticancer effects against various cancer types, the clinical application of LBPs is hindered by their suboptimal stability, low bioavailability, and poor pharmacokinetic properties13. Nanodrug delivery systems, leveraging advantages such as superior stability, excellent biocompatibility, sustained-release characteristics, and targeting capabilities, have been widely applied in diverse fields including drug delivery, immunomodulation, neuroprotection, and antitumor therapy14.
This study aims to investigate the mechanism by which LBPs-based nano-systems influence the progression of breast cancer through intervention in the Hippo–YAP1 signaling pathway. We intend to evaluate the regulatory effects of LBPs and LBPs-loaded poly(lactic-co-glycolic acid) (PLGA) nanoparticles (LBPs NPs) on YAP1 expression, as well as their synergistic impact on inhibiting malignant cellular behaviors, metabolic reprogramming, and the induction of ferroptosis. We will test the following hypothesis: LBPs NPs suppress YAP1 activity, thereby synergistically inducing metabolic disruption and ferroptosis, and ultimately inhibiting breast cancer metastasis.
Results
LBPs suppress YAP1 overexpression-induced acquisition of malignant phenotypes in MCF 10A cells
Given that the Hippo-YAP1 signaling pathway is a key driver of breast cancer development, this study successfully established a YAP1 overexpression cell model using MCF 10A cells (Fig. 1A). Cell colony formation assays demonstrated that YAP1 overexpression significantly promoted the cell proliferation, while LBPs treatment effectively counteracted this pro-proliferative effect induced by YAP1 overexpression (Fig. 1B,C). Importantly, flow cytometry demonstrated that neither intervention significantly affected the percentage of apoptotic cells (Fig. 1D). F-actin immunofluorescence staining and Transwell assays revealed that YAP1 overexpression induced cytoskeletal remodeling and enhanced invasive capacity (Fig. 1E–H). In contrast, LBPs significantly suppressed these YAP1 overexpression-induced phenotypic changes.

LBPs suppress the malignant phenotype of human mammary epithelial cells induced by YAP1 overexpression. (A) RT-qPCR analysis of the relative mRNA expression of YAP1 in MCF 10A cells after transfection with the YAP1-overexpression plasmid. Data are presented as the mean ± SD (n = 3), *** P < 0.001. (B,C) Colony formation assays were used to evaluate the proliferative capacity of MCF 10A cells after LBPs treatment or transfection with a YAP1-overexpression plasmid. Data are presented as the mean ± SD (n = 3), *** P < 0.001. (D) Flow cytometry was employed to quantify apoptosis in MCF 10A cells following LBPs treatment or transfection with a YAP1-overexpression plasmid. (E,F) Actin-Tracker Green-488 probes were employed to visualize F-actin architecture in MCF 10A cells following LBPs treatment or transfection with the YAP1-overexpression plasmid. Data are presented as the mean ± SD (n = 3), *** P < 0.001. Scale bar, 50 μm. (G,H) Transwell assays were performed to evaluate the invasive capacity of MCF 10A cells after LBPs treatment or transfection with the YAP1-overexpression plasmid. Data are presented as the mean ± SD (n = 3), *** P < 0.001. Scale bar, 200 μm.
LBPs block YAP1 overexpression-induced enhancement of mitochondrial function and energy metabolism in MCF 10A cells
Changes in mitochondrial membrane potential (MMP) and intracellular ATP levels were monitored using JC-1 and ATP fluorescent probes, respectively. JC-1 staining revealed that compared to the control group, MCF 10A cells in the YAP1 overexpression plasmid (YAP1-OE) group exhibited bright red fluorescence aggregates within mitochondria, while showing diminished green fluorescence intensity, indicating elevated MMP. LBPs treatment significantly suppressed this effect induced by YAP1 overexpression (Fig. 2A,B). Furthermore, ATP level quantification demonstrated that YAP1 overexpression markedly promoted cellular ATP synthesis, an effect similarly counteracted by LBPs (Fig. 2C,D). Collectively, these results demonstrate that YAP1 overexpression enhances mitochondrial function and energy metabolism in MCF 10A cells, and LBPs effectively block this YAP1 overexpression-mediated enhancement of mitochondrial function and energy metabolism.

LBPs abrogate YAP1-overexpression-induced augmentation of mitochondrial function and upregulation of related factor expression in human mammary epithelial cells. (A,B) MMP changes were assessed with the JC-1 fluorescent probe in MCF 10A cells following LBPs treatment or transfection with the YAP1-overexpression plasmid. Data are presented as the mean ± SD (n = 3), *** P < 0.001. Scale bar, 50 μm. (C,D) Representative fluorescence images illustrating ATP probe expression after transfection of pCMV-Mito-AT1.03 into MCF 10A cells that had been subjected to LBPs treatment or transfected with the YAP1-overexpression plasmid. Data are presented as the mean ± SD (n = 3), *** P < 0.001. Scale bar, 50 μm. (E,F) RT-qPCR analysis of the relative mRNA expression of YAP1 and TAZ in MCF 10A cells following LBPs treatment or transfection with the YAP1-overexpression plasmid. Data are presented as the mean ± SD (n = 3), *** P < 0.001. (G) Western blot analysis of YAP1, TAZ, MMP2, MMP9, Vimentin protein expression in MCF 10A cells following LBPs treatment or transfection with the YAP1-overexpression plasmid. (H–L) The gray blots were analyzed with ImageJ software (n = 3). *** P < 0.001.
LBPs suppress protein expression of YAP1, TAZ, and malignant hallmark proteins in MCF 10A cells
To further investigate the mechanism by which LBPs inhibit YAP1 expression, we performed Western blot and Real-time RT-PCR (RT-qPCR) analyses. The results demonstrated that YAP1 overexpression significantly increased both the mRNA and protein expression levels of YAP1 and its downstream effector transcriptional coactivator with PDZ-binding motif (TAZ). In contrast, LBPs significantly suppressed the mRNA and protein expression of both YAP1 and TAZ (Fig. 2E–I). Furthermore, we found that YAP1 overexpression markedly upregulated the protein expression of malignant hallmark proteins (Vimentin, MMP9, MMP2), whereas LBPs significantly inhibited the expression of all three proteins (Fig. 2G, J–L). Collectively, these experimental findings indicate that YAP1 overexpression induces the acquisition of malignant phenotypic characteristics in normal mammary epithelial cells, including aberrant proliferation, apoptosis resistance, cytoskeletal remodeling, enhanced invasive capacity, and upregulated expression of malignant hallmarks, while concurrently enhancing mitochondrial function and energy metabolism. Conversely, LBPs demonstrate potential anticancer activity by inhibiting YAP1 expression and its downstream functions.
LBPs inhibit the proliferation and promote the apoptosis of MDA-MB-231 cells
To further investigate the role of YAP1 in breast cancer cells, we established a YAP1-knockdown MDA-MB-231 cell model. RT-qPCR results indicated that YAP1 small interference (si)RNA1 demonstrated the most potent silencing effect and was therefore selected for subsequent experiments (Fig. 3A). Colony formation assays revealed that, compared with the control group, YAP1 overexpression significantly promoted cell proliferation. In contrast, YAP1-overexpressing cells treated with LBPs (LBPs + YAP1-OE) exhibited significantly inhibited proliferation. Furthermore, YAP1 knockdown effectively suppressed cell proliferation, an effect consistent with that of LBPs treatment (Fig. 3B,C). TUNEL apoptosis assays demonstrated that YAP1 knockdown significantly increased the apoptosis rate compared to the siRNA-NC group, mirroring the effect observed in the LBPs-treated group. Moreover, the combination of LBPs treatment and YAP1 knockdown resulted in a more pronounced increase in apoptosis. Notably, compared to the YAP1-OE group, the addition of LBPs treatment significantly elevated the apoptosis rate (Fig. 3D,E).

LBPs suppress YAP1-overexpression-driven malignant progression of breast cancer cells. (A) RT-qPCR analysis of the relative mRNA expression of YAP1 in MDA-MB-231 cells after transfection with YAP1-siRNA. Data are presented as the mean ± SD (n = 3), *** P < 0.001, ** P < 0.01. (B,C) Colony formation assays were used to evaluate the proliferative capacity of MDA-MB-231 cells after LBPs treatment or transfection with a YAP1-overexpression plasmid or YAP1-siRNA. Data are presented as the mean ± SD (n = 3), *** P < 0.001. ns: not significant. (D,E) Representative TUNEL staining images of MDA-MB-231 cells after LBPs treatment, transfection with the YAP1-overexpression plasmid, or YAP1-siRNA. Red fluorescence indicates TUNEL-positive cells and blue fluorescence denotes nuclei. Data are presented as the mean ± SD (n = 3), *** P < 0.001, ** P < 0.01, ns: not significant. Scale bar, 200 μm. (F,G) Actin-Tracker Green-488 probes were employed to visualize F-actin architecture in MDA-MB-231 cells after LBPs treatment, transfection with the YAP1-overexpression plasmid, or YAP1-siRNA. Data are presented as the mean ± SD (n = 3), *** P < 0.001. ns: not significant. Scale bar, 50 μm. (H,I) Transwell assays were performed to evaluate the invasive capacity of MDA-MB-231 cells after LBPs treatment, transfection with the YAP1-overexpression plasmid, or YAP1-siRNA. Data are presented as the mean ± SD (n = 3), *** P < 0.001, ** P < 0.01, ns: not significant. Scale bar, 200 μm.
LBPs significantly suppress cytoskeletal remodeling and invasive capacity in MDA-MB-231 cells
F-actin staining with a green fluorescent probe was employed to visualize the morphology and distribution of intracellular actin filaments. The results demonstrated that compared to the control group, the YAP1-OE group exhibited not only significantly enhanced F-actin fluorescence intensity but also concomitant pseudopodia formation. In contrast, the LBPs + YAP1-OE group showed significantly reduced fluorescence intensity. Furthermore, significantly decreased F-actin fluorescence intensity was also observed in both the YAP1-siRNA group and the LBPs-treated group (Fig. 3F,G). Given that cytoskeletal remodeling serves as a central driver for the acquisition of invasive capabilities by tumor cells, we further evaluated cellular invasive potential using Transwell assays. The results revealed that cell invasion was significantly enhanced in the YAP1-OE group compared to the control, whereas it was markedly suppressed in the LBPs + YAP1-OE group. Notably, either YAP1 knockdown alone or LBPs treatment alone effectively inhibited cell invasion. Moreover, the combination of both interventions demonstrated a stronger synergistic inhibitory effect (Fig. 3H,I).
LBPs suppress mitochondrial function and energy metabolism in MDA-MB-231 cells
JC-1 fluorescent probe staining revealed that, compared to the control group, MDA-MB-231 cells in the YAP1-OE group exhibited enhanced red fluorescence intensity within mitochondria and significantly reduced green fluorescence intensity, indicating an elevated mitochondrial membrane potential. Conversely, the siRNA targeting YAP1 (YAP1-siRNA) and LBPs + YAP1-OE groups displayed the opposite trend (diminished red fluorescence and enhanced green fluorescence) (Fig. 4A,B). ATP generation assays demonstrated a significant increase in ATP production in the YAP1-OE group relative to the control, whereas ATP levels were reduced in both the YAP1-siRNA group and the LBPs + YAP1-OE group (Fig. 4C,D). Collectively, these results demonstrate that LBPs counteract the YAP1 overexpression-mediated enhancement of mitochondrial function and energy metabolism.

LBPs abrogate YAP1-overexpression-induced augmentation of mitochondrial function and upregulation of related factor expression in breast cancer cells. (A,B) MMP changes were assessed with the JC-1 fluorescent probe in MDA-MB-231 cells after LBPs treatment, transfection with the YAP1-overexpression plasmid, or YAP1-siRNA. Data are presented as the mean ± SD (n = 3). *** P < 0.001, ns: not significant. Scale bar, 50 μm. (C,D) Representative fluorescence images illustrating ATP probe expression after transfection of pCMV-Mito-AT1.03 into MDA-MB-231 cells that had been subjected to LBPs treatment or transfected with the YAP1-overexpression plasmid, or YAP1-siRNA. Data are presented as the mean ± SD (n = 3). *** P < 0.001, ns: not significant. Scale bar, 50 μm. (E,F) RT-qPCR analysis of the relative mRNA expression of YAP1 and TAZ in MDA-MB-231 cells following LBPs treatment or transfection with the YAP1-overexpression plasmid or YAP1-siRNA. Data are presented as the mean ± SD (n = 3), ***P < 0.001, **P < 0.01, *P < 0.05, ns: not significant. (G) Western blot analysis of YAP1, TAZ, ACSL4 and GPX4 protein expression in MDA-MB-231 cells following LBPs treatment or transfection with the YAP1-overexpression plasmid or YAP1-siRNA. (H–K) The gray blots were analyzed with ImageJ software (n = 3). ***P < 0.001, **P < 0.01, *P < 0.05, ns: not significant. (L,M) Detection of mitochondrial Fe2+ levels using Mito-FerroGreen probe. Data are presented as the mean ± SD (n = 3), ***P < 0.001, ns: not significant. Scale bar, 100 μm.
LBPs induce ferroptosis by modulating YAP1/TAZ expression in MDA-MB-231 cells
RT-qPCR and Western blot analyses revealed that the YAP1-OE group exhibited significantly upregulated mRNA and protein expression levels of both YAP1 and TAZ. LBPs treatment effectively suppressed the transcriptional and protein expression of YAP1/TAZ (Fig. 4E–I). Furthermore, Western blot results demonstrated significantly decreased Acyl-CoA Synthetase Long Chain Family Member 4 (ACSL4) protein expression and significantly increased GPX4 protein expression in the YAP1-OE group, concomitant with reduced Fe2+ fluorescence intensity. Conversely, the LBPs + YAP1-OE group displayed the opposite trends (Fig. 4G, J–M). Given that GPX4 and ACSL4 are key regulators of ferroptosis, this study reveals that LBPs, by inhibiting YAP1 expression, downregulate GPX4, upregulate ACSL4 protein expression, and promote Fe2+ accumulation. These findings collectively suggest that LBPs promote ferroptosis via the YAP1 signaling axis.
Characterization of LBPs NPs
The above study confirmed that LBPs exhibit significant anticancer activity in both normal mammary cells and breast cancer cells by inhibiting YAP1 expression. Based on this finding, we developed an LBPs-loaded nanodrug delivery system aimed at overcoming its current application limitations and enhancing bioavailability. The encapsulation content and unencapsulated amount of LBPs were determined by ultraviolet spectrophotometry, and their encapsulation efficiency (EE%) and drug loading capacity (DL%) were calculated. The results showed that the EE% was 87.13%, and the DL% was 8.8%. Furthermore, the LBPs NPs exhibited an average particle size of 301.5 nm, a zeta potential of −18.9 mV, and a polydispersity index (PDI) of 0.319, indicating a uniform nanoscale distribution and favorable colloidal stability (Fig. 5A,B). Transmission electron microscope (TEM) results demonstrated that both empty PLGA NPs (PLGA NPs) and LBPs NPs exhibit a regular, well-defined spherical morphology, exhibiting excellent dispersion without significant aggregation (Fig. 5C). The observed particle sizes were consistent with the average particle size measured by the dynamic light scattering (DLS) instrument. Cellular uptake assay was conducted to evaluate the permeability of nanoparticles in MDA-MB-231 cells. The results revealed a stronger fluorescence signal in the LBPs NP group compared to free LBPs, which was predominantly localized within the cytoplasm. This indicates that LBPs NPs may enhance cellular uptake, leading to increased intracellular accumulation of the drug and consequently inducing a more potent antitumor effect (Fig. 5D).

Characterization of LBPs-NPs and their capacity to suppress YAP1-overexpression-induced malignant progression. (A,B) Size distribution of PLGA NPs and LBPs NPs. (C) TEM images of PLGA NPs and LBPs NPs. (D) Representative fluorescence microscopy images of MDA-MB-231 cells treated with FITC-LBPs and FITC-LBPs NPs. The blue signal represents nucle stained with DAPI and the green signal represents FITC. Scale bar, 30 μm. (E,F) Colony formation assays were used to evaluate the proliferative capacity of MDA-MB-231 cells after LBPs or LBPs NPs treatment or transfection with a YAP1-overexpression plasmid. Data are presented as the mean ± SD (n = 3), *** P < 0.001. (G) Flow cytometry was employed to quantify apoptosis in MDA-MB-231 cells following LBPs or LBPs NPs treatment or transfection with a YAP1-overexpression plasmid. (H,I) Actin-Tracker Green-488 probes were employed to visualize F-actin architecture in MDA-MB-231 cells following LBPs or LBPs NPs treatment or transfection with the YAP1-overexpression plasmid. Data are presented as the mean ± SD (n = 3), *** P < 0.001. Scale bar, 40 μm. (J,K) Transwell assays were performed to evaluate the invasive capacity of MDA-MB-231 cells after LBPs treatment or transfection with the YAP1-overexpression plasmid. Data are presented as the mean ± SD (n = 3), ***P < 0.001, **P < 0.01. Scale bar, 200 μm.
LBPs NPs inhibit proliferation, cytoskeletal remodeling, and invasion while promoting apoptosis by blocking YAP1 expression in MDA-MB-231 cells
To evaluate the antitumor efficacy of LBPs NPs, we examined their effects on breast cancer cell proliferation, apoptosis, cytoskeletal remodeling, and invasive capacity. Colony formation assays and flow cytometry revealed that LBPs NPs significantly inhibited colony formation and induced a higher proportion of apoptotic cells compared to free LBPs. Furthermore, LBPs NPs effectively counteracted the oncogenic effects induced by YAP1 overexpression (Fig. 5E–G). Additionally, LBPs NPs demonstrated markedly superior inhibitory effects on actin polymerization and invasion compared to free LBPs, and also abrogated the promoting effects of YAP1 overexpression on cytoskeletal reorganization and invasion (Fig. 5H–K).
LBPs NPs suppress the expression of YAP1 and TAZ while promoting the expression of p-YAP1 and ferroptosis in MDA-MB-231 cells
To further investigate the anticancer mechanism of LBPs NPs, Prussian blue staining was performed. The results revealed that both free LBPs and LBPs NPs significantly induced Fe2+ accumulation, with LBPs NPs exhibiting a more pronounced effect (Fig. 6A,B). Western blot analysis demonstrated that, compared to free LBPs, LBPs NPs significantly suppressed the protein expression of YAP1, TAZ, and the key ferroptosis inhibitor GPX4, while simultaneously upregulating the protein expression level of p-YAP1 and the ferroptosis marker ACSL4. Furthermore, the functional effects of YAP1 overexpression were significantly inhibited by LBPs NPs (Fig. 6C–H). Furthermore, treatment with the ferroptosis inhibitors (Ferrostatin-1) following LBPs NPs administration markedly up-regulated GPX4 protein expression and down-regulated ACSL4 protein expression. These results indicate that both free LBPs and LBPs NPs exert anticancer activity by inducing ferrous iron accumulation and promoting ferroptosis, a process mediated at least in part through the suppression of YAP1 signaling and the enhancement of YAP1 phosphorylation. Immunofluorescence analysis revealed that both LBPs and LBPs NPs significantly reduced the expression of YAP1 in the nucleus and cytoplasm, with the effect of LBPs NPs being more pronounced (Fig. 6I,J).

LBPs NPs suppressed the expression of YAP1 and TAZ, and promoted the expression of p-YAP1 and ferroptosis. (A,B) Prussian blue staining for the detection of non-heme Fe2+ in MDA-MB-231 cells after LBPs treatment or transfection with the YAP1-overexpression plasmid. Data are presented as the mean ± SD (n = 3), ***P < 0.001, *P < 0.05. Scale bar, 50 μm. (C) Western blot analysis of YAP1, p-YAP1, TAZ, ACSL4 and GPX4 protein expression in MDA-MB-231 cells following LBPs or LBPs NPs treatment or transfection with the YAP1-overexpression plasmid. (D–H) The gray blots were analyzed with ImageJ software (n = 3). **P < 0.01, ***P < 0.001. (I) Western blot analysis of ACSL4 and GPX4 protein expression in MDA-MB-231 cells following the treatment of LBPs NPs or LBPs NPs plus Ferrostatin-1. (J,K) The gray blots were analyzed with ImageJ software (n = 3). ***P < 0.001. (L) Immunofluorescence staining analysis of YAP1 expression in MDA-MB-231 cells following LBPs or LBPs NPs treatment. (M) Quantification of YAP1 expression levels. Data are presented as the mean ± SD (n = 3), ***P < 0.001, **P < 0.01, *P < 0.05. Scale bar, 50 μm.
Discussion
To meet their proliferation and growth demands, cancer cells exhibit a higher requirement for iron than healthy cells and are more susceptible to death induced by iron homeostasis imbalance, a process termed ferroptosis15. Currently, ferroptosis has been implicated in various diseases, including cancer, and targeting ferroptosis is emerging as a promising novel strategy in cancer therapy16. In this study, leveraging the established anti-tumor properties of LBPs, we successfully developed LBPs-loaded PLGA nanoparticles. We further investigated their molecular mechanisms in blocking the acquisition of malignant phenotypes by normal mammary cells through suppression of YAP1 expression, and disruption of energy metabolism to induce ferroptosis in breast cancer cells. We demonstrate that LBPs inhibit cell proliferation, invasion, and cytoskeletal remodeling, while promoting apoptosis, inducing mitochondrial dysfunction, metabolic dysfunction, and ferroptosis. This is achieved primarily by suppressing YAP1 expression and promoting its phosphorylation (p-YAP1), leading to YAP1 functional inactivation. Notably, the LBP NPs exhibited significantly more potent anti-cancer efficacy than free LBPs.
The Hippo pathway is an evolutionarily conserved signaling cascade whose core regulatory module comprises the MST/LATS kinase cascade and the YAP/TAZ-TEAD transcription complex. Specifically, upon Hippo pathway activation, phosphorylated LATS1/2 kinases phosphorylate the transcriptional coactivators YAP and its paralog TAZ. This phosphorylation leads to their cytoplasmic retention and subsequent proteasomal degradation, thereby inhibiting their oncogenic activities17. Conversely, when the pathway is inactive, dephosphorylated YAP/TAZ translocate to the nucleus where they bind to transcription factors TEAD1-4. This complex activates the transcription of downstream target genes that promote proliferation, confer anti-apoptotic properties, drive cell growth and survival, and ultimately facilitate tumorigenesis and development18,19.
Current research indicates that aberrantly elevated YAP/TAZ activity drives accelerated growth in breast cancer cells, concomitant with increased expression of malignant phenotype markers (Vimentin, MMP2, MMP9) and promotes tumor progression20. Our cellular-level findings align with these conclusions. The results demonstrate that YAP1 overexpression induces malignant phenotypic acquisition in normal mammary epithelial cells, characterized by elevated protein expression, aberrant proliferation, cytoskeletal remodeling, and enhanced invasive capacity, alongside augmented mitochondrial function and energy metabolism. Furthermore, studies in cancer research confirm that elevated YAP/TAZ expression and enhanced nuclear localization are significantly correlated with aggressive features in lung cancer (such as advanced TNM stage and lymph node metastasis) and poor patient prognosis7. Qadir et al. further substantiated this at the tissue level, demonstrating that elevated YAP1 expression in breast cancer tissues significantly correlates with tumor progression, metastasis, and reduced patient survival21. Consistent with these findings, our results indicate that YAP1 overexpression promotes cell proliferation and invasion, suppresses apoptosis, enhances mitochondrial function, and induces cytoskeletal remodeling. This collectively delineates the mechanistic role of YAP1 in driving malignant transformation of normal mammary epithelial cells and facilitating breast cancer progression. The Hippo pathway regulates ferroptosis through multiple mechanisms. Studies have shown that cell density and cell-cell contacts have also been demonstrated to be important regulators of YAP/TAZ-mediated ferroptosis22. Notably, the role of YAP/TAZ in ferroptosis exhibits cell-type specificity. For instance, in renal carcinoma cells, YAP depletion confers resistance to ferroptosis, while YAP overexpression sensitizes cancer cells to ferroptosis23. Conversely, in hepatocellular carcinoma (HCC) cells, YAP/TAZ function as suppressors of ferroptosis. They induce the expression of SLC7A11 in a TEAD-dependent manner, thereby enhancing cellular antioxidant defenses and enabling HCC cells to resist sorafenib-induced ferroptosis. Overall, the susceptibility of cancer cells to ferroptosis depends on the activity status of the Hippo pathway. Generally, Hippo pathway inhibition and YAP activation enhance cancer cell sensitivity to ferroptosis24. This study further substantiates the role of YAP in regulating ferroptosis in breast cancer: YAP1 overexpression significantly upregulated GPX4 protein expression while downregulating ACSL4 protein expression. This was accompanied by reduced intracellular Fe2+ fluorescence intensity, collectively indicating enhanced ferroptosis resistance in breast cancer cells. These changes demonstrate a critical role for YAP1 in the ferroptosis mechanism of breast cancer cells.
Natural products are gaining increasing attention in the field of cancer prevention and treatment and have become a significant direction for anti-cancer drug development. Lycium barbarum polysaccharides, natural macromolecular water-soluble polysaccharides extracted from Lycium barbarum, possess diverse biological activities and broad pharmacological effects, including immunomodulation, hypolipidemic activity, anti-aging properties, and anti-tumor effects. Studies have demonstrated that LBPs significantly inhibit tumor growth in BALB/c mice and reduce the protein expression levels of PMI, ABCG2, PI3K, and AKT in mice with colon cancer25. Research by Miao et al. confirmed that LBPs inhibit the proliferation of human gastric cancer cells MGC-803 and SGC-7901 by arresting them in the G0/G1 and S phases, respectively26. Based on the favorable antitumor activity of LBPs, LBPs NPs were developed in this study, exhibiting a high encapsulation efficiency of 87.13% and a drug loading of 8.8%. Compared to other nanoparticles reported in the literature, such as Zein/Lycium barbarum polysaccharide/chlorogenic acid composite nanoparticles (EE = 85.66%)27 and temozolomide loaded PLGA nanoparticles (EE = 70.02%)28, the LBPs NPs demonstrate a significant advantage in terms of encapsulation efficiency. Functional assay results demonstrated that both free LBPs and LBP NPs effectively inhibit breast cancer cell proliferation, invasion, and cytoskeletal remodeling while promoting apoptosis. It is noteworthy that the antitumor efficacy of LBPs NPs was significantly superior to that of free LBPs. This enhanced effect is primarily attributed to their ability to facilitate cellular uptake, leading to increased intracellular drug accumulation and consequently, a more potent antitumor response. Previous research has indicated that LBPs can effectively reduce cell viability and induce cell death accompanied by G0/G1 phase arrest in breast cancer cells through ferroptosis induction, characterized by downregulation of the light-chain subunit of cystine/glutamate antiporter system Xc- and GPX4 expression10. This study revealed that treatment with LBPs or LBPs NPs significantly decreased GPX4 and increased ACSL4 protein levels, whereas LBPs NPs’ effects were suppressed upon co-administration of Ferrostatin-1. More importantly, PLGA encapsulation of LBPs significantly enhanced their ferroptosis-inducing capacity. We hypothesized that this improved anti-cancer efficacy stems not only from increased bioavailability but also involves modulation of the Hippo signaling pathway, particularly through the induction of YAP1 phosphorylation. Experimental evidence confirmed that both LBPs and LBP NPs promoted YAP1 phosphorylation at Ser127 and sequestered it in the cytoplasm. As a key event in Hippo pathway activation, this directly inactivates YAP1 function. Taken together, these findings indicate that the anti-cancer effects of LBPs are mediated, at least in part, through activation of the Hippo pathway and enhanced YAP1 phosphorylation.
This study confirms the oncogenic role of YAP1 in promoting malignant transformation of normal mammary epithelial cells, enhancing mitochondrial function and energy metabolism, boosting proliferation and invasion capabilities in breast cancer cells, and conferring ferroptosis resistance. Both LBPs and LBPs NPs effectively counteract the oncogenic effects mediated by YAP1 overexpression. Notably, this study found that the combination of siRNA and LBPs suppressed YAP1 expression—at both the mRNA and protein levels—more effectively than free LBPs alone. This suggests that the combined use of the two may produce an “additive” or even “synergistic” effect, thereby more efficiently inhibiting the YAP1 signaling pathway. The underlying molecular mechanisms merit further investigation in subsequent studies.
Studies demonstrate that YAP1 overexpression promotes malignant transformation in normal mammary epithelial cells, an effect effectively blocked by LBPs. In breast cancer cells, LBPs similarly suppress cell proliferation, invasion, and cytoskeletal remodeling, while promoting apoptosis, mitochondrial dysfunction, energy metabolism dysregulation, and ferroptosis. These effects are mediated through the inhibition of YAP1 expression and an alteration of its phosphorylation status. Importantly, PLGA nanoparticle encapsulation enhances the drug utilization efficiency of LBPs, resulting in significantly superior anti-cancer efficacy of LBP NPs compared to free LBPs. Collectively, LBP NPs exert potent anti-tumor effects by inhibiting the Hippo-YAP1 signaling pathway, inducing changes in energy, and triggering ferroptosis.
Limitations
However, this study is not exempt from limitations. While our work identifies the suppression of YAP1 by LBPs NPs, as a critical step, their direct upstream target and functional connection to the Hippo pathway require further elucidation. All experimental data were obtained from in vitro experiments and single cell line, which cannot fully recapitulate the complex tumor microenvironment in humans, the interplay with the immune system, or systemic pharmacokinetics. The release and stability data of the nanosystem, as well as whether it can effectively target the tumor site in vivo, along with its biodistribution, metabolic pathways, and potential toxicity, remains to be elucidated.
Based on these limitations, subsequent research should include validation in animal models. Advanced imaging techniques, such as fluorescence in vivo imaging and positron emission tomography, combined with toxicological assessments, are necessary to comprehensively predict clinical efficacy and evaluate the in vivo behavior and safety profile. The critical next steps will involve investigating the release kinetics, stability, in vivo targeting efficiency, biodistribution, metabolic pathways, and potential toxicity of the nanosystem.
Materials and methods
Preparation of LBPs NPs
LBPs-loaded PLGA-PEG-COOH nanoparticles were prepared using the emulsion-solvent evaporation method. Briefly, 100 mg of LBPs and 200 mg of PLGA-PEG-COOH were completely dissolved in 5 mL of chloroform. A 3% (w/v) polyvinyl alcohol (PVA) aqueous solution was prepared. The organic solution was then added to the PVA solution, followed by sonication for 5 min. The resulting emulsion was transferred to a three-necked round-bottom flask and stirred magnetically for 4 h. Subsequently, the nanoparticles were washed three times using an ultrafiltration tube and concentrated for future use. Cells were treated with free LBPs and LBPs NPs at a final concentration of 100 µM each.
Characterization of LBPs NPs
To determine particle size and Zeta-potential, the sample was used for the preparation of 1 mg/mL suspension and the measurements were performed on a Zetasizer Ultra analyzer (Malvern Panalytical, Britain) using a standardized protocol.
Encapsulation efficiency and drug loading
The samples were centrifuged at 11,000 RPM and 4 °C for 45 min. The supernatant was carefully collected, and the absorbance at the characteristic absorption peak was measured using a ultraviolet spectrophotometer to determine the concentration of free drug in the supernatant. The EE% and DL% were then calculated based on the respective formulas.
where Wtotal is the total weight of LBPs input and Wfree is the weight of the free, unencapsulated LBPs, and Wcarrier is the mass of the PLGA carrier.
Transmission electron microscope
Shape and surface morphology of nanoparticles were examined with TEM (FEI, USA). A copper grid was placed on a piece of adhesive tape, and 30 µL of the sample was pipetted onto the surface of the grid, followed by incubation for 10 min. Excess sample was then removed by wicking with filter paper from the edge of the grid. Subsequently, 30 µL of phosphotungstic acid stain was applied and allowed to stand for 2 min. After staining, the excess stain was absorbed using filter paper. The grid was then dried under an infrared lamp for 10 min. Finally, the sample was observed using TEM.
Cell culture and transfection
Human mammary epithelial cells (MCF 10 A, ZQ0451) and (MDA-MB-231, ZQ0118) obtained from Shanghai Zhong Qiao Xin Zhou Biotechnology Co., Ltd. (https://www.zqxzbio.com/). According to the manufacturers’ instructions, MCF 10 A cells were cultured in specialized human mammary epithelial cell growth medium, while MDA-MB-231 cells were maintained in DMEM supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin solution. The effective concentration of Ferrostatin-1 (F408509, Aladdin) for MDA-MB-231 cells is 5 µM29.
The YAP1 overexpression plasmid and siRNA targeting YAP1 were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China). The sequences are as follows: YAP1-siRNA (Sense 5’-GGUCAGAGAUACUUCUUAATT-3’, Antisense 5’-UUAAGAAGUAUCUCUGACCTT-3’), NC-siRNA (sense 5’-UUCUCCGAACGUGUCACGUTT-3’, antisense 5’-ACGUGACACGUUCGGAGAATT-3’). 2.5 µg of the YAP1 overexpression plasmid was transfected into cells using Lipo8000™ (Cat No. C0533; Beyotime, China) and incubated for 24 h, with the empty vector serving as the negative control (NC). For siRNA transfection, the cells were first seeded in a 6-well plate (2 × 105 cells/well) and incubated at 37 °C and 5% CO2 for 24 h. Then, 100 pmol siRNA was added per well and 4 µL Lipo8000™ were added in each well for transfection at room temperature for 24 h. The cells were harvested for subsequent Western blot assays to confirm the efficiency of the transfections. After successful transfection, the cells were incubated at 37 °C and 5% CO2 for 24 h prior to subsequent experiments.
RT-qPCR
Total RNA was extracted from cells using the Trizol method. RNA purity and concentration were respectively assessed using a NanoDrop ND-2000 spectrophotometer and a Qubit 3.0 fluorometer. According to the manufacturer’s instructions of the RevertAid First Strand cDNA Synthesis Kit (00752219, Thermo Fisher Scientific, China), the reaction mixture was prepared. Each group contained 5 µg of RNA in a total reaction volume of 20 µL. The mixture was then incubated under the following conditions: 25 °C for 5 min, 42 °C for 60 min, and 70 °C for 5 min. Finally, the synthesized cDNA was stored at −20 °C. Real-time RT-PCR (RT-qPCR) was performed using PowerUp™ SYBR™ Green Master Mix (01118369, Thermo Fisher Scientific, China) in a 20 µL reaction volume. The reaction was pre-denatureed at 95 °C for 2 min; denatured at 95 °C for 15 s, annealed at 60 °C for 60 s, and extended at 72 °C for 20 s for 40 cycles. GAPDH was used as the internal reference gene. Primers were synthesized by Sangon Biotech (Shanghai, China; https://www.sangon.com/). The primer sequences were as follows:
YAP1-F: 5’-ATGAGCAGCTACAGTGTCC-3’.
YAP1-R: 5’-GGTTGATAGTATCACCTGTATCCA-3’.
TAZ-F: 5’-TCTGGACCAAGTACATGAACC-3’.
TAZ-R: 5’-ATTGGACACGGTGATGAGG-3’.
GAPDH-F: 5’-CAAGGTCATCCATGACAACTTTG-3’.
GAPDH-R:5’ -GTCCACCACCCTGTTGCTGTAG-3’.
Gene expression was evaluated using the comparative threshold cycle method (2−ΔΔCt), with data normalized to the housekeeping gene and the control group serving as the calibrator.
Colony formation assay
Following treatment according to the experimental groups, cells were seeded in culture dishes at a density of approximately 200 cells per dish. 2 mL of culture medium were added, and the dishes were incubated at 37 °C in a humidified atmosphere of 5% CO2 for 14 days to allow colony formation. The supernatant was then removed, and the cells were washed twice with PBS. After fixation with 4% formaldehyde for 20 min at room temperature, the cells were stained with 0.5% crystal violet solution for 20 min, washed twice with PBS, and air-dried. Colonies were visualized and manually counted.
Apoptosis assay
Following drug treatment or genetic modification, cells were cultured for 24 h in an incubator. After incubation, cells were digested using trypsin without EDTA for 5 min. The cell pellet was collected after washing and centrifugation. Cells were gently resuspended in 500 µL of Binding Buffer to prepare a single-cell suspension. Subsequently, 5 µL of Annexin V-FITC was added and mixed, followed by the addition of 5 µL of Propidium Iodide and mixing. The suspension was incubated for 10 min at room temperature in the dark. Finally, apoptosis was analyzed by flow cytometry.
Mitochondrial membrane potential (MMP) assay
Changes in MMP were assessed using a JC-1 assay kit(C2006, Beyotime Biotechnology, China). Following drug treatment or genetic modification, cells were cultured for 24 h in an incubator. The culture medium was aspirated and discarded. After washing the cells with PBS, 1 mL of complete medium and JC-1 staining working solution were added and mixed thoroughly, followed by incubation at 37 °C for 20 min. The supernatant was aspirated and discarded, and the cells were washed twice with JC-1 staining buffer. After adding culture medium, cells were observed using a fluorescence microscope.
Mitochondria ATP production assay
Changes in intracellular ATP concentration were measured using an ATP fluorescent probe (D2606, Beyotime Biotechnology, China). Cells subjected to drug treatment or genetic modification were seeded into 12-well plates and cultured for 24 h in a incubator at 37 °C with 5% CO2. After adding 1 µg ATP fluorescent probe and 1.6 µL Lipo8000™ transfection reagent, cells were maintained for an additional 24 h. Subsequently, images were acquired using a fluorescence microscope.
Mito-FerroGreen staining
Changes in mitochondrial Fe2+ levels were assessed using Mito-FerroGreen (M489, Dojindo, China). Following drug treatment or genetic modification, cells were plated onto culture dishes and incubated for 24 h at 37 °C under 5% CO2. After incubation, the medium was discarded, and cells were washed with HBSS buffer. Subsequently, 5 µmol/L Mito-FerroGreen working solution was added, and cells were incubated for an additional 30 min. The supernatant was then aspirated and discarded, and the washing step with HBSS buffer was repeated. Finally, cells were imaged using a fluorescence microscope.
Actin-Tracker Green-488 staining
Actin-Tracker Green-488 (C2201S, Beyotime Biotechnology, China) was used to label and visualize the morphology and distribution of intracellular F-actin. Cells subjected to drug treatment or genetic modification were seeded onto microscope slides and incubated for 24 h. Subsequently, cells were washed with PBS and fixed with 3.7% formaldehyde solution for 20 min at room temperature. Following fixation, cells were washed three times with immunostaining washing buffer. Actin-Tracker Green-488 working solution was applied to the slides, followed by incubation for 60 min at room temperature in the dark. After incubation, cells were washed again with immunostaining washing buffer. After air-drying, the slides were mounted using an anti-fade mounting medium containing DAPI. The distribution of F-actin was then examined using a fluorescence microscope.
Cellular internalization of NPs
Following trypsinization, the cells were seeded on microscope slides and cultured in a 37 °C CO2 incubator for 24 h. The cells were then treated with fluorescently labeled LBPs and LBPs NPs for 6 h. After incubation, the culture medium was removed, and the wells were washed three times with PBS. the cells were mounted using an antifade mounting medium containing DAPI. Images were captured using a fluorescence microscopy. All procedures were carried out under light-protected conditions.
Transwell assay
Following drug treatment or genetic modification, the cells were seeded into 12-well plates and incubated for 24 h. Cells were then resuspended in serum-free medium at a density of 1 × 105 cells/mL. Subsequently, 200 µL of the cell suspension was added to the upper chamber of a Transwell insert. The lower chamber was filled with 600 µL of complete medium. The Transwell assembly was incubated for 48 h. After incubation, the insert was removed, the medium was aspirated, and cells were washed twice with PBS. Cells were fixed with 4% paraformaldehyde for 10 min and stained with 0.1% crystal violet solution (Cat# G1062; Solarbio, China) for 20 min. Following two additional PBS washes, non-invaded cells on the upper chamber were gently removed using a cotton swab. Invaded cells on the lower surface were then quantified and imaged using an inverted light microscope.
TUNEL assay
Apoptosis was detected using a one-step TUNEL apoptosis assay kit (C1090, Beyotime Biotechnology, China). Following drug treatment or genetic modification, cells were seeded onto glass microscope slides and incubated for 24 h. Cells were washed with PBS and fixed with 4% paraformaldehyde for 30 min. After washing, cells were permeabilized with PBS containing 0.3% Triton X-100 for 5 min at room temperature. 50 µL of TUNEL reaction solution was then applied to the samples, followed by incubation at 37 °C in the dark for 60 min. Following three washes, the slides were mounted using an anti-fade mounting medium and TUNEL-positive cells were examined using a fluorescence microscope.
Prussian blue staining
Intracellular Fe²⁺ was detected using a Prussian Blue Staining Kit (G1429, Solarbio, China). Following drug treatment or genetic modification, cells were seeded onto microscope slides and incubated for 24 h. After incubation, Perls’ staining working solution was applied to completely cover the cells, and slides were incubated in a humidified chamber at 37 °C for 20 min. Slides were then washed three times with distilled water. The incubation solution was applied, followed by a 10-min incubation. Subsequently, slides were washed three times with PBS, for 60 s per wash. The enhancer working solution was applied, and slides were incubated at 37 °C for 10 min. After washing, slides were counterstained for 3 min. Slides were then immersed in distilled water for 10 min. Finally, slides were dehydrated through a graded ethanol series, cleared in xylene, mounted with neutral resin, and examined under a light microscope.
Cellular immunofluorescence
The cells were fixed with 4% paraformaldehyde for 10 min and then permeabilized with 0.5% Triton X-100 for 15 min at 37 °C. After incubation with goat serum for 30 min, cells were incubated overnight with the following antibodies: YAP1 (1:100; bs-3605R; Bioss, Beijing, China). The second day, the cells were incubated with Alexa Fluor® 488 Conjugate (1:100; ZF-0511; ZSGB-BIO, China) secondary antibodies at 37 °C for 1 h and sealed with antifade mounting medium containing DAPI, observed under a fluorescence microscope.
Western blot assay
Total protein extraction kit (KGB5303; KeyGEN BioTECH, Nanjing, China) and BCA Protein Assay Kit (PC0020; Solarbio, Beijing, China) were used to extract proteins and determine protein concentrations. Protein was separated by 9% polyacrylamide gels and transferred to PVDF membranes. The membranes were incubated with blocking solution containing TBST (TBS with Tween-20) and 5% milk powder for 1 h at room temperature. After that, the membranes were incubated with primary antibodies: MMP2 (1:1000; Cat No. 10373-2-AP; Proteintech, Wuhan, China), MMP9 (1:1000; AF5228; Affinity, Liyang, China), Vimentin (1:2000; bs-8533R; Bioss, Beijing, China), TAZ (1:1000; bs-12367R; Bioss, Beijing, China), YAP1 (1:1000; bs-3605R; Bioss, Beijing, China), p-YAP1(1:1000; AF3328; Affinity, Liyang, China), ACSL4 (1:1000; DF12141, Affinity, Liyang, China), GPX4 (1:1000; Cat No. 67763-1-Ig; Proteintech, Wuhan, China), β-actin (1:10000; Cat No.20536-1-AP; Proteintech, Wuhan, China). After overnight incubation at 4 °C, the secondary antibody Horseradish Peroxidase (HRP)-conjugated goat-anti-rabbit IgG (ZB-2301, ZSGB-Bio) or HRP-conjugated goat-anti-mouse IgG (ZB-2305, ZSGB-Bio) was used to incubate the PVDF membranes for 1 h at room temperature. Finally, the protein signals were detected signals with electrochemiluminescence solution.
Statistical analysis
For all experiments, the group assignment was randomized. Furthermore, the collection and analysis of outcome data were performed by researchers blinded to the experimental groups. All statistical analyses were conducted using SPSS 27.0 software. Data are presented as the mean ± standard deviation (SD) from at least three independent experiments (n ≥ 3). Differences between groups were compared with one-way ANOVA followed by Bonferroni post hoc test. The P value less than 0.05 indicates a statistically significant difference.
Data availability
All data generated or analysed during this study are included in this published article.
References
Akram, M., Iqbal, M., Daniyal, M. & Khan, A. U. Awareness and current knowledge of breast cancer. Biol. Res. 50, 33 (2017).
Xiong, X. et al. Breast cancer: pathogenesis and treatments. Signal. Transduct. Target. Therapy. 10, 49 (2025).
Luo, Y. et al. Ferroptosis in cancer therapy: Mechanisms, small molecule Inducers, and novel approaches. Drug. Des. Devel. Ther. 18, 2485–2529 (2024).
Lei, G., Zhuang, L. & Gan, B. The roles of ferroptosis in cancer: Tumor suppression, tumor microenvironment, and therapeutic interventions. Cancer cell. 42, 513–534 (2024).
Yu, F. X., Zhao, B. & Guan, K. L. Hippo pathway in organ size Control, tissue Homeostasis, and cancer. Cell 163, 811–828 (2015).
Liu, X. et al. FOXP4 is a direct YAP1 target that promotes gastric cancer stemness and drives metastasis. Cancer Res. 84, 3574–3588 (2024).
Zanconato, F., Cordenonsi, M. & Piccolo, S. YAP/TAZ at the roots of cancer. Cancer cell. 29, 783–803 (2016).
Gu, Y. et al. CYLD regulates cell ferroptosis through Hippo/YAP signaling in prostate cancer progression. Cell Death Dis. 15, 79 (2024).
Li, H., Wu, Y., Ma, Y. & Liu, X. Interference with ENO2 promotes ferroptosis and inhibits Glycolysis in clear cell renal cell carcinoma by regulating Hippo–YAP1 signaling. Oncol. Lett. 28, 443 (2024).
Du, X. et al. A novel anticancer property of lycium barbarum polysaccharide in triggering ferroptosis of breast cancer cells. J. Zhejiang Univ. Sci. B. 23, 286–299 (2022).
Chen, Q., Shi, R., Jiang, D., Liu, W. & Jia, Z. Lycium barbarum polysaccharide inhibits gastric cancer cell proliferation, migration and invasion by down-regulation of MMPs and suppressing epithelial-mesenchymal transition. Int. J. Clin. Exp. Pathol. 10, 7369–7374 (2017).
Zhang, X. J., Yu, H. Y., Cai, Y. J. & Ke, M. Lycium barbarum polysaccharides inhibit proliferation and migration of bladder cancer cell lines BIU87 by suppressing Pi3K/AKT pathway. Oncotarget 8, 5936–5942 (2017).
Bo, R. et al. The immunological activity of lycium barbarum polysaccharides liposome in vitro and adjuvanticity against PCV2 in vivo. Int. J. Biol. Macromol. 85, 294–301 (2016).
Li, B. et al. Nano-drug co-delivery system of natural active ingredients and chemotherapy drugs for cancer treatment: A review. Drug Deliv. 29, 2130–2161 (2022).
Yang, J. et al. Metformin induces ferroptosis by inhibiting ufmylation of SLC7A11 in breast cancer. J. Exp. Clin. Cancer Res.: CR. 40, 206 (2021).
Stockwell, B. R. et al. Ferroptosis: A regulated cell death nexus linking Metabolism, redox Biology, and disease. Cell 171, 273–285 (2017).
Cunningham, R. & Hansen, C. G. The Hippo pathway in cancer: YAP/TAZ and TEAD as therapeutic targets in cancer. Clin. Sci. (London England: 1979). 136, 197–222 (2022).
Lin, K. C., Park, H. W. & Guan, K. L. Regulation of the Hippo pathway transcription factor TEAD. Trends Biochem. Sci. 42, 862–872 (2017).
Lei, Q. Y. et al. TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the Hippo pathway. Mol. Cell. Biol. 28, 2426–2436 (2008).
Koohestanimobarhan, S., Salami, S., Imeni, V., Mohammadi, Z. & Bayat, O. Lipophilic Statins antagonistically alter the major epithelial-to-mesenchymal transition signaling pathways in breast cancer stem-like cells via Inhibition of the mevalonate pathway. J. Cell. Biochem. 120, 2515–2531 (2019).
Qadir, J. et al. Increased YAP1 expression is significantly associated with breast cancer progression, metastasis and poor survival. Future Oncol. (London England). 17, 2725–2734 (2021).
Sun, T. & Chi, J. T. Regulation of ferroptosis in cancer cells by YAP/TAZ and Hippo pathways: The therapeutic implications. Genes Dis. 8, 241–249 (2021).
Yang, W. H. et al. The Hippo pathway effector YAP promotes ferroptosis via the E3 ligase SKP2. Mol. Cancer Research: MCR. 19, 1005–1014 (2021).
Wu, J. et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 572, 402–406 (2019).
Ma, L. et al. Lycium barbarum polysaccharide reverses drug resistance in oxaliplatin-resistant colon cancer cells by inhibiting PI3K/AKT-dependent phosphomannose isomerase. Front. Pharmacol. 15, 1367747 (2024).
Miao, Y. et al. Growth Inhibition and cell-cycle arrest of human gastric cancer cells by lycium barbarum polysaccharide. Med. Oncol. (Northwood Lond. Engl.). 27, 785–790 (2010).
Ma, Y. L. et al. Zein/Lycium barbarum polysaccharide/chlorogenic acid composite nanoparticles: construction, characterization, physicochemical and biological properties. Int. J. Biol. Macromol. 318, 145030 (2025).
Şahin, Ş., Kaya-Tilki, E., Baysal, M. & Öztürk, A. A. Preparation and evaluation of Temozolomide loaded PLGA nanoparticles for the treatment of glioblastoma multiforme. Sci. Rep. 15, 36194 (2025).
Wu, S. et al. Aldo-keto reductase 1B10 (AKR1B10) suppresses sensitivity of ferroptosis in TNBC by activating the AKT/GSK3β/Nrf2/GPX4 axis. Front. Biosci. (Landmark edition). 30, 36615 (2025).
Acknowledgements
Not applicable.
Funding
This study was supported by Mechanistic Investigation of Lycium barbarum Polysaccharide-Induced Effects on Human Breast Cancer Cells via the Hippo Signaling Pathway (2023AAC03719).
Author information
Authors and Affiliations
Contributions
Conceptualization and Methodology: [Fangfang Zhao]; Data curation and Formal analysis: [Sen Yang]; Resources and Software: [Ting He]; Visualization and Investigation: [Lijuan Yang]; Project administration and Supervision: [Ting Mo]; Validation: [Juying Kang]; Writing-review & editing: [Xiangping Sun]; Funding acquisition [Hui Cao]; Writing-original draft: [Yang Yu]
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhao, F., Yang, S., He, T. et al. LBPs NPs suppress breast cancer progression by inhibiting YAP1 expression to induce ferroptosis and alter energy metabolism. Sci Rep 16, 9257 (2026). https://doi.org/10.1038/s41598-025-34454-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-34454-w
