
Abstract
The risk of suicide in patients with major depressive disorder (MDD) poses a major concern, with studies suggesting that genetics may be a contributing factor. Although there are many transcriptomic studies on postmortem brain tissue related to suicidal behavior, the blood transcriptional mechanisms of suicidal ideation (SI) remain unknown. This study utilized a weighted gene coexpression network analysis (WGCNA) approach to investigate the associations between gene coexpression modules and SI in individuals with MDD using peripheral blood RNA-seq data from 75 MDD patients with SI (MDD_SI), 82 MDD patients without SI (MDD_nSI), and 149 healthy controls (HC). An ANCOVA was conducted to assess differences in gene coexpression modules among groups, with age and sex included as covariates. The gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) databases were used to annotate module functions. Results indicated that the magenta module (associated with RNA splicing processes) differentiated MDD_SI from MDD_nSI (p = 0.021), while the green module (related to immune and inflammatory responses) distinguished MDD_SI from HC (p = 0.004). Additionally, three modules showed differences between MDD_nSI and HC: magenta (p = 0.009), brown (related to innate immunity and mitochondrial metabolism; p = 0.001), and turquoise (associated with energy metabolism and neurodegeneration; p = 0.005). Our findings highlight that gene expression regulation, immune response, and inflammation may be linked to SI in patients with MDD, while pathways associated with innate immunity, energy metabolism, mitochondrial function, and neurodegeneration appear to be more broadly related to MDD.
Similar content being viewed by others


Introduction
Major depressive disorder (MDD) is a serious and debilitating psychiatric disorder that affects approximately 121 million individuals globally1. Patients with MDD face an elevated risk of suicide, a leading cause of death worldwide that results in an estimated 1 million fatalities annually2. Among individuals who died by suicide with a psychiatric diagnosis, up to 38.9% had previously been diagnosed with MDD3. Suicidal ideation (SI), an early warning sign of potential suicidal behavior, frequently occurs in patients with MDD and presents a crucial target for timely intervention4,5. However, the biological mechanisms that underlie SI in MDD patients remain largely unexplored and poorly understood6.
Genetic factors have been suggested to play a role in suicide7,8,9, but focusing solely on DNA-level studies has limitations, as suicide risk is shaped not only by genetics but also by environmental factors and personal circumstances10. Recent advances in transcriptomics offer promising avenues for understanding the complex biological processes associated with MDD and suicide by examining RNA-level gene expression, which provides insights into gene structure, function, and regulatory networks across the genome11. Transcriptomic studies using postmortem brain tissue have shed light on gene expression patterns associated with MDD and suicide. For example, Pantazatos et al. (2017) identified 34 differentially expressed genes (DEGs) linked to MDD and suicide, particularly genes involved in oligodendrocyte differentiation and ATPase activity12. Similarly, Pandey et al. (2019) observed increased expression of cytokines, inflammasomes, and Toll-like receptors (TLRs) in brain samples from patients with MDD who died by suicide13. Furthermore, Sha et al. (2023) reported that inflammatory and neurodevelopmental pathways are associated with suicide in postmortem brain tissue14. However, these studies ignored the high interconnection between genes with similar functions and focused only on the identification of DEGs.
Gene coexpression modules, which represent groups of interconnected genes with shared functions, provide a more nuanced understanding of complex biological mechanisms15. Weighted gene coexpression network analysis (WGCNA) has been utilized to cluster genes with similar expression into a gene coexpression module, and this approach has been widely applied to biological processes related to neurological and psychiatric disorders, including MDD16,17,18. Previous studies have employed the WGCNA algorithm to analyze the transcriptome of human postmortem brain tissue from suicide decedents and found that suicidal behavior is related to nitric oxide synthase activation, the estrogen signaling pathway, and synaptic dysfunction15,19. However, suicide is a complex process, and the biological mechanisms of SI and suicidal behavior may not be completely consistent4. Detecting the transcriptome in clinically depressed individuals with SI, may uncover RNA expression patterns specifically related to SI. To our knowledge, only one study to date has explored the gene coexpression modules in clinical patients with MDD to reveal the biological processes and pathways underlying SI20. This study indicated that SI-associated gene modules were enriched for functions related to defending against microbial infections, inflammation, and adaptive immune responses20. Furthermore, evidence suggests that over 80% of gene expression in peripheral blood is similar to that in the brain21. Given the accessibility of peripheral blood, it may provide a practical means of exploring the transcriptomic landscape of SI, potentially circumventing the limitations of postmortem brain tissue studies.
Identifying gene coexpression modules associated with SI might be pivotal in clinical translational studies for developing blood-based biomarkers that could identify individuals at risk of suicide early. Thus, the aim of this study was to employ the WGCNA method22 to identify the gene coexpression modules associated with SI using peripheral blood RNA-seq data. Subsequently, functional enrichment analysis of the modules was conducted to elucidate their biological functions, with the goal of uncovering transcriptomic mechanisms associated with SI in MDD patients.
Results
Demographics and relevant clinical measurements for the participants are shown in (Table 1). There was no difference in age among the three groups, but sex (F = 9.16, p = 0.010) was significantly different among the three groups. The MDD patients with SI (MDD_SI) and MDD patients without SI (MDD_nSI) groups did not differ in onset age, total duration, onset number, or HAMA score, but as expected, they differed in HAMD score (t = 35.6, p < 0.001).
The data are presented as the mean (SD) unless otherwise stated.
MDD_nSI major depressive disorder without suicidal ideation, MDD_SI major depressive disorder with suicidal ideation, HC healthy control, HAMD hamilton rating scale for depression, HAMA hamilton rating scale for anxiety.
Gene coexpression network construction
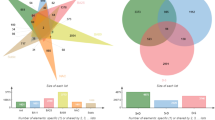
In this study, we identified 61,806 mRNA transcripts through alignment with the reference genome. After removing transcripts with low expression levels, 10,884 genes were retained for further analysis. Through WGCNA, a total of 19 coexpression modules were constructed, with the number of genes in each module ranging from 37 to 2955. Notably, the grey module comprised 734 genes and did not belong to any specific module (Table S1). Four modules were significantly different among the three groups (Fig. 1). Post hoc analysis revealed that one module differed between the MDD_SI and MDD_nSI groups (MEmagenta: p = 0.021), three modules differed between the MDD_nSI and healthy controls (HC) groups (MEmagenta: p = 0.009, MEbrown: p = 0.001, MEturquoise: p = 0.005), and one module differed between the MDD_SI and HC groups (MEgreen, p = 0.004).

Differences in gene coexpression modules among the MDD-SI, MDD-nSI, and HCs groups. MDD_nSI major depressive disorder without suicidal ideation, MDD_SI major depressive disorder with suicidal ideation, HC healthy control, ME module eigengene.
Functional annotation of genes in the coexpression modules
The GO and KEGG functional annotation and pathway enrichment analyses were performed to reveal the biological functions of the genes within the modules. For modules with more than 15 significant terms, only the top 15 are presented (Fig. 2, Table S2). The magenta module was mainly enriched in nuclear RNA speck processing and regulation of alternative metabolic processes, such as RNA splicing (enrichment = 0.056, FDR-p < 0.001) and regulation of mRNA processing (enrichment = 0.065, FDR-p = 0.013), and no KEGG pathway significant after FDR multiple correct in this module. The brown module was mainly associated with the activation of innate immune-related biological processes and mitochondrial metabolism pathways, such as the innate immune response-activating signaling pathway (enrichment = 0.187, FDR-p < 0.001), the FoxO signaling pathway (enrichment = 0.191, FDR-p < 0.001), and the thyroid hormone signaling pathway (enrichment = 0.198, FDR-p < 0.001). The turquoise module was primarily related to the regulation of metabolism and energy production, including pathways such as oxidative phosphorylation (enrichment = 0.522, FDR-p < 0.001), proton motive force-driven ATP synthesis (enrichment = 0.645, FDR-p < 0.001), and thermogenesis (enrichment = 0.375, FDR-p < 0.001). It was also enriched in pathways related to multiple types of neurodegenerations, including Parkinson’s disease (enrichment = 0.368, FDR-p < 0.001), Huntington’s disease (enrichment = 0.340, FDR-p < 0.001), and Alzheimer disease (enrichment = 0.286, FDR-p < 0.001). The green module was mainly enriched in immune and inflammatory responses, including pathways such as cell and leukocyte activation involved in the immune response (enrichment = 0.162, FDR-p < 0.001), the B-cell receptor signaling pathway (enrichment = 0.214, FDR-p < 0.001), Th17 cell differentiation (enrichment = 0.185, FDR-p = 0.001), the JAK-STAT signaling pathway (enrichment = 0.151, FDR-p = 0.001), the chemokine signaling pathway (enrichment = 0.146, FDR-p = 0.001), and Fc gamma R-mediated phagocytosis (enrichment = 0.216, FDR-p < 0.001).

The GO and KEGG functional enrichment analysis of gene coexpressed modules. GO gene ontology, KEGG Kyoto encyclopedia of genes and genomes.
Hub genes of the differential modules
The PPI networks were constructed for the top-ranking genes within the magenta, brown, green, and turquoise modules. These networks were visualized using Cytoscape software, as shown in (Fig. 3). The genes with the highest connectivity were the hub genes for these modules and were identified using the cytoHubba plugin (Table 2). Eukaryotic translation initiation factor 4A3 (EIF4A3), serine and arginine rich splicing factor 11 (SRSF11), and splicing factor 3B subunit (SF3B1) were identified in the magenta module, which was enriched in RNA splicing and translation initiation (Fig. 3A). Tumor protein p53 (TP53), E1A binding protein p300 (EP300), TNF receptor associated factor 6 (TRAF6), and histone deacetylase 1 (HDAC1) were identified in the brown module and were enriched in the repair of damaged DNA, regulation of immune responses and inflammation (Fig. 3B). Finkel-Biskis-Jinkins murine osteosarcoma viral oncogene homolog (FAU), ribosomal protein S3 (RPS3), RPS11, RPL23, and RPS8 were identified in the turquoise module, which is enriched in cellular processes, including regulation of apoptosis, mRNA translation, and protein synthesis (Fig. 3C). Spleen tyrosine kinase (SYK), Lck/Yes-related novel tyrosine kinase (LYN), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit delta (PIK3CD), protein tyrosine phosphatase nonreceptor type 6 (PTPN6), signal transducer and activator of transcription 3 (STAT3), tyrosine kinase 2 (TYK2), interleukin 1 beta (IL1B), cluster of differentiation 8 alpha (CD8A), and integrin subunit beta 2 (ITGB2) were identified in the green module; these genes are enriched in inflammation and immune response and regulate signaling cascades in immune cells such as B cells, T cells, and macrophages (Fig. 3D).

Protein-protein interaction networks and hub genes of gene coexpression modules.
The relationships between differential modules and depressive symptoms
Furthermore, in the analysis of the relationships between depressive symptoms and gene modules, only the magenta module was significantly related to HAMD scores in the MDD without SI group (r = 0.23, p = 0.037; Fig. 4).

Relationships between gene coexpression networks and depressive symptoms. MDD_nSI major depressive disorder without suicidal ideation, MDD_SI major depressive disorder with suicidal ideation, ME module eigengene, HAMD hamilton rating scale for depression, HAMA hamilton rating scale for anxiety.
Discussion
In this study, we used transcriptional data to investigate the differences in blood gene expression among the MDD_SI, MDD_nSI, and HC groups. We found that two modules were linked to the MDD_SI, while three modules were specifically associated with the MDD_nSI. GO and KEGG enrichment revealed that genes within the MDD_SI modules were primarily involved in gene expression regulation, immune response, and inflammatory. On the other hand, the MDD_nSI modules were found to be associated with functions related to innate immunity, energy metabolism, mitochondrial function, and neurodegeneration.
Over the past two decades, a growing body of research has supported a theory that immune and inflammatory processes play a significant role in the pathogenesis of MDD and suicide23,24. Our results suggested that MDD_SI exhibit alterations in immune-related pathways, including cell and leukocyte activation, B-cell receptor signaling, Th17 cell differentiation, JAK-STAT signaling, and chemokine signaling pathways. These findings align with previous studies suggesting that MDD patients at high suicide risk may experience immune dysregulation, affecting both innate and adaptive immunity. This dysregulation involves key cell types related to inflammatory response and memory cells essential for long-term immune defense25,26. Altered Th17 cell differentiation, observed in both animal models and humans, has been linked to the onset of depression27. Imbalances in T helper cell distribution in the bloodstream have been associated with suicide28. Increased IL1B expression was observed in MDD patients with a high risk of suicide29,30. A meta-analysis further demonstrated significantly greater levels of the proinflammatory cytokines interleukin (IL)-1β and IL-6 in suicide victims than in nonsuicidal patients and HC across blood, cerebrospinal fluid, and postmortem brain samples31. Moreover, Holmes et al.32 demonstrated heightened microglial activation in MDD_SI but not in those MDD_nSI32. Another study also suggested that the low-grade inflammation associated with MDD and suicidality may involve microglial priming and macrophage recruitment in the brain33. Activated microglia might increase cytokine production within the central nervous system, influencing noradrenergic or serotonergic neurotransmission and potentially contributing to suicidality34.
Our findings revealed RNA splicing, reflecting transcriptional mechanisms related to gene expression regulation, are associated with MDD_SI. Posttranscriptional processes, including alternative splicing and RNA editing, have been reported to contribute to suicide risk in individuals with MDD35. In particular, the hub genes EIF4A3, SRSF11, and SF3B1 identified in the brown module are known to be involved in the regulation of RNA splicing. Previous research has highlighted the connection between RNA splicing and suicidal behavior36. Furthermore, a postmortem brain tissue study demonstrated that aberrant alternative splicing and RNA editing of the 5HT2C receptor and tryptophan hydroxylase are associated with suicidal behaviors, highlighting RNA splicing as a potentially modifiable mechanism in suicide pathology37.
Our findings suggest a correlation between energy metabolism and MDD. Researches have highlighted the significance of energy metabolism disturbances in the pathophysiology of MDD1,38. Notably, disturbances in energy metabolism, such as altered glucose metabolism39, mitochondrial dysfunction40, and abnormal ATP levels41, have been documented both in animal models and in clinical studies. A recent meta-analysis further proposed that abnormal extracellular ATP secretion, especially by astrocytes, is associated with depression, suggesting that targeting ATP may be an innovative antidepressant strategy42.
Cellular processes such as apoptosis regulation, mRNA translation, protein synthesis may also be related to the onset and development of MDD. Our study indicates that multiple pathways associated with neurodegenerative diseases are linked to MDD. Clinical studies suggest that MDD increased susceptibility to secondary neurodegenerative diseases such as Alzheimer’s disease43 and Parkinson’s disease44, which is support the findings of this study. We speculate that there is a shared neurobiological basis between MDD and neurodegenerative diseases. In addition, the expression of TP53, a hub gene in the brown module known for its role as a tumor suppressor and promotor of apoptosis, was altered under cellular stress. Increased apoptosis has been reported in animal models of stress-induced depression and in MDD patients45,46,47. Moreover, RPL23 suppresses apoptosis, and its downregulation is associated with antidepressant efficacy48,49.
This study has several limitations. First, the sample size was limited, and expanding the sample size in future studies could enhance the robustness of the results. Second, the WGCNA aggregation of modules into a single value might obscure single gene-level details, and these outcomes lack experimental validation. Third, although we have included sex as a covariate, differences in sex across the three groups remain, and there may be an impact on the transcriptomic results. Finally, the current study did not assess the severity of SI or account for suicidal behavior. Future research should increase the sample size and assess SI and suicidal behavior independently to better understand the similarities and differences in their transcriptomic mechanisms.
In the present study, WGCNA was applied to construct gene-weighted coexpression network from peripheral blood to identify differentially expressed gene modules among the MDD_SI, MDD_nSI, and HC groups. The findings provide further evidence that SI in MDD patients is associated with gene expression regulation, immune response, and inflammatory, whereas that in MDD_nSI is related to pathways involving innate immunity, energy metabolism, mitochondrial function, and neurodegeneration. Our results enhance the understanding of the molecular mechanisms underlying suicide risk in MDD, offering potential insights into diagnostic biomarkers or therapeutic targets for managing suicide risk in MDD patients.
Materials and methods
Participants
Study participants aged 18–65 and patients were recruited from the Mental Health Center of West China Hospital at Sichuan University. The participants provided written informed consent as approved by the Ethics Committee of West China Hospital of Sichuan University (No. [2016] 170). The diagnosis of MDD was confirmed by trained psychiatrists based on the Structured Clinical Interview for DSM-IV (SCID)50. All patients did not use any antidepressants for at least 3 months before enrollment and had taken medication for no more than 1 week. The 17-item Hamilton Depression Rating Scale (HAMD 17)51 and the Hamilton Anxiety Rating Scale (HAMA)52 were used to measure the severity of depressive and anxiety symptoms, respectively. The inclusion criteria for healthy controls were that they had no DSM-IV diagnosis, no history of suicidal ideation or behavior, and no history of mental disorders or suicidal behavior in their first-degree relatives. Patients with serious physical illness and long-term substance use were also excluded.
MDD patients were divided into two subgroups: those with/without SI based on HAMD item 3 (suicide). The item evaluates SI as follows: 0 represents absent, 1 represents feeling that life is not worth living, 2 indicates wishes for death or thoughts of self-injury, 3 represents suicidal ideations or gestures, and 4 indicates attempted suicide. In this study, participants with a score of 2 or above indicated the presence of SI, as was the case in previous studies53,54,55. Finally, three groups were included in this study: 75 MDD_SI patients, 82 MDD_nSI patients, and 149 HC.
RNA extraction and quality control
Peripheral blood samples (3 ml) were collected from participants using Tempus Blood RNA Tubes (Applied Biosystems, Foster City, CA, United States). The tubes were inverted for 10 s to ensure thorough mixing and then stored at −80 °C for further analysis. Total RNA was isolated using the MagMAX Stabilized Blood Tubes RNA Isolation Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s instructions. RNA concentration was measured with a NanoDrop ND-2000 spectrophotometer.
RNA sequencing and data processing
A total of 1 µg RNA was used for RNA sequencing. The mRNA was isolated from the total RNA using oligo (dT) beads and then fragmented into small segments with lengths between 200 and 500 nt. The fragmented mRNA served as a template for the synthesis of first-strand complementary DNA (cDNA), followed by the generation of second-strand cDNA. The double-stranded cDNA was purified and subjected to end repair, dA tailing, and ligation to Illumina sequencing adapters. PCR amplification was subsequently carried out to generate the cDNA library. The library was sequenced on a NovaSeq 6000 System (by Illumina) using a 2 × 150 bp (PE) configuration according to the manufacturer’s instructions. The raw reads were subjected to a filtration process to yield clean reads. The cleaned reads were then mapped onto the reference genome using HISAT2 software (v 2.0.1). The RNA sequencing procedures were conducted by Genewiz (Suzhou, P. R. China).
RNA sequencing data processing was conducted using the R package EdgeR56. Transcripts exhibiting counts per million (CPM) less than 1 across more than 5% of the total samples were discarded to eliminate low expression. Then, normalization was executed through the utilization of the trimmed mean of M-values (TMM) method57. To measure sample anomalies and establish overall associations, a biological coefficient of variation (BCV) plot was constructed58. Dispersions were estimated by employing the quantile-adjusted conditional maximum likelihood (qCML) strategy58.
Weighted coexpression network construction
The WGCNA algorithm, which follows a scale-free distribution, was applied to construct the coexpression network module in all samples22,59. The pickSoftThreshold function was applied to calculate the soft-threshold β based on the scale-free topology. A threshold of 8 was chosen when the fit index reached 0.85 (Figure S1A). The adjacency matrix was transformed into a topological overlap matrix (TOM) with the TOMsimilarity function60. Then, genes with similar expression patterns were clustered into the same color modules, with a minimum module size of 30 genes, while genes in the gray module were excluded from further analysis22,61. The module eigengene (ME), which represents the overall expression level of genes within a specific module, was calculated as the first principal component of the gene expression data.
Functional enrichment analysis of genes in the modules
We conducted a functional enrichment analysis by employing ClusterProfiler (v3.9) in the R package to determine further biological implications associated with the identified gene modules. This analysis used the biological process (BP) category of gene ontology (GO)62 and Kyoto encyclopedia of genes and genomes (KEGG) databases63. Pathways with a p-value < 0.05 after false discovery rate (FDR) correction were deemed to be significantly different. If more than 15 significant terms were identified, only the top 15 terms were presented.
Construction of protein-protein networks and identification of hub genes
To analyze the crucial genes in each module, genes from each module were mapped to the STRING database, which is instrumental in protein‒protein networks (PPIs)64. The transcriptional regulatory networks for each module were then established and visualized using the CytoHubba plug-in in Cytoscape software (v 3.7.2), and genes with the maximum degree of connectivity were identified as the hub genes.
Statistical analysis
Statistical analysis was conducted using the R software environment. One-way ANOVA was utilized to compare age, while the chi-square test was used to compare sex differences among the MDD_nSI, MDD_SI, and HC groups. Student’s t test was employed to assess the differences in onset age, onset number, total disease duration, HAMA scores, and HAMD scores between the MDD_nSI and MDD_SI groups. ANCOVA was performed to compare the averages among the MDD_nSI, MDD_SI, and HC groups, with age and sex included as covariates. Post hoc multiple comparisons were carried out using Tukey’s test with the R package multcomp. Additionally, Spearman’s correlation was utilized to investigate the relationships between the eigengenes of gene coexpression modules and HAMD or HAMA scores.
Data availability
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: HRA009854) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human/browse/HRA009854.
References
Malhi, G. S., Mann, J. J. & Depression Lancet 392, 2299–2312 https://doi.org/10.1016/S0140-6736(18)31948-2 (2018).
Turecki, G. & Brent, D. A. Suicide and suicidal behaviour. Lancet 387, 1227–1239. https://doi.org/10.1016/S0140-6736(15)00234-2 (2016).
Arsenault-Lapierre, G., Kim, C. & Turecki, G. Psychiatric diagnoses in 3275 suicides: a meta-analysis. Bmc Psychiatr. 4 https://doi.org/10.1186/1471-244X-4-37 (2004).
Klonsky, E. D., Pachkowski, M. C., Shahnaz, A. & May, A. M. The three-step theory of suicide: description, evidence, and some useful points of clarification. Prev. Med. 152, 106549. https://doi.org/10.1016/j.ypmed.2021.106549 (2021).
Cai, H. et al. Prevalence of suicidal ideation and planning in patients with major depressive disorder: a meta-analysis of observation studies. J. Affect. Disord. 293, 148–158 https://doi.org/10.1016/j.jad.2021.05.115 (2021).
Ling, Z., Qing, T. & Chunming, X. Epigenetic insight into the suicidal biomarker of depression with suicide ideation: a narrative review. Neuroscience 560, 48–55 https://doi.org/10.1016/j.neuroscience.2024.09.029 (2024).
Li, Q. S. et al. Genome-wide association study meta-analysis of suicide death and suicidal behavior. Mol. Psychiatr. 28, 891–900 https://doi.org/10.1038/s41380-022-01828-9 (2023).
Bakian, A. V. et al. A population-wide analysis of the familial risk of suicide in Utah, USA. Psychol. Med. 53, 1448–1457 https://doi.org/10.1017/S0033291721003020 (2023).
DiBlasi, E., Kang, J. & Docherty, A. R. Genetic contributions to suicidal thoughts and behaviors. Psychol. Med. 51, 2148–2155 https://doi.org/10.1017/S0033291721001720 (2021).
Orsolini, L. et al. Understanding the complex of suicide in depression: from research to clinics. Psychiatr. Investig. 17, 207–221. https://doi.org/10.30773/pi.2019.0171 (2020).
Dong, Z. & Chen, Y. Transcriptomics: advances and approaches. Sci. China Life Sci. 56, 960–967 https://doi.org/10.1007/s11427-013-4557-2 (2013).
Pantazatos, S. P. et al. Whole-transcriptome brain expression and exon-usage profiling in major depression and suicide: evidence for altered glial, endothelial and ATPase activity. Mol. Psychiatr. 22, 760–773. https://doi.org/10.1038/mp.2016.130 (2017).
Pandey, G. N., Rizavi, H. S., Bhaumik, R. & Ren, X. Innate immunity in the postmortem brain of depressed and suicide subjects: role of toll-like receptors. Brain Behav. Immun. 75, 101–111. https://doi.org/10.1016/j.bbi.2018.09.024 (2019).
Sha, Q. et al. Integrative transcriptome- and DNA methylation analysis of brain tissue from the temporal Pole in suicide decedents and their controls. Mol. Psychiatr. https://doi.org/10.1038/s41380-023-02311-9 (2023).
Zhao, Y., Wang, L., Wu, Y., Lu, Z. & Zhang, S. Genome-wide study of key genes and scoring system as potential noninvasive biomarkers for detection of suicide behavior in major depression disorder. Bioengineered 11, 1189–1196 https://doi.org/10.1080/21655979.2020.1831349 (2020).
Ciobanu, L. G. et al. Co-expression network analysis of peripheral blood transcriptome identifies dysregulated protein processing in endoplasmic reticulum and immune response in recurrent MDD in older adults. J. Psychiatr Res. 107, 19–27. https://doi.org/10.1016/j.jpsychires.2018.09.017 (2018).
Zhao, B. et al. Identification of key modules and genes associated with major depressive disorder in adolescents. Genes 13 https://doi.org/10.3390/genes13030464 (2022).
Yao, P. A., Sun, H. J. & Li, X. Y. Identification of key genes in late-onset major depressive disorder through a co-expression network module. Front. Genet. 13, 1048761 https://doi.org/10.3389/fgene.2022.1048761 (2022).
Peng, S., Zhou, Y., Xiong, L. & Wang, Q. Identification of novel targets and pathways to distinguish suicide dependent or independent on depression diagnosis. Sci. Rep. 13, 2488. https://doi.org/10.1038/s41598-023-29101-1 (2023).
Sun, S. et al. Brain and blood transcriptome profiles delineate common genetic pathways across suicidal ideation and suicide. Mol. Psychiatr. https://doi.org/10.1038/s41380-024-02420-z (2024).
Liew, C. C., Ma, J., Tang, H. C., Zheng, R. & Dempsey, A. A. The peripheral blood transcriptome dynamically reflects system wide biology: a potential diagnostic tool. J. Lab. Clin. Med. 147, 126–132. https://doi.org/10.1016/j.lab.2005.10.005 (2006).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559. https://doi.org/10.1186/1471-2105-9-559 (2008).
Petho, B. et al. Investigation of peripheral inflammatory biomarkers in association with suicide risk in major depressive disorder. Front. Psychiatr. 15, 1321354. https://doi.org/10.3389/fpsyt.2024.1321354 (2024).
Courtet, P. et al. Neuroinflammation in suicide: toward a comprehensive model. World J. Biol. Psychiatr. 17, 564–586. https://doi.org/10.3109/15622975.2015.1054879 (2016).
Maes, M. et al. The immune profile of major dysmood disorder: proof of concept and mechanism using the precision nomothetic psychiatry approach. Cells 11 https://doi.org/10.3390/cells11071183 (2022).
Vasupanrajit, A., Jirakran, K., Tunvirachaisakul, C., Solmi, M. & Maes, M. Inflammation and nitro-oxidative stress in current suicidal attempts and current suicidal ideation: a systematic review and meta-analysis. Mol. Psychiatr. 27, 1350–1361. https://doi.org/10.1038/s41380-021-01407-4 (2022).
Wang, H. et al. MicroRNA-messenger RNA regulatory network mediates disrupted TH17 cell differentiation in depression. Front. Psychiatr. 13, 824209. https://doi.org/10.3389/fpsyt.2022.824209 (2022).
Schiweck, C. et al. Depression and suicidality: a link to premature T helper cell aging and increased Th17 cells. Brain Behav. Immun. 87, 603–609. https://doi.org/10.1016/j.bbi.2020.02.005 (2020).
Schiweck, C. et al. Childhood trauma, suicide risk and inflammatory phenotypes of depression: insights from monocyte gene expression. Transl Psychiatr. 10, 296. https://doi.org/10.1038/s41398-020-00979-z (2020).
Bastos, C. R. et al. Polymorphism in CRHR1 gene affects the IL-1beta levels in suicidal attempters. J. Psychiatr. Res. 86, 34–38 https://doi.org/10.1016/j.jpsychires.2016.11.009 (2017).
Black, C. & Miller, B. J. Meta-analysis of cytokines and chemokines in suicidality: distinguishing suicidal versus nonsuicidal patients. Biol. Psychiatr. 78, 28–37. https://doi.org/10.1016/j.biopsych.2014.10.014 (2015).
Holmes, S. E. et al. Elevated translocator protein in anterior cingulate in major depression and a role for inflammation in suicidal thinking: a Positron emission tomography study. Biol. Psychiatr. 83, 61–69. https://doi.org/10.1016/j.biopsych.2017.08.005 (2018).
Torres-Platas, S. G., Cruceanu, C., Chen, G. G., Turecki, G. & Mechawar, N. Evidence for increased microglial priming and macrophage recruitment in the dorsal anterior cingulate white matter of depressed suicides. Brain Behav. Immun. 42, 50–59. https://doi.org/10.1016/j.bbi.2014.05.007 (2014).
Steiner, J. et al. Immunological aspects in the neurobiology of suicide: elevated microglial density in schizophrenia and depression is associated with suicide. J. Psychiatr. Res. 42, 151–157. https://doi.org/10.1016/j.jpsychires.2006.10.013 (2008).
Pantazatos, S. P. et al. Isoform-level brain expression profiling of the spermidine/spermine N1-acetyltransferase1 (SAT1) gene in major depression and suicide. Neurobiol. Dis. 79, 123–134. https://doi.org/10.1016/j.nbd.2015.04.014 (2015).
Pereira, C. A., Reis-de-Oliveira, G., Pierone, B. C., Martins-de-Souza, D. & Kaster, M. P. Depicting the molecular features of suicidal behavior: a review from an omics perspective. Psychiatr. Res. 332, 115682 https://doi.org/10.1016/j.psychres.2023.115682 (2024).
Grohmann, M. et al. Alternative splicing and extensive RNA editing of human TPH2 transcripts. PLoS One 5, e8956 https://doi.org/10.1371/journal.pone.0008956 (2010).
Sforzini, L. et al. Transcriptomic profiles in major depressive disorder: the role of immunometabolic and cell-cycle-related pathways in depression with different levels of inflammation. Mol. Psychiatr. https://doi.org/10.1038/s41380-024-02736-w (2024).
Yao, J. K. & Reddy, R. D. Metabolic investigation in psychiatric disorders. Mol. Neurobiol. 31, 193–203 (2005).
Haj-Mirzaian, A. et al. Anxiety- and depressive-like behaviors are associated with altered hippocampal energy and inflammatory status in a mouse model of Crohn’s disease. Neuroscience 366, 124–137 https://doi.org/10.1016/j.neuroscience.2017.10.023 (2017).
Fang, W. et al. APOE4 genotype exacerbates the depression-like behavior of mice during aging through ATP decline. Transl Psychiatr. 11, 507 https://doi.org/10.1038/s41398-021-01631-0 (2021).
Wang, K. et al. The neurobiological mechanisms and therapeutic prospect of extracellular ATP in depression. CNS Neurosci. Ther. 30, e14536. https://doi.org/10.1111/cns.14536 (2024).
Petkus, A. J. et al. 20-year depressive symptoms, dementia, and structural neuropathology in older women. Alz. Dement. https://doi.org/10.1002/alz.13781 (2024).
Badenoch, J. B. et al. Neuroanatomical and prognostic associations of depression in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatr. https://doi.org/10.1136/jnnp-2023-333007 (2024).
Lucassen, P. J. et al. Stress, depression and hippocampal apoptosis. CNS Neurol. Disord Drug Targ. 5, 531–546 (2006).
Liu, W. et al. The role of neural plasticity in depression: from hippocampus to prefrontal cortex. Neural Plast. 2017 (6871089). https://doi.org/10.1155/2017/6871089 (2017).
Mahmood, S., Evinova, A., Skerenova, M., Ondrejka, I. & Lehotsky, J. Association of EGF, IGFBP-3 and TP53 gene polymorphisms with major depressive disorder in Slovak population. Cent. Eur. J. Public. Health. 24, 223–230 https://doi.org/10.21101/cejph.a4301 (2016).
Grzenda, A., Siddarth, P., Laird, K. T., Yeargin, J. & Lavretsky, H. Transcriptomic signatures of treatment response to the combination of escitalopram and memantine or placebo in late-life depression. Mol. Psychiatr. 26, 5171–5179 https://doi.org/10.1038/s41380-020-0752-2 (2021).
Borsini, A. et al. Omega-3 polyunsaturated fatty acids protect against inflammation through production of LOX and CYP450 lipid mediators: relevance for major depression and for human hippocampal neurogenesis. Mol. Psychiatr. 26, 6773–6788 https://doi.org/10.1038/s41380-021-01160-8 (2021).
First, M., Spitzer, R., Gibbon, M. & Williams, J. Structured clinical interview for DSM-IV axis I disorders: Patient edition (SCIDI/P. Version 2.0.) (1995).
Ostwald, P. F. A rating scale for depression. Am. J. Psychother. 14, 817–818 (1960).
Thompson, E. Hamilton rating scale for anxiety (HAM-A). Occup. Med. 65, 601–601 https://doi.org/10.1093/occmed/kqv054 (2015).
Lan, X. et al. Association between cognition and suicidal ideation in patients with major depressive disorder: a longitudinal study. J. Affect. Disord. 272, 146–151. https://doi.org/10.1016/j.jad.2020.03.141 (2020).
Qin, K. et al. Transcriptional patterns of brain structural covariance network abnormalities associated with suicidal thoughts and behaviors in major depressive disorder. Biol. Psychiatr. https://doi.org/10.1016/j.biopsych.2024.01.026 (2024).
Szanto, K. et al. Emergence, persistence, and resolution of suicidal ideation during treatment of depression in old age. J. Affect. Disord. 98, 153–161. https://doi.org/10.1016/j.jad.2006.07.015 (2007).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. https://doi.org/10.1093/bioinformatics/btp616 (2010).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25. https://doi.org/10.1186/gb-2010-11-3-r25 (2010).
Robinson, M. D. & Smyth, G. K. Small-sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics 9, 321–332. https://doi.org/10.1093/biostatistics/kxm030 (2008).
Wei, J. et al. Dysconnectivity of the brain functional network and abnormally expressed peripheral transcriptional profiles in patients with anxious depression. J. Psychiatr. Res. https://doi.org/10.1016/j.jpsychires.2024.01.021 (2024).
Boutros, P. C. & Okey, A. B. Unsupervised pattern recognition: an introduction to the whys and wherefores of clustering microarray data. Brief. Bioinform. 6, 331–343 (2005).
Reyes, J. M. et al. Differing molecular response of young and advanced maternal age human oocytes to IVM. Hum. Reprod. 32, 2199–2208 https://doi.org/10.1093/humrep/dex284 (2017).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Gene Ontol. Consort. Nat. Genet. 25, 25–29 (2000).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Szklarczyk, D. et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49, D605–D612. https://doi.org/10.1093/nar/gkaa1074 (2021).
Acknowledgements
We would like to express our gratitude to all the participants in this study. Special thanks go to Editage (https://www.editage.cn) for providing English language editing services.
Funding
This work was supported by the Ministry of Science and Technology of the People’s Republic of China (No. 2022ZD0211700), the China Postdoctoral Science Foundation (No. GZC20231802), and the 135 Project from West China Hospital of Sichuan University (Nos. 2023HXFH006, 2023HXFH040).
Author information
Authors and Affiliations
Contributions
M.W. designed the experiments and wrote the manuscript. M.W., HL.X., and JX.W. conducted the statistical analysis. M.W., HL.X., YK.D., Y.D., HH.F., LS.Z., RJ.N., and X.Y. contributed to collecting the data. X.Y. and XH.M. have revised the manuscript. All authors reviewed and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The current study was performed in accordance with the Declaration of Helsinki. All participants provided written informed consent as approved by the Ethics Committee of West China Hospital of Sichuan University (No. [2016] 170).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, M., Xiang, H., Wei, J. et al. Identification of blood transcriptome modules associated with suicidal ideation in patients with major depressive disorder. Sci Rep 15, 1067 (2025). https://doi.org/10.1038/s41598-025-85431-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-85431-2
