
Abstract
The identification and typing of bacteria are very expensive and time-consuming due to their growth times, and the expertise needed. MALDI-TOF MS represents a fast technique, reproducible with molecular approaches. This technique is still poorly applied in Legionella surveillance with estimation occurring only at the genus level. The aim of this study was to compare three sample preparation methods: direct smear (DS), extended direct smear (EDS), and full extraction (E), using MALDI Biotyper, developing an in-house library. Moreover, Hierarchical cluster analysis (HCA) was compared to mip and rpoB gene sequencing. The dataset was composed of 104 isolates belonging to six Legionella species. The isolates were identified with a sensitivity of 97.11% for DS, 98.08% for EDS, and 95.19% for E. The error rates were 2.88% for DS, 1.92% for EDS, and 4.90% for E, with no significant differences among them. The HCA confirmed the relationship among the isolates reported in the phylogenetic trees. An improvement in sensitivity was obtained using an in-house library. The results suggest the use of a fast and inexpensive DS method, combined with instrument and in-house library for routine Legionella surveillance. HCA analysis could be useful for screening isolates, before undertaking expensive and time-laborious molecular techniques.
Similar content being viewed by others


Introduction
In recent years, matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) has become a reference method for the routine identification of bacterial isolates in clinical microbiology laboratories around the world. The analysis of ribosomal protein patterns is an emerging, innovative, rapid, and inexpensive technique for species-level microbial identification, through the analysis of ribosomal protein patterns. This technique has changed and improved the routine of clinical practice, by replacing most traditional techniques and combining them with the novel molecular approaches1,2,3,4. This technique is based on the production of a strain-specific spectral pattern of the whole cell mass that provides a species-specific ‘fingerprint’5. This technology does not require expensive consumables and has shown high reproducibility in both intra-laboratory and inter-laboratory tests6. The analysis provides reliable results in less than 5 min, which has favored its implementation and further development7,8.
Several microorganisms among Gram-negative (e.g., Citrobacter spp., Enterobacter spp., and Escherichia spp.,etc.) and Gram-positive bacteria (e.g., Staphylococcus aureus and Streptococcus spp.) have been identified over the years, due to improvements in the method9. Moreover, the identification of pathogenic fungi has been implemented using an upgrade of the processing software and the spectral database of the reference strain10. Although the use of MALDI-TOF MS is widespread across the world, few data have been published regarding its application for the identification of Legionella spp. in clinical and environmental samples11,12,13,14,15,16. Moreover, in Italy, to the best of our knowledge, MALDI-TOF MS is not widely used in clinical or environmental laboratories where routine Legionella surveillance is carried out17.
Legionella is a genus of Gram-negative bacteria that are ubiquitous in both natural and man-made fresh water. The presence of bacteria in water distribution systems, as well as in soil and mud, is associated with a wide range of factors that promote their survival and proliferation18,19,20. The main factors associated with the Legionella presence and proliferation are water and chemical components (e.g., hardness, secondary disinfectant, total chlorine residual, and total organic carbon), pipeline characteristics (e.g., corrosion of materials, low flow-rate, and water pressure) and physical factors (e.g., temperature, pH, dissolved oxygen concentration). Moreover, its persistence in the environment is associated with natural hosts such as amoebae, protozoa, and biofilm formation. Legionella is associated with human infection through the inhalation of contaminated water aerosols or droplets, causing pneumonic and non-pneumonic forms called Legionellosis. A high impact of the disease is reported in elderly, smokers, and immunocompromised people, often with fatal outcomes21. Legionellosis is becoming an important public health concern due to its increasing incidence and high impact on health costs22. During diagnosis as well as during mandatory environmental surveillance, the isolation of Legionella is based on culture techniques23,24.
Despite the international and national guidelines on Legionella prevention and control, the culture technique remains poorly used in clinical practice. This occurred due to the difficulty of isolating microorganisms from clinical specimens, especially from the upper respiratory tract (e.g., nasopharyngeal or throat swab samples), the long duration of bacterial isolation and the expertise required for culture (e.g., media containing yeast extract and activated charcoal for Legionella growth). Moreover, the low sensitivity and cross-reactivity of common techniques used for Legionella identification (e.g., serological tests for serogroup identification of Legionella pneumophila), have been reported. In the same manner, also during epidemiological investigation, the isolation and identification of Legionella involve similar obstacles24,25, due to the high number of samples to process, the long incubation time and the presence of disinfection treatments, wich can interfere with Legionella growth. Several sensitive and specific methods such as real-time PCR, loop-mediated isothermal amplification (LAMP), Legiolert, direct fluorescent antibody (DFA) testing, indirect immunofluorescence assay (IFA), sequence-based typing (SBT), and amplification of the specific target gene (e.g. macrophage infectivity potentiator gene), have been developed over the years for water sample analysis as well as for isolates identification. These techniques are expensive and require a qualified team and sophisticated technologies. Moreover, considering the high abundance of Legionella in water environments and the increasing number of Legionella species reported in the last few years (66 Legionella species described), rapid, good, and reliable Legionella identification is the principal goal in clinical and environmental practice26,27.
The MALDI-TOF MS approach met these requirements. In recent years, several methodologies or mass spectrometry platforms have been developed1. One of the main points is represented by the appropriate sample pretreatment methods. In fact, in the clinical microbiology laboratory, three different sample preparation methods are routinely applied with some advantages and disadvantages.
The main methods used are: the direct colony transfer method, also known as the direct smear method (DS), and the on-target extraction method, also known as the extended direct smear method (EDS), which is frequently used to identify common Gram-negative, Gram-positive, and mucinous bacteria and the in-tube extraction method, also known as the full extraction method (E) reserved for hard-to-identify microorganisms (i.e., fungi and mycobacteria).
The selection of an appropriate and efficient method is essential for all applications since, technicians, frequently work with unknown bacteria28,29.
The aim of this study was to evaluate the best preparation method among DS, EDS, and E, for improving the MALDI Biotyper identification for Legionella spp. The results obtained were used to study the relationships among the isolates using hierarchical cluster analysis (HCA), and subsequently compared with the phylogenetic tree generated from the gene sequencing results. Finally, an in-house library, was tested on isolates with low identification scores or not-identified, according to the instrument’s database. This study suggest a quick and easy workflow to introduce on Legionella surveillance routine, able to increase the identification process and simultaneously trace the relationships among the isolates.
Results
Identification by mip and rpoB gene sequencing and phylogenetic analysis
The results obtained by mip and rpoB gene sequencing for the dataset, in relation to the reference strains present in the GenBank database, showed perfect concordance (100% match).
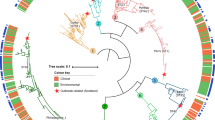
The species detected by the sequencing analysis of both genes, were: 36/104 (34.61%) L. anisa, 6/10 (45.77%) L. londiniensis, 20/104 (19.23%) L. nautarum, 31/104 (29.81%) L. rubrilucens, 10/104 (9.62%) L. taurinensis, and 1/104 (0.96%) L. pneumophila. The phylogenetic tree elaborated using the mip and rpoB sequences, revealed the presence of six clades, in which the reference strains were strictly related to the environmental ones (Figs. 1 and 2).

Phylogenetic tree based on mip gene sequencing of the three representative environmental isolates and closely related species of the genus Legionella. Branch labels show the substitutions per site calculated by Bayesian inference using the Markov Chain Monte Carlo (MCMC) method38,39. Bar 0.03 substitutions per nucleotide position.

Phylogenetic tree based on rpoB gene sequencing of the three representative environmental isolates, and closely related species of the genus Legionella. Branch labels show the substitutions per site calculated by Bayesian inference using the MCMC method38,39. Bar 0.02 substitution per nucleotide position.
MALDI Biotyper results and comparison with gene sequencing analysis
The identifications obtained for the dataset are reported in Table 1.
Correct identification (high confidence + low confidence score) of the isolates was achieved for 97.11%, 98.07% and 95.19% for DS, EDS and E methods, respectively.
A comparison between the three preparation methods returned no significant differences among them. In particular, the p-values between the pairs were as follows: DS vs. EDS showed a p-value = 1.00, DS vs. E displayed a p-value = 1.00, and EDS vs. E showed a p-value = 0.89.
Considering the identification among the species present in the data set, using the three sample preparation methods, showed for L. anisa a high identification score for the main number of isolates, with only one identified with a low identification score using the DS method. For L. londiniensis, using DS and E methods, all six isolates were correctly identified with high and low confidence score, while a decrease in identification rate was observed for the EDS method. L. nautarum and L. pneumophila displayed a reliable identification rate for all methods, mainly with high confidence scores. Ten isolates from the dataset showed reliable results with high confidence scores, reporting a match with Legionella sp HWL_078 HWH. Among L. rubrilucens, only a few isolates were identified with high confidence scores, while the others were identified with low confidence score or remained unidentified. The comparison between MALDI Biotyper results and gene sequencing identification data, is summarized in Table 2.
These MALDI-TOF MS results agreed with the sequencing identification for all the species, except for L. taurinensis and L. rubrilucens. In detail, the ten environmental isolates of L. taurinensis, belonging to our dataset, for both genes reported a match of 100% with the reference strain of L.taurinensis ATCC700508T. Considering that these isolates, as previously described, were identified by MALDI Biotyper as Legionella sp HWL_078 HWH, it is possible that the strain present in the instrument database was not correctly identified by manufacturer, and could belong to L. taurinensis species. Regarding L. rubrilucens, despite the MALDI Biotyper results, the gene sequencing confirms the identification of all the isolates. The sensitivity, confidence interval and error rate among the methods for each Legionella species were calculated. The DS method displayed a sensitivity of 97.12% (95% CI: 0.93–0.99) and an error rate of 2.88%, while the EDS method showed a sensitivity of 98.08% (95% CI: 0.94–0.99) and an error rate of 1.92%, due to a L. rubrilucens isolate not identified by the DS method. The E method showed a sensitivity of 95.19% (95% CI: 0.91–0.99) and the highest error rate (4.90%) among the different methods, associated with the un-identification of five isolates of L. rubrilucens. The sensitivity of the MALDI Biotyper in the identification of L. rubrilucens strains could be improved considering the cut-off proposed by Martiny et al.30. It attributes a reliable identification also when the difference in the log (score) values between the first- and second-best matches is ≥ 0.3. Applying this new cut-off, the sensitivity arises the value of 99.04%, 100% and 99.04% for DS, EDS and E, respectively. Specificity among the methods and Legionella species, was not measured considering the presence in the dataset of Legionella isolates already identified by gene sequencing (absence of false-positive results). To solve the un-identification returned for L. rubrilucens strains (n = 31), new MSPs acquired from environmental L. rubrilucens isolates, genotyped by WGS (i.e., IM34) were used, developing an in-house library. The robustness of the in-house library was tested by subjecting all the environmental L. rubrilucens isolates (n = 31), to a second acquisition. The use of new MSP affected the sensitivity of the technique, that arised the value of 100%. In particular, among the tecniques, the following trends were observed: 21/31 (67.74%) for DS, 24/31 (77.42%) for EDS and 25/31 (80.65%) for E, with high confidence scores. The remaining isolates were identified with low confidence score with 10/31 (32.26%) for DS, 7/31 (22.58%) for EDS and 6/31 (19.35%) for E. Moreover, to understand the problem with L. rubrilucens identification, the only spectrum present in the Bruker’s database was compared with the ones acquired for environmental isolates. This comparison showed differences in terms of the mass /charge ratio (m/z) and peak intensity, that will be further investigated (Fig. 3).

Comparison between MSPs produced for the L. rubrilucens environmental strain IM34 and L. rubrilucens DSM118847T present in Bruker’s database (a). All detected peaks of the reference strains are blue lines; the peaks of the environmental isolates are indicated with green, yellow, or red lines. The colors represent the similarity in terms of mass/charge (m/z) between the strains: green for the same m/z, yellow for a similar m/z, and red for a different m/z. (b) represents the interval from 6000 to 7000 m/z, with the main differences observed.
Hierachical cluster analysis (HCA) among the isolates: MALDI Biotyper vs. gene sequencing phylogenetic tree
A dendrogram based on HCA of MSPs, obtained for each isolate of the dataset, generated a tree-like structure containing six main clades, representing L. nautarum, L. pneumophila, L. londiniensis, L. anisa, L. taurinensis and L. rubrilucens (Fig. 4).

Dendrogram based on whole-cell MALDI-TOF MS (Maldi Biotyper System) of 18 representative environmental isolates and other Legionella strains present in the instrument library.
All the isolates belonging to L. nautarum and L. pneumophila are strictly related to the corresponding reference strains, present in Bruker’s database (L. nautarum DSM21805T and L. pneumophila ATCC33152T and DSM7513T, respectively). Interesting data were found for the clade composed of L. londiniensis, L. anisa and L. rubrilucens.
In detail, in the L. rubrilucens and L. londiniensis clades, the unique reference MSP present in Bruker’s database (L. londiniensis 191108_01 OOV and L. rubrilucens DSM 11884T, respectively), was located far from the main group, represented by the environmental isolates (indicated as MCH for L.rubrilucens and VS for L. londiniensis), without differences among the techniques. For L. anisa, the instrument’s database is composed of ten reference and environmental strains. Two major clades were observed: one represented by the main number of reference strains with VS59 and VS57 and the second represented by L.anisa 37,715 BBR with the MTH3. Again, no differences were found between the techniques.
To better understand these distances, a new dendrogram was developed using MSPs generated from L. rubrilucens, L. londiniensis and L. anisa, and added into an in-house library. Figure 5 shows the reduction in distance for L. londiniensis and L. rubrilucens, where all isolates formed a single clade with the in-house library (L. londiniensis VS43, and L. rubrilucens IM34). However, for the L. anisa clade, an improvement in correlation was not observed, considering that the environmental strain, L. anisa SC21, remains strictly correlated with the clade composed of L. anisa 37715 BBR and MTH3.

Dendrogram based on whole-cell MALDI-TOF MS (MALDI Byotyper System) using the Biotyper database and in-house library based on three representative environmental isolates, analyzed with three methods: DS, EDS, E.
Discussion
MALDI-TOF MS is considered an important technology able to improve bacterial identification, with more efficient, rapid and accurate systems.
Several studies have demonstrated its high reproducibility in both intra-laboratory and inter-laboratory tests6, starting from different preparation methods according to the manufacturer’s suggestions, that are able to improve the identification level obtained6,29. Moreover, the choice of method depends on the experience and practices of personnel/laboratories, and the internal validation protocol used by the operators. However, in routine laboratories, it is not possible to perform all of these procedures, to save time, personnel and costs29.
In this study, we evaluated the performance of the three sample preparation methods (DS, EDS, and E) used to enhance Legionella identification through MALDI Biotyper. Moreover, comparisons were performed with mip gene sequencing, the Legionella identification reference method, and the rpoB gene, according to Pascale et al.31. Good and reliable identification was obtained for the main numbers of isolates, independent of the methods, while a low number of isolates were identified using the E method. The sensitivity of the instrument among methods and species showed a high confidence score with the DS and EDS, while a slight decrease was reported with the E method. Considering the results obtained, L. rubrilucens exhibited an interesting pattern. Most of the isolates were correctly identified (high and low confidence scores) by the DS and EDS methods. Five isolates in the dataset remained unidentified using the E method, demonstrating a decrease of sensitivity only for this species. The second acquisition performed using the L. rubrilucens MSPs, inserted in the in-house library, increased the identification score from red or yellow to green, increasing the identification level and the sensitivity. An explanation could be provided by the spectra peaks distribution, which refers to only one reference strain present in Bruker’s database. The differences in the number of peaks obtained by comparing the L. rubrilucens environmental isolates with those of the Bruker strain, other than evident changes in the m/z and intensity values, could affect the identification rate. The improvement in terms of the identification score achieved using the in-house library confirms our hypothesis, which is in line with the results obtained by Chalupova et al., which showed that spectra quality could affect identification10.
The same evidence explains the differences in the L. rubrilucens clade, found using the HCA analysis. The reduction in the distance inside the clade, among the isolates and the new MSP provided by the in-house library, with respect to that presented in the instrument database, confirms how low quality spectrum acquisition, associated with the extraction method used, and laborious spectra processing could interfere with good and reliable identification. Regarding L. londinensis, the distance detected between environmental isolates and the MSP in the instrument database, was overcome by introducing the new MSP to the in-house library.
Despite the good identification provided by the instrument for L. anisa, the HCA revealed two main clades that remain well separated, despite the use of the in-house library. This could be attributed to the presence of changes in the spectra’s peaks distribution (data not shown) in the instrument database of both references and environmental strains. Indeed, the isolates in our dataset are strictly related to the environmental instrument isolates, demonstrating that there is high variability inside the species with respect to the most well-preserved type strains used to build manufacture’s library. The issue of spectra stability could also affect the reference strains, as addressed in several studies7,32. Some authors well described that storage, and the repeated freezing and thawing cycles, influence the qualitative features of the spectra, inducing a decrease in identification score, which is mainly attributed to the mass shift of some peaks7.
Among L. pneumophila, as well as for L. nautarum, the rate of identification was high, from 75 to 100%, with few differences among the techniques.
No significant differences were found among the sample method preparations in terms of the level of identification achieved; therefore, as the best method, we suggest the use of the DS, an easier and faster method that does not require specialized expertise. Moreover, looking at the score acquired for our dataset, we can confirm the proposal of Martin et al. on the possibility of decreasing the instrument’s cutoff from 2.0 to 1.6 to obtain good and reliable identification30.
Regarding the concordance of results obtained using the MALDI Biotyper and gene sequencing, it was demonstrated that the two techniques yield the same level of identification. The phylogenetic tree developed using mip and rpoB sequences, showed the presence of five main clades, illustrating a group of monophyletic species (L. rubrilucens and L. taurinensis) and four other well-separated clades, one for each species present in the dataset. The relationships among the species were reliable according to the HCA results. Therefore, the dendrogram based on the ribosomial proteins profile could be useful for quick and rapid analysis of the relationships among the isolates.
In the presence of Legionella identification, MALDI Biotyper can help to discriminate the species, especially in the presence of non-L.pneumophila species, which are generally identified only using molecular techniques, permitting to assess the relationship among the isolates quickly and easily. MALDI-TOF MS could be used in a screening phase, especially in the presence of many samples, as occurs during clusters and epidemic events. The HCA provided by the instrument could support the first estimation of correlations among the isolates, highlighting the links that need to be investigated in depth using molecular approaches, such as the WGS analysis, reducing the time and cost.
This study is the first to compare the identification performance of three different sample preparation methods used for Legionella identification, with results in line with those of Wang et al.29. for a large microorganisms dataset. The results obtained suggest that it is possible to use directly the DS method to obtain fast and reliable identification of the Legionella species tested. Moreover, it points out the key role of a well-established and standardized in-house library, developed using the best extraction methods, to be used in parallel with the instrument database, which according to Drevinek et al. can improve identification7.
In addition, according to Moliner et al. and Drevinek et al., increasing the number of MSPs in an in-house library, rather than evaluating spectra quality, is the first step in improving instrument identification power7,11. These aspects are urgently needed and, at present, constitute the main limitation of our work, representing a point for future research.
In conclusion, the role of Legionella in public health emphasizes the importance of its surveillance in conventional and unconventional artificial environments, which represent a reservoir of infections33,34.
The life and colonization of Legionella in water distribution systems are still under study and new valuable information must be acquired for human safety starting from a good and reliable identification step. The approach used in this study could improve the knowledge regarding Legionella identification, especially in accordance with the new Directive on water for human consumption35. Overall, our study could improve the identification of Legionella in mandatory routine water quality assessments in all types of buildings, not only in conventional facilities (e.g. hospitals). The introduction of a sensitive and reliable, easy, and low-cost identification system could support not only routine laboratories but also improve the actual common practice, which is still focused only on the presence or absence of bacteria. Moreover, a rapid study of the relationships among the strains in water distribution systems, using HCA represents the first approach to assess changes in bacterial ecology and establish the mechanism of bacterial water survival.
Methods
The isolates tested in the study (n = 104) were obtained from the biobank produced in the laboratory (MAbLab Colture Collection), during the Legionella environmental surveillance program. According to the Legionella guidelines and the recent Italian legislation on water for human consumption, Legionella surveillance in priority and non-priority buildings is mandatory24,25,36.
Legionella culture and isolates selection
The hot and cold water samples were analyzed for Legionella isolation, according to ISO 11731:201723. Briefly, the membrane filtration technique was performed using 1L of water samples, using a 0.22 μm polyethersulfone membrane (Sartorius, Bedford, MA, USA). The untreated, filtered, heated, and acid-treatments aliquots from 0.2 to 0.1 mL of samples, were seeded on the selective medium glycine-vancomycin-polymyxin B-cycloheximide (GVPC) (Thermo Fisher Diagnostic, Basingstoke, UK), and then incubated at 35 ± 2 °C with 2.5% CO2. Legionella growth was assessed every two days, until 15 days of incubation. Typical colonies with morphological features ascribable to the Legionella genera were subcultured on buffered charcoal yeast extract (BCYE) agar supplemented with L-cysteine (cys+) and without L-cysteine (cys-) (Thermo Fisher Diagnostics, Basingstoke, UK). The colonies that were able to grow only on Legionella BCYE cys + agar, with no growth on Legionella BCYE cys-, were used for phenotypical and genotypic identification and then frozen at – 20 °C using a glycerol mixture (Biolife, Milan, Italy).
To perform gene sequencing and MALDI-TOF MS technique, all the isolates in glycerol stocks, belonging to the MAbLab collection were seeded (25 µl) on BCYE cys + plates, and incubated under aerobic conditions at 35 °C in a 2.5% CO2 atmosphere, for 24–72 h. The time of growth is species related. The isolates grown, were then subcultured in new plates of BCYE cys + and incubated for another 24 h.
Identification by gene sequencing analysis
A total of 104 isolates selected based on culture results, were analyzed by the mip and rpoB gene sequencing protocol. The DNA was extracted using the InstaGene Purification Matrix (Bio-Rad, Hercules, CA), and the concentrations were determined using an Eppendorf Biophotometer with Traycell (Hellma, Milano, Italy). PCR analysis of all the isolates was performed to determine the gene sequences of mip and rpoB as described by Ratcliff et al. and Ko et al., respectively37,38.
Briefly, mip gene amplification was performed using degenerate primers modified by M13 tailing to avoid noise in the DNA sequence [Legmip_f_M13F TGTAAAACGACGGCCAGTGGGRATTVTTTATGAAGATGARAYTGG and Legmip_r_M13R CAGGAAACAGCTATGACCTCRTTNGGDCCDATNGGNCCDCC]. PCR was conducted in a 50 µL reaction volume containing DreamTaq Green PCR Master Mix 2X (Thermo Fisher Diagnostic Basingstoke, UK) and 40 pmol of each primer; 100 ng of the DNA extracted from the presumed Legionella colonies was added as the template.
Instead, rpoB gene amplification was performed in a 50 µL reaction volume containing 100 ng of template DNA and 40 pmol of each primer (RL1 5’- GATGATATCGATCAYCTDGG – 3’; RL25’- TTCVGGCGTTTCAATNGGAC). PCR products were visualized by electrophoresis on a 2% agarose gel. Following purification by the ExoSAP-ITTM PCR Product Cleanup kit (Applied Biosystems, Foster City, CA), the mip and rpoB amplicons were sequenced using BigDye Chemistry and analyzed on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA). The raw sequencing data were assembled using CLC Main Workbench 7.6.4 software (QIAgen, Hilden, Germany). The sequences of the mip gene deposited in the publicly available Legionella mip gene sequence database, created by the ESCMID Study Group for Legionella Infections (ESGLI), were utilized for comparison via a similarity analysis tool. Although this database is under development and not yet accessible from the outside, UKHSA database curators can still access it internally (Legionella-sbt@ukhsa.gov.uk). Species-level identification was performed on a similarity score of > 98% 37,39. The rpoB gene sequences obtained from several culture collections were compared to those of type strains deposited in the National Center for Biotechnology Information (NCBI) from several culture collections, as: the American Type Culture Collection (ATCC), the National Collection of Type Cultures, the Central Public Health Laboratory (NCTC), The National Institute of Technology and Evaluation (NBRC), and the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSM). In relation to the new Legionella classification scheme targeting the rpoB gene, which was developed on a 329 bp gene fragment and proposed by Pascale et al., species-level identification was performed on a similarity score of > 95.2% 31.
The mip and rpoB gene sequences generated were submitted to GenBank. The provided accession numbers were as follows: from ID PP400560 to PP400663 and from ID PP400456 to PP400559 for mip and rpoB, respectively.
Phylogenetic analyses based on mip and rpoB gene sequencing
The phylogenetic trees for the mip and rpoB sequences were built using multiple sequence alignment (MSA), to estimate the relationships among the 104 isolates. Manual editing of the sequences was carried out, and the sequences were trimmed to the same length with respect to the reference sequences. BLAST searches were used to obtain the top ten results for the best identification.
The nucleotide sequences were aligned by the multiple sequence comparison by log-expectation (MUSCLE) algorithm40, which was performed in the Geneious Prime genome browser implemented with 2023.0.4 software (http://www.geneious.com)41, retaining the default settings. Phylogenetic trees were constructed with the aligned sequences that were passed to the Bayesian Evolutionary Analysis Utility (BEAUti) (v. 1.10.4)42, setting HKY as the substitution model, Yule as the speciation process, and 10.000.000 as the length of the chain43; the other parameters were kept by default. The generated file was used as input for the Bayesian Evolutionary Analysis by Sampling Trees (BEAST) software (v. 1.10.4)44, which generated various samples of phylogenetic trees. TreeAnnotator software (v. 1.10.4), which permits to elaborate the best tree based on the information provided by the samples of trees previously obtained, was used. This tree was subsequently visualized and edited by figTree software (v. 1.4.4).
Sample preparation methods for MALDI-TOF MS
All the isolates (n = 104) were identified using MALDI Biotyper (Bruker Daltonics, Bremen, Germany). Three biological and three technical replicates were used in this phase of the study. A comparison among three sample preparation methods: (i) direct smear (DS), (ii) extended direct smear (EDS) and (iii) full extraction (E), was performed for all isolates following the manufacturer’s instructions. Thereafter, each biological replicate biomass was harvested and spotted in triplicate on the MALDI Biotyper target. Considering the preparation methods, the isolates were then processed as follows: for the DS method, the spots were overlaid with 1 µl of α-Cyano-4-hydroxycinnamic acid (HCCA), and allowed to air dry before the measurement; for the EDS method, the spots were covered with 1 µl of 70% v/v formic acid. Once dry, 1 µl of HCCA was added to each of the spots. The E method, it was performed using the full protein extraction protocol, developed by Bruker (instrument manual). Briefly, some biomass was suspended in 300 µL of MS-grade water, and then 900 µL of absolute ethanol (99% v/v) was added. The suspension was mixed carefully and centrifuged for 2 min at 15,000 ×g. The supernatant was discarded, and the centrifugation was repeated. The pellet was resuspended in 30 µL of 70% v/v formic acid, and then 30 µL of acetonitrile was added by pipetting the solution up and down two or three times. At the end, the solution was again centrifuged and 1 µL of the surnatant was spotted on the target plate in triplicate. After drying, the spots were covered with 1 µl of HCCA, and the analysis was performed. Spectra acquisition and processing were performed by Microflex LT mass spectrometer (2000–20,000 Da, linear positive mode) and MALDI Biotyper Compass 4.1.1 software, using the manufacturer’s library (version BDAL 7854), which containing 48 Legionella main spectra (MSPs) of reference and environmental strains.
The identification result was considered reliable when the log(score) was ≥ 2.00 (“high confidence level” or identification at the species level) or between 1.70 and 1.99 (“low confidence level” or identification at the genus level). In addition, in the case of a score between 1.50 and 2.00, we could obtain correct species identifications if the first three proposed results were identical or in those cases in which the best match was at least 1.60 and the difference in the log(score) values between the first- and second-best matches was ≥ 0.30. The environmental isolates present in the data set were named as: VT, TIN, VS, MTH, and MCH.
Statistical analysis
R Statistical Software (version 4.4.1, “Race for Your Life” R Foundation for Statistical Computing, Vienna, Austria) was used to conduct the statistical analysis. In particular, the normality of the variables was assessed using the Shapiro‒Wilk test. The Friedman test, a non-parametric method, was conducted to compare three or more related or paired groups. Additionally, the Wilcoxon test, was employed as a post-hoc analysis to compare two paired or matched samples. In conclusion, the Bonferroni correction, a statistical adjustment used to reduce the likelihood of obtaining false-positive results during multiple comparisons, was applied. All the statistical tests were set to have a significance level of (p) < 0.05.
Determination of the sensitivity and confidence interval (CI) at the 95% level of significance were performed using Microsoft Excel software version 2108 for Windows 10 (Microsoft Office LTSC Professional Plus 2021, USA).
MALDI Biotyper new spectra acquisition for an in-house library
To develop an in-house library, the extracted environmental isolates (two isolates for each species), were spotted eight times on MALDI Biotyper target plate, and measured three times (technical replicates), following the manufacturer’s suggestions. The 24 raw spectra acquired were loaded into flexAnalysis v3.4 software (Bruker Daltonics, Bremen, Germany) for spectral cleaning and quality control, following the manufacturer’s instructions. Briefly, the spectra were subjected to baseline subtraction, smoothing, removal of outlier peaks, anomalies and spectra with flat lines, to mantain the differences between the largest and the smallest masses ≤ 500 ppm. After spectral quality control, a minimum of 20 processed spectra for each strain are required to create an MSP. The produced MSPs were inserted into our in-house library, by using MALDI Biotyper Compass Explorer software (v.4.1.1).
Hierarchical cluster analysis (HCA) developed using MALDI Biotyper database and in-house library
The dendrogram based on HCA was developed using MALDI Biotyper Compass Explorer software (v.4.1.1), using the environmental MSPs of the dataset (noted as VT, TIN, VS, MTH, and MCH) and the MALDI Biotyper database. Regarding the MSPs present in the database of the instrument, there is only one MSP for L. londiniensis, L. rubrilucens, L. sp HWL_078 HWH and L. nautaturm, two MSPs for L. pneumophila serogroup 1, while for L. anisa the library contains ten different MSPs, belonging to several reference strains (ATCC and DSMZ), other than Bruker’s environmental isolates. Considering the high numbers of MSPs obtained for our dataset using the three sample preparation methods (n = 312), this may interfere with the clarity of the dendrogram. For graphical purposes, three of the 104 isolates from each species were selected as example. The selected 18 isolates processed through the three sample preparation methods (DS, EDS and E) were displayed on the HCA dendrogram .
The accession numbers of mip and rpoB gene sequences are from ID PP400560 to PP400663 and from ID PP400456 to PP400559, respectively.
Data availability
The datasets used and analyzed during the current study are available from the corresponding author upon reasonable request. The accession numbers of mip and rpoB gene sequences are from ID PP400560 to PP400663 and from ID PP400456 to PP400559, respectively.
References
Oviaño, M. & Rodríguez-Sánchez, B. MALDI-TOF mass spectrometry in the 21st century clinical microbiology laboratory. Enfermedades Infecciosas y Microbiologia Clinica 39, 192–200 Preprint at (2021). https://doi.org/10.1016/j.eimc.2020.02.027
Maier, T., Klepel, S., Renner, U. & Kostrzewa, M. Fast and reliable MALDI-TOF MS-based microorganism identification. Nat. Methods 3, i–ii (2006). https://doi.org/10.1038/nmeth870
Croxatto, A., Prod’hom, G. & Greub, G. Applications of MALDI-TOF mass spectrometry in clinical diagnostic microbiology. FEMS Microbiol. Rev. 36, 380–407 (2012).
Singhal, N., Kumar, M., Kanaujia, P. K. & Virdi, J. S. MALDI-TOF mass spectrometry: an emerging technology for microbial identification and diagnosis. Front. Microbiol. 6, 1–16 (2015).
Wahl, K. L. et al. Analysis of microbial mixtures by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal. Chem. 74, 6191–6199 (2002).
Mellmann, A. et al. High interlaboratory reproducibility of matrix-assisted laser desorption ionization-time of flight mass spectrometry-based species identification of nonfermenting bacteria. J. Clin. Microbiol. 47, 3732–3734 (2009).
Drevinek, M., Dresler, J., Klimentova, J., Pisa, L. & Hubalek, M. Evaluation of sample preparation methods for MALDI-TOF MS identification of highly dangerous bacteria. Lett. Appl. Microbiol. 55, 40–46 (2012).
Fox, A. Mass spectrometry for species or strain identification after culture or without culture: Past, present, and future. J. Clin. Microbiol. 44, 2677–2680 Preprint at (2006). https://doi.org/10.1128/JCM.00971-06
Edwards-jones, V. et al. Rapid Discrimination between Methicillin-Sensitive and Methicillin-Resistant Staphylococcus Aureus by Intact Cell Mass Spectrometry.
Chalupová, J., Raus, M., Sedlářová, M. & Šebela, M. Identification of fungal microorganisms by MALDI-TOF mass spectrometry. Biotechnol. Adv. 32, 230–241 Preprint at (2014). https://doi.org/10.1016/j.biotechadv.2013.11.002
Moliner, C. et al. Rapid identification of Legionella species by mass spectrometry. J. Med. Microbiol. 59, 273–284 (2010).
Fujinami, Y. et al. Rapid discrimination of Legionella by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Microbiol. Res. 166, 77–86 (2011).
Gaia, V., Casati, S. & Tonolla, M. Rapid identification of Legionella spp. by MALDI-TOF MS based protein mass fingerprinting. Syst. Appl. Microbiol. 34, 40–44 (2011).
Svarrer, C. W. & Uldum, S. A. The occurrence of Legionella species other than Legionella pneumophila in clinical and environmental samples in Denmark identified by mip gene sequencing and matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Clin. Microbiol. Infect. 18, 1004–1009 (2012).
Dilger, T., Melzl, H. & Gessner, A. Rapid and reliable identification of waterborne Legionella species by MALDI-TOF mass spectrometry. J. Microbiol. Methods. 127, 154–159 (2016).
Trnková, K. et al. MALDI-TOF MS analysis as a useful tool for an identification of Legionella pneumophila, a facultatively pathogenic bacterium interacting with free-living amoebae: a case study from water supply system of hospitals in Bratislava (Slovakia). Exp. Parasitol. 184, 97–102 (2018).
Pascale, M. R. et al. Evaluation of MALDI–TOF mass spectrometry in diagnostic and environmental surveillance of Legionella species: a comparison with culture and mip-gene sequencing technique. Front. Microbiol. 11, 1–12 (2020).
Rowbotham, T. J. Preliminary report on the pathogenicity of Legionella pneumophila for freshwater and soil amoebae. J. Clin. Pathol. 33, 1179–1183 (1980).
Fields, B. S. The molecular ecology of legionellae. Trends Microbiol. 4, 286–290 (1996).
Diederen, B. M. W. Legionella spplegionnairesnaires’ disease. J. Infect. 56, 1–12 (2008).
European Centre for Disease Prevention and Control (ECDC). Legionnaires’ Disease. In: ECDC. Annual Epidemiological Report for 2021. (2023). https://www.ecdc.europa.eu/sites/default/files/documents/legionnaires-disease-annual-epidemiological-report-2021.pdf
Viasus, D., Gaia, V., Manzur-Barbur, C. & Carratalà, J. Legionnaires’ disease: update on diagnosis and treatment. Infect. Dis.Ther. 11, 973–986 Preprint at (2022). https://doi.org/10.1007/s40121-022-00635-7
UNI EN ISO 11731:2017 Water Quality - Enumeration of Legionella. (2017). https://www.iso.org/standard/61782.html
Italian National Institute of Health. Guidelines for Prevention and Control of Legionellosis. Approvate in Conferenza Stato-Regioni Seduta Del 7 Maggio 2015. Italy. (2015). (2015).
European Parliament the Council of the European Union. Directive (EU) 2020/2184 of the European parlliament and of the Council of 16 December 2020 on the quality of Water intended for human consumption. Official J. Eur. Union. 2019, 1–61 (2020).
Cristino, S. et al. Characterization of a Novel species of Legionella isolated from a Healthcare Facility: Legionella resiliens sp. nov. Pathogens 13, 250 (2024).
Girolamini, L. et al. Legionella bononiensis sp. nov., isolated from a hotel water distribution system in northern Italy. Int. J. Syst. Evol. Microbiol. 72, 1–8 (2022).
Van Belkum, A., Welker, M., Pincus, D., Charrier, J. P. & Girard, V. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry in clinical microbiology: What are the current issues? Ann. Lab. Med. 37, 475–483 Preprint at (2017). https://doi.org/10.3343/alm.2017.37.6.475
Wang, J. et al. Evaluation of three sample preparation methods for the identification of clinical strains by using two MALDI-TOF MS systems. J. Mass Spectrom. 56(2), e4696 (2020). https://doi.org/10.1002/jms.4696.
Martiny, D., Dediste, A., Vandenberg, O. & Vandenberg, O. Comparison of an in-house method and the commercial sepsityper kit for bacterial identification directly from positive blood culture broths by matrix-assisted laser desorption-ionisation time-of-flight mass spectrometry. Eur. J. Clin. Microbiol. Infect. Dis. 31, 2269–2281 (2012).
Pascale, M. R. et al. New Insight regarding Legionella non-Pneumophila species Identification: comparison between the traditional mip gene classification Scheme and a newly proposed Scheme Targeting the rpoB Gene. Microbiol. Spectr. 22;9(3):e0116121 (2021). https://doi.org/10.1128/Spectrum.01161-21.
Horneffer, V., Haverkamp, J., Janssen, H. G. & Notz, R. MALDI-TOF-MS analysis of bacterial spores: wet heat-treatment as a new releasing technique for biomarkers and the influence of different experimental parameters and microbiological handling. J. Am. Soc. Mass. Spectrom. 15, 1444–1454 (2004).
Mazzotta, M., Salaris, S., Pascale, M. R., Girolamini, L. & Cristino, S. Occurrence of Legionella spp. In man-made water sources: Isolates distribution and phylogenetic characterization in the Emilia-Romagna region. Pathogens 3;10(5):552. (2021). https://doi.org/10.3390/pathogens10050552.
Girolamini, L. et al. Sit bath systems: a new source of Legionella infection. PLoS One. 15, 1–19 (2020).
Italian Republic. Legislative Decree 23.02.2023, n.18, Implementation of the Water Quality Directive 2184/2020/EC Relative to Water Quality Intended for Human Consuption. OJ. of the Italian Republic n.55, 06.03. (2023). (2023).
D.Lgs.31. Legislative Decree 02.02.2001, n.31. Implementation of TheWater Quality Directive 98/83/EC Relative to Water Quality Intended for Human Consumption. OJ. of the Italian Republic n. 52, 3.03. (Italy, 2001). (2001).
Ratcliff, R. M., Lanser, J. A., Manning, P. A. & Heuzenroeder, M. W. Sequence-based classification scheme for the genus Legionella targeting the mip gene. J. Clin. Microbiol. 36, 1560–1567 (1998).
Ko, K. S. et al. Application of RNA polymerase β-subunit gene (rpoB) sequences for the molecular differentiation of Legionella species. J. Clin. Microbiol. 40, 2653–2658 (2002).
Fry, N. K. et al. Identification of Legionella spp. by 19 European reference laboratories: results of the European Working Group for Legionella Infections External Quality Assessment Scheme using DNA sequencing of the macrophage infectivity potentiator gene a. Clin. Microbiol. Infect. 13, 1119–1124 (2007).
Edgar, R. C. Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32. MUSCLE, 1792–1797 (2004).
Kearse, M. et al. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649 (2012).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012).
Gernhard, T. The conditioned reconstructed process. J. Theor. Biol. 253, 769–778 (2008).
Drummond, A. J. & Rambaut, A. BEAST: bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214 (2007). https://doi.org/10.1186/1471-2148-7-214
Acknowledgements
The authors would like to thank Dr. Miriam Cordovana for her support on MALDI Biotyper database.
Funding
This work received no specific funds from any funding agency in the public, commercial or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
L.G. , P.C., and S.C. carried out the conceptualization, investigation, and writing of original draft preparation; L.G., P.C., F.M., M.R.P., and S.S. performed sample collection and analysis; L.C., C.D., M.L.S. and A.G. performed molecular and phylogenetic analysis. All authors reviewed and approved the submitted version of manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Girolamini, L., Caiazza, P., Marino, F. et al. Identification of Legionella by MALDI Biotyper through three preparation methods and an in-house library comparing phylogenetic and hierarchical cluster results. Sci Rep 15, 2162 (2025). https://doi.org/10.1038/s41598-025-85251-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-85251-4
