
Abstract
Circular RNAs (circRNAs) are widely involved in diverse biological processes of cancers. Nonetheless, the potential function of hsa_circ_0008305 in hepatocellular carcinoma (HCC) remains largely unknown. This study aims to elucidate the role and underlying mechanism of hsa_circ_0008305 in HCC. Our findings reveal that the novel circRNA hsa_circ_0008305 (circPTK2) is significantly upregulated in HCC tissues, with its elevated expression being positively correlated with advanced tumor T stage and vascular invasion. The circular characteristics and subcellular localization of hsa_circ_0008305 was determined by RNase R treatment and RNA nucleocytoplasmic separation. Further functional assays, including CCK8, EdU, colony formation assays, scratch-healing, transwell assays, and Xenograft tumor models were conducted to explore the biological functions of circPTK2. The regulatory mechanisms of circPTK2 were elucidated through RNA sequencing, enrichment analysis, and dual luciferase reporter assay. Our findings indicate that circPTK2 is stably localized in the cytoplasm. Functionally, circPKT2 promoted the HCC cells proliferation, migration, and invasion both in vitro and vivo. Mechanistically, circPTK2 was found to positively regulates the expression of AKR1C3 by acting as a sponge for miR-379-5p. Inhibition of miR-379-5p significantly mitigates the biological effects induced by circPTK2. AKR1C3 is identified as a direct target of miR-379-5p, and silencing AKR1C3 overturns the promotion progression effects of miR-379-5p inhibitor. In conclusion, our results revealed that circPTK2 facilitates the malignant progression of HCC via sponging miR-379-5p to up-regulate AKR1C3 expression.
Similar content being viewed by others


Introduction
Hepatocellular carcinoma (HCC) ranks sixth among the most common malignancies globally, and is the third leading cause of cancer-related mortality1,2. Curative interventions, such as surgical resection, radiofrequency ablation, and liver transplantation are available for patients diagnosed with early-stage HCC3. However, the majority of HCC cases are identified at an advanced stage3. Despite recent therapeutic advancements, including the development of targeted drugs, immunotherapy, engineered vaccine, and the combined application of nanoparticles with immune checkpoint inhibitors, the 5-year survival rate is still poor and remains below 12% for advanced HCC4,5,6. The lack of early diagnostic biomarkers, coupled with the highly proliferative and invasive nature of the disease, are primary factors contributing to the poor prognosis of HCC7,8. Therefore, it is imperative to identified key driver genes and further investigate the molecular mechanisms underlying the onset and progression of HCC.
Circular RNAs (circRNAs) is generated from reverse splicing of precursors mRNA (pre-mRNA), resulting in a single-stranded closed-loop structure that lacks a 5’ cap and 3 ‘poly (A)9. Due to their unique structure and biological characteristics, circRNAs play a significant regulatory role in various biological processes, including growth, differentiation, senescence, and apoptosis10,11. Recent studies have reported that circRNAs are abnormally expressed in various tumors and are closely associated with the tumorigenesis and progression12. In HCC, certain circRNAs, such as circPIAS1, circGPR137B, and circCCAR1, have been identified as key players in processes including cell proliferation, apoptosis, invasion, metastasis, immune escape, metabolism, and drug resistance10,13,14. Despite significant advancements in understanding the biology of circRNAs, numerous circRNAs in HCC remain to be elucidated. Therefore, it is crucial to identify functional circRNAs and explore their intrinsic molecular mechanisms in HCC.
In this study, we identified a novel circular RNA, circPTK2 (circbase ID: hsa_circ_0008305), which is remarkably upregulated in HCC tissues. circPTK2 generated through the reverse splicing of parent gene PTK2 transcript NM_00119964915. A previous study reported that circPTK2 inhibits TGF-β-induced EMT and tumor cell invasion by regulating TIF-1 γ through miR-429/miR-200b-3p pathway in non-small cell lung cancer (NSCLC)15. Furthermore, circ-PTK2 plays a crucial role in enhancing cell migration and invasions in ovarian cancer through the miR-639 and FOXC1 regulatory cascade16. In bladder cancer, circPTK2 markedly facilitates migration, metastasis, and resistance to gemcitabine by binding to PABPC1 and enhancing the stability of SETDB1 protein17. Additionally, circPTK2/miR-136/NFIB axis is implicated in the progression and development of triple-negative breast cancer18. However, the role of circPTK2 in HCC remains largely unexplored. This study aims to explore the potential role of circPTK2 in the malignant behavior of HCC, and to further elucidate whether its involvement in HCC progression is mediated through miR-379-5p/AKR1C3 axis.
Results
circPTK2 is highly expressed in HCC tissues
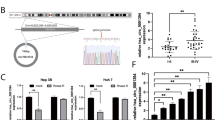
To identify the unique circRNAs related to HCC development, we analyzed the microarray dataset GSE97332 from the GEO database to screened the differential circRNAs between HCC tissues and normal liver tissues. According to the cutoff criteria of | log2FC | > 1.5 and p < 0.05, a total of 441 circRANs were differentially expressed between HCC and normal tissue samples, 234 of them were upregulated and 207 were downregulated(Fig. 1A). We found hsa_circ_0008305 (circPTK2) was significantly upregulated in HCC tissues (log2FC = 1.98, p = 3.39E-04)(Fig. 1B). Moreover, the high expression of circPTK2 in HCC was identified in GSE94508 dataset (tissue samples) and GSE16678 dataset (plasma samples)(Fig. 1C,D). The results of qRT-PCR(N = 72) also showed that circPTK2 is highly expressed in cancerous tissues compared with that in para-cancerous tissues (Fig. 1E). The waterfall distribution plot of the logFC among the 72 patients manifested that circPTK2 was highly expressed in 67% individuals(Fig. 1F). Moreover, higher expression of circPTK2 was found to be associated with a more advanced T stage(Fig. 1G), microvascular invasion(Fig. 1H), and larger tumor diameter(Fig. 1I). Subsequently, we performed Fluorescence in situ hybridization(FISH) assay in HCC tissues, which also confirmed that circPTK2 expression in HCC tissues was markedly higher than that in adjacent tissues(Fig. 1J). Additionally, circPTK2 was highly expressed in multiple HCC cells compared to that in normal liver cell line L02 (Fig. 1K). Among which the expression in HCC-LM3 and MHCC 97-H was relatively higher, so we selected HCC-LM3 and MHCC 97-H for subsequent experiments(Fig. 1K).

circPTK2 is highly expressed in HCC tissues and cells. (A) Volcano plot shows the expression profiles of circRNAs in HCC tissues from GSE97332. (B–D) circPTK2 is highly expressed in HCC samples based on GSE97332(B), GSE94508(C), GSE166678(D). (E) Expression analysis of circPTK2 in 72 paired HCC samples by qRT-PCR (Mean ± SD, n = 72, ***p < 0.001). (F) The overall distribution of the fold change (tumor tissues vs. adjacent tissues) of circPTK2 among 72 HCC patients. (G) The expression level of circPTK2 at different tumor stages(Mean ± SD, *p < 0.05). (H) The expression level of circPTK2 in samples with vascular invasion and without microvascular invasion(Mean ± SD, *p < 0.05). (I) The expression level of circPTK2 in samples within different tumor size (≤ 5 cm and > 5 cm)(Mean ± SD, ***p < 0.001). (J) circPTK2 in HCC tissues and adjacent normal tissues detected by FISH. (K) The relative expression of circPTK2 in HCC cell lines and normal live cell (Mean ± SD, n = 3, **p < 0.01, ***p < 0.001). (L) Schematic illustration displays the origination and circularization site sequence of circPTK2 according to circBase and Dideoxy sequencing. (M) Compared with liner PTK2, circPTK2 is more resistant to R Nase R treatment in HCC cells (Mean ± SD, n = 3, ns: no significance, ***p < 0.001). (N) The half-life of circPTK2 is longer than that of linear transcriptome PTK2 after actinomycin D(2ug/mL) treatment (Mean ± SD, n = 3, ***p < 0.001). (O–P) nuclear-cytoplasmic fractions (O) and FISH (P) confirmed that circPTK2 is mainly distributed in cytoplasm.
To further clarify the correlation between the expression level of circPTK2 and the clinicopathological features of HCC patients, we divided the HCC patients (n = 72) into circPTK2 high expression group and low expression group based on the median expression value of circPTK2(median value = 0.32). Then, Chi-squared or Fisher’s exact tests was used for statistical analysis. The results indicated that the circPTK2 expression was closely related to tumor size (p = 0.004), T stage(p = 0.002), hepatitis B virus infection(p = 0.024) and microvascular invasion (p = 0.007), but not to age, gender, and other factors (Table 1), suggesting that high expression of circPTK2 in HCC tissues may contribute to the tumor malignant progression.
circPTK2 stably presents in HCC cytoplasm
According to gene information of circBase (http://www.circbase.org), we found that hsa_circ0008305 was originated from the parental gene protein tyrosine kinase 2 (PTK2) (Chr8:14799572–141840625, NM_001199649), seven exons (exon 8–14, 584nt) were spliced back to form a cycle structure. circPTK2 was specifically amplified by convergent primers, and the cyclic junction was confirmed by Dideoxy sequencing(Fig. 1L). RNase R and Actinomycin D treatment was used to identify the circular structure. circPTK2 treated with RNase R was more resistant to degradation than linear PTK2(Fig. 1M). The circPTK2 showed a longer half-life period of up to 24 h after actinomycin D treatment, while linear PTK2 was only 2–6 h (Fig. 1N). The nucleoplasmic separation and FISH assays suggested that circPTK2 was mainly distributed in the cytoplasm, but little in the nucleus (Fig. 1O–P). These results suggested that circPTK2 is mainly present in HCC cytoplasm and more stable than linear PTK2 due to its circular structure.
circPTK2 accelerates the HCC cells proliferation and tumor growth in vivo and and in vitro
To investigate the molecular function of circPTK2 in HCC cells, we transfected the circPTK2 knockdown(sh-circPTK2 ) and overexpression (OE-circPTK2) vectors into HCC-LM3 and MHCC97-H, respectively.The results of RT-qPCR showed the expression of circPTK2 was downregulated more than 70% in sh-circPTK2 groups compared to that in control groups (Figure S1A). The circPTK2 overexpression plasmid was transfected into HCC cells, and notably elevated circPTK2 expression was observed in the OE-circPTK2 group compared with the OE-NC group (Figure S1B). Then, CCK8, EdU assay, and colony formation assays was conducted to detect cell proliferation in different transfection groups. The results of CCK revealed that the cell proliferation activity was significantly decreased in circPTK2 silencing groups, but increased in circPTK2 overexpression groups (Fig. 2A,B). For the EdU assay, the proportion of EdU positive cells in circPTK2 knockdown groups was significantly lower than that in control groups (Fig. 2C), while overexpression circPTK2 caused an opposite effect in these cells (Fig. 2D). Plate clone assay suggested that the clone number of sh-circPTK2 cells was significantly less than that in sh-NC group (Fig. 2E), while the number in circPTK2 overexpression group was significantly increased (Fig. 2F). These results suggest circPTK2 promotes the proliferation of HCC cells in vitro.

circPTK2 promotes HCC cells proliferation and tumor growth in vitro and vivo. (A) Cell viability of HCC-LM3 and MHCC 97-H cells transfected with sh-circPTK2 or shRNA-NC was evaluated by CCK8 (Mean ± SD, n = 6, ** p < 0.01, ***p < 0.001). (B) Cell viability of HCC-LM3 and MHCC 97-H cells transfected with overexpression plasmids (OE-circPTK2 or OE-NC) was detected by CCK8 (Mean ± SD, n = 6, **p < 0.01, ***p < 0.001). (C) DNA replication capacity of HCC-LM3 and MHCC 97-H transfected with sh-circPTK3 or shRNA-NC examined by EdU assays (Mean ± SD, n = 3, **p < 0.01, ***p < 0.001), scale bar: 100 μm. (D) DNA replication capacity of HCC-LM3 and MHCC 97-H transfected with circPTK2 overexpression vector or negative control (Mean ± SD, n = 3, ***p < 0.001), scale bar: 100 μm. (E) Colony formation assays detected the cell growth ability of HCC-LM3 and MHCC 97-H transfected with sh-circPTK2 or shRNA-NC (Mean ± SD, n = 3, ***p < 0.001). (F) Colony formation assays analyzed the cell growth ability of HCC-LM3 and MHCC 97-H transfected with OE-circPTK2 or OE-NC (Mean ± SD, n = 3, ***p < 0.001). (G) Subcutaneous tumors using HCC-LM3 cells infected with LV-sh-circPTK2 or LV-sh-NC. (H) Tumor weight and tumor volume were measured in sh-circPTK2 and sh-NC groups(Mean ± SD, n = 5, ***p < 0.001). (I) Subcutaneous tumors using HCC-LM3 cells infected with LV-OE-circPTK2 or LV-OE-NC. (J) Tumor weight and tumor volume were measured in OE-circPTK2 and OE-NC groups (Mean ± SD, n = 5, **p < 0.01, ***p < 0.001). (K) The expression of Ki- 67 was evaluated using IHC in sh-circPTK2 and sh-NC. Scale bar: 50 μm. (L) The expression of Ki- 67 was detected using IHC in OE-circPTK2 and OE-NC. Scale bar: 50 μm.
Subsquently, stable HCC-LM3 cells transfected with sh-circPTK2 or OE-circPTK2 plasmids were injected into nude mice to establish a xenograft tumor model. From the appearance of tumors, we found the tumors generated from sh-circPTK2 lentiviruses treated cells were significant smaller than those derived from sh-NC cells(Fig. 2G), knockdown of circPTK2 significantly suppressed the tumor volume and weight(Fig. 2H). Nevertheless, HCC-LM3 cells infected with OE-circPTK2 lentiviruses exhibited a remarkable increase in tumor volume and weight compared to those in nagative control group (Fig. 2I,J). Additionally, Immunohistochemical staining of Ki-67 showed lower proliferation rate in sh-circPTK2 groups, but higher proliferation rate in OE-circPTK2 group (Fig. 2K,L). In general, these results indicate circPTK2 promotes the HCC cells proliferation and tumor growth in vivo.
circPTK2 promotes HCC cells migration and invasion
Scratch-healing and transwell assays were conducted to verify the role of circPTK2 in the migration and invasion of HCC cells. The results of scratch-healing assays showed that the cell migration ability of HCC-LM3 and MHCC 97-H were remarkably suppressed by circPTK2 downregulation(Fig. 3A), while circPTK2 overexpression promoted cell migration in these cells(Fig. 3B). Transwell migration assays came to the same results as scratch-healing experiment(Fig. 3C,D). Moreover, transwell invasion assay showed that sh-circPTK2 inhibited the invasion of HCC cells (Fig. 3E), while overexpression of circPTK2 had an opposite effect on HCC-LM3 and MHCC 97-H cells (Fig. 3F).

circPTK2 increases the migration and invasion capability of HCC cells. (A,B) Cell migration ability of HCC-LM3 and MHCC 97-H cells transfected with sh-circPTK2 plasmid(A), or OE-circPTK2 plasmid(B), assessed by scratch-healing assays; Mean ± SD, n = 3, **p < 0.01, ***p < 0.001, scale bar:100 μm. (C,D) The transwell assays without Matrigel evaluate the migration ability of HCC-LM3 and MHCC 97-H after circPTK2 knockdown(C) or overexpressing(D); Mean ± SD, n = 3, **p < 0.01, ***p < 0.001. (E,F) The invasion capability of HCC cells transfected with sh-circPTK2 plasmid(E), or OE-circPTK2 plasmid(F), was examined using transwell assays with Matrigel; Mean ± SD, n = 3, **p < 0.01, ***p < 0.001, vs. corresponding control plasmids.
AKR1C3 is identified as a downstream regulatory gene of circPTK2
To further explore the regulatory mechanism of circPTK2 in HCC progression, we first conducted RNA sequencing (RNA-seq) in HCC-LM3 cell, which infected with sh-circPTK2 or negative control (sh-NC) plasmids, respectively. The differential genes was screened between the two groups in accordance with the criteria of |log2FC > = 1| and p < 0.05. The results showed 576 genes were down-regulated and 122 genes were up-regulated in the circPTK2 knockdown group(Fig. 4A). Then, GO and KEGG enrichment analysis was conducted based on these differential genes. The GO enrichment analysis showed circPTK2 mainly involved in “angiogenesis”, “cell substrate adhesion”, “cell differentiation”, “cell migration”, etc. (Figure S2A). The KEGG enrichment analysis demonstrated circPTK2 was significantly enriched in “pathways in cancer”, “Focal adhesion”, “Regulation of actin cytoskeleton”, “PI3K-AKT signaling pathway“(Figure S2B). These results indicate that circPTK2 is relevant to tumor proliferation, migration, and invasion.

AKR1C3 can be regulated by circPTK2 and associates with poor prognosis in HCC. (A) Volcano plot displays the differential genes when silencing circPTK2 in HCC-LM3 cell via RNA sequencing. (B) A heatmap shows the top 20 down-regulated genes after circPTK2 knockdown in HCC cell. (C) Western blot analysis verifies the protein level of AKR1C3 in HCC-LM3 and MHCC 97-H transfected with sh-circPTK2 or OE-circPTK2 vectors (Mean ± SD, n = 3, ***p < 0.001). (D) Spearman correlation analysis showing the association between circPTK2 and AKR1C3 expression in HCC tissues (n = 72). (E) The expression level of AKR1C3 in HCC tissues and normal tissues based on TCGA and GTEx databases (***p < 0.001). (F) The diagnostic accuracy of AKR1C3 estimated by receiver operating characteristic (ROC) curves. (G) Expression analysis for AKR1C3 in 72 paired HCC specimens by qRT-PCR (Mean ± SD, n = 72, ***p < 0.001). (H) The overall distribution of the fold change (tumor tissues vs. adjacent tissues) of AKR1C3 among 72 patients with HCC. (I) The protein level of AKR1C3 in 8 paired tumor tissues and adjacent normal tissues measured by Western blot.(J–L) The high expression of AKR1C3 significantly associated with poor OS (J), PFI (K), and DSS(L) based on Kaplan–Meier analysis.
The cluster heatmap displayed the most 20 downregulated genes in the sh-circPTK2 groups (Fig. 4B). We found that AKR1C3 was remarkably decreased in cricPTK2 knockdown groups compared to that in control groups (log2FC=-3.53, q = 2.22e-48)(Fig. 4B). The results of Western blot were further confirmed the regulatory effect of circPTK2 on AKR1C3 expression, that AKR1C3 expression was dramatically decreased in sh-circPTK2 cells, while increased in circPTK2 overexpression groups when compared with the corresponding control group (Fig. 4C). Spearman correlation analysis revealed a positive relationship (r = 0.629, p < 0.001) between circPTK2 and AKR1C3 expression in HCC tissues(Fig. 4D). The above results suggest that circPTK2 positively regulates the AKR1C3 expression both in mRNA and protein level.
Based on TCGA and GTEx databases, we found AKR1C3 was significantly up-regulated in 374 HCC tissues compared to that in 160 normal tissues (Fig. 4E). AKR1C3 expression exhibited high diagnostic accuracy for HCC patients with AUC = 0.9 in the ROC curves analysis (Fig. 4F). Then qRT-PCR and Western blot was used to further validate the differential expression of AKR1C3 in HCC specimens. qRT-PCR analysis showed that AKR1C3 was upregulated in the cancerous tissues, and it was highly expressed in 85% of HCC patients (Fig. 4G,H). Eight paired HCC tissues and para-carcinoma tissues were randomly selected for Western blot analysis, of which six were elevated cancerous tissues(Fig. 4I). In addition, Kaplan-Meier analyses revealed high AKR1C3 levels was positively associated with poorer OS (Fig. 4J), PFI (Fig. 4K), and DSS (Fig. 4L). Finally, Univariate binary logistic regression analysis was used to evaluate the correlation between AKR1C3 and clinicopathological features, the result showed that high expression of AKR1C3 showed higher T stage(p = 0.046) and AFP level (p = 0.028), worse histological grade(p = 0.014), and more vascular invasion(p = 0.006) (Table S2). These results suggest that high expression of AKR1C3 in HCC tissues is associated with malignant progression and poor prognosis.
circPTK2 promotes HCC progression via AKR1C3
We also found the AKR1C3 protein expression was elevated in multiple HCC cells (Fig. 5A). To detect the role of AKR1C3 in proliferation, migration and invasion of HCC cells, we transfected the HCC-LM3 and MHCC 97-H with AKR1C3 siRNA((si-AKR1C3#1, si-AKR1C3#2, si-AKR1C3#3)). The protein level of AKR1C3 was significantly lower after transfection with AKR1C3 siRNA than after transfection with NC siRNA (Fig. 5B). si-AKR1C3#1 was selected for the subsequent functional experiments. The results of loss-of-function experiments showed AKR1C3 knockdown reduced cell viability (Fig. 5C) and DNA replication ability (Fig. 5D). Moreover, the AKR1C3-silencing markedly attenuated the cell migration and invasion capacity of HCC cells (Fig. 5E–G).

AKR1C3 promotes HCC cells proliferation, migration and invasion. (A) The expression level of AKR1C3 in HCC cells detected by Western blot. (B) The knockdown efficacy of AKR1C3 in HCC-LM3 and MHCC 97-H after transfecting siRNA. (C) Cell viability of HCC-LM3 and MHCC 97-H cells transfected with si-AKR1C3 or si-NC was evaluated by CCK8 (Mean ± SD, n = 6, ** p < 0.01, ***p < 0.001). (D) DNA replication capacity of HCC-LM3 and MHCC 97-H transfected with si-AKR1C3 or si-NC examined by EdU assays (Mean ± SD, n = 3, **p < 0.01), scale bar: 100 μm. (E) Cell migration ability of HCC-LM3 and MHCC 97-H cells transfected with si-AKR1C3 assessed by scratch-healing assays; Mean ± SD, n = 3, ***p < 0.001, scale bar:100 μm. (F) The transwell assays without Matrigel evaluate the migration ability of HCC-LM3 and MHCC 97-H after AKR1C3 knockdown; Mean ± SD, n = 3, ***p < 0.001. (G) The invasion capability of HCC cells transfected with si-AKR1C3 was examined using transwell assays with Matrigel; Mean ± SD, n = 3, ***p < 0.001.
To further investigate whether circPTK2 regulates the biological behaviors of HCC cells via AKR1C3, we transfected the AKR1C3 overexpression plasmids into circPTK2 stable knockdown cells to perform functional rescue assay. The results of Western blot showed that knockdown of circPTK2 obviously decreased the protein expression of AKR1C3 in HCC-LM3 and MHCC 97-H cells, while this decrease could be reversed by AKR1C3 overexpression (Fig. 6A). Then, the cell proliferation was detected by CCK8, EdU and colony formation assays, and the results suggested circPTK2 silencing inhibited HCC cell proliferation, cell clone formation, and DNA replication.whereas AKR1C3 overexpression could reverse this suppression effects(Fig. 6B–G). Moreover, AKR1C3 upregulation in HCC-LM3 and MHCC 97-H partially restored their migration and invasion abilities induced by circPTK2 silencing(Fig. 6H–K). In general, these results suggested knockdown of circPTK2 suppresses HCC cells proliferation, migration, and invasion via down-regulating the expression of AKR1C3.

circPTK2 promotes HCC cells proliferation, migration, and invasion via AKR1C3. (A) The protein level of AKR1C3 in HCC-LM3 and MHCC 97-H cells transfected with sh-circPTK2 plasmid alone or co-transfected with AKR1C3 overexpression vectors. (B,C) Cell viability of HCC-LM3(B) and MHCC 97-H (C) transfected with sh-circPTK2 plasmid alone or co-transfected with OE-AKR1C3 plasmids, measured by CCK8 (Mean ± SD, n = 6, ***p < 0.001). (D,E) Clone forming capacity of HCC-LM3 (D) and MHCC 97-H (E) cells co-transfected with sh-circPTK2 and OE-AKR1C3, evaluated by colony formation assays (Mean ± SD, n = 3, ***p < 0.001). (F,G) EdU assays estimated the DNA replication capacity of HCC-LM3(F) and MHCC 97-H (G) cells transfected with sh-circPTK2 plasmid alone or co-transfected with OE-AKR1C3. Mean ± SD, n = 3, ***p < 0.001, scale bar: 100 μm. (H–J) The cells migration of HCC-LM3 and MHCC 97-H co-transfected with sh-circPTK2 and OE-AKR1C3, assessed by scratch-healing experiments (H,I) and transwell migration assays (J). Mean ± SD, n = 3, **p < 0.01, ***p < 0.001, scale bar:100 μm. (K) The invasion ability of HCC-LM3 and MHCC 97-H co-transfected with sh-circPTK2 plasmid and OE-AKR1C3 vector, measured by transwell invasion assays. Mean ± SD, n = 3, **p < 0.01, ***p < 0.001.
miR-379-5p is deemed as an intermediate bridge of circPTK2 and AKR1C3
Numerous studies have confirmed that circRNAs located in the cytoplasm can serve as miRNA sponges via combining with argonaute2 (AGO2) protein and forming RNA-induced silencing complex (RISCs), and further regulate the downstream genes19. Thus, circPTK2 might regulate AKR1C3 expression via binding to miRNA. In order to validate this hypothesis, we performed RNA immunoprecipitation (RIP) assay, in which the anti-AGO2 was used to enrich the RNA, and the circPTK2 expression was detected by qRT-PCR. The results showed that circPTK2 was significantly enriched in the anti-AGO2 group compared to that in control anti-IgG group, suggesting circPTK2 may act as a sponge for miRNAs (Fig. 7A). To determine intermediate miRNAs between circPTK2 and AKR1C3, we first applied the online prediction tools (miRDB, Targetscan, and mirDIP) to predict potential miRNA-targeted AKR1C3. Venn analysis indicated the miR-3922-3p, miR-4703-3p, miR-379-5p, miR-3136-5p, miR-184, miR-3176, and miR-5701 contains the binding sites for AKR1C3 (Fig. 7B). Then, miRanda v3.3a was used to examine whether the above seven miRNAs could interact with circPTK2. The results showed miR-379-5p, miR-3922-3p, and miR-4703-3p contains binding sites for circPTK2 (Fig. 7C). Subsequently, we analyzed the differential expression of the three miRNAs (miR-379-5p, miR-3922-3p, and miR-4703-3p) based on TCGA datasets, and found that miR-379-5p was significantly down-regulated in HCC tissues compared with normal tissues (Figure S3A), while miR-3922-3p was upregulated in HCC tissues (Figure S3B), and no significance was observed in miR-4703 (Figure S3C). Subsequently, we analyzed the expression of miR-379-5p in 72 paired HCC tissues and adjacent tissues using qRT-PCR and discovered it was lower expression in HCC tissues than that in paracancerous tissues (Figure S3D-E). Moreover, miR-379-5p was markedly downregulated in HCC cell lines compared to that in normal liver cell (L02) (Figure S3F). Additionally, Spearman correlation analysis showed the miR-379-5p expression in HCC tissues was negatively correlated with AKR1C3 (R=−0.67, p < 0.001) and circPTK2(R=−0.512, p < 0.001)(Fig. 7D). Thus, it is reasonable to speculate that that circPTK2 may regulate the expression of AKR1C3 by sponging miR-379-5p.

Identification of miRNA binding to circPTK2 that regulates the proliferation, migration and invasion in HCC cells. (A) RIP assays validated the combination of circPTK2 with anti-AGO2 in HCC-LM3 and MHCC 97-H cells (Mean ± SD, n = 3, ***p < 0.001). (B) Venn plot showed the miRNAs combination to AKR1C3 predicted by miRDB, Targetscan, and mirDIP. (C) miRNAs combination sites with circPTK2 forecast by miRanda v3.3a. (D) Spearman correlation analyses showing the relationships of miR-379-5p and AKR1C3 (left), and circPTK2 (right) in HCC tissues (n = 72). (E) Biotin-labeled miRNA pull-down assay confirmed the interaction of miR-379-5p with circPTK2 and AKR1C3 (Mean ± SD, n = 3, ***p < 0.001). (F) The co-location of miR-379-5p and circPTK2 in HCC cells judged by FISH. (G) Luciferase reporter assay detected the luciferase activity in HEK-293T cells co-transfected with miRNA-379-5p mimic (or mimic-NC) and circPTK2-wt (or circPTK2-mut) luciferase reporter vectors. Mean ± SD, n = 3, ***p < 0.001. (H) Cell viability of HCC-LM3 and MHCC 97-H cells transfected with sh-circPTK2 alone or co-transfected with miR-379-5p inhibitor, measured by CCK8(Mean ± SD, n = 6, ***p < 0.001). (I) EdU assays estimates the DNA replication capacity of HCC-LM3 and MHCC 97-H cells transfected with sh-circPTK2 alone or co-transfected with miR-379-5p inhibitor. Mean ± SD, n = 3, **p < 0.01, ***p < 0.001, scale bar: 100 μm. (J,K) The cells migration of HCC-LM3 and MHCC 97-H co-transfected with sh-circPTK2 and miR-379-5p inhibitor, assessed by scratch-healing experiments (J) and transwell migration assays (K). Mean ± SD, n = 3, ***p < 0.001, scale bar:100 μm. (L) The invasion ability of HCC-LM3 and MHCC 97-H transfected with sh-circPTK2 and miR-379-5p inhibitor, measured by transwell invasion assays. Mean ± SD, n = 3, ***p < 0.001.
The interaction between circPTK2 and miR-379-5p was identified by Biotin-labeled miRNA pull-down assay, Fluorescence in situ hybridization (FISH) co-location and luciferase reporter assay. For the Biotin-labeled miRNA pull-down assay, we found compared with control groups, circPTK2 and AKR1C3 was preferentially enriched in biotin-labeled miR-379-5p transfection groups (Fig. 7E), suggesting that miR-379-5p may interact simultanously with circPTK2 and AKR1C3. The FISH assays further confirmed that circPTK2 and miR-379-5p were co-located in the cytoplasm (Fig. 7F). Afterwards, the binding sites between circPTK2 and miR-379-5p was verified via luciferase reporter assay, the results indicated that the miR-379-5p mimic significantly weaken luciferase activity of circPTK2-wt (p < 0.001), while did not affect the luciferase activity of circPTK2-mut (Fig. 7G).
miR-379-5p overturns the effects of circPTK2 on the proliferation, migration and invasion of HCC cells
To further clarify the role of circPTK2 and miR-379-5p in HCC cells proliferation, migration and invasion, we transfected miR-379-5p inhibitor into circPTK2 stable knockdown HCC cells. The results showed that the proliferation and DNA replication capability was increased in co-tansfection groups than those in only transfected sh-circPTK2, suggesting that miR-379-5p inhibitor could recover the suppressive effect on cell proliferation mediated by circPTK2 silencing (Fig. 7H,I). Furthermore, the inhibitory effects of circPTK2 knockdown on migration and invasion of HCC cells were abolished by miR-379-5p down-regulation (Fig. 7J–L).
miR-379-5p inhibition promotes HCC progression by targeting AKR1C3
To clarify the regulatory effect of miR-379-5p on AKR1C3 expression, we conducted the Western blot and found that miR-379-5p mimic remarkably down-regulated the AKR1C3 expression, while miR-379-5p inhibitor increased AKR1C3 protein level(Fig. 8A). Then, TargetScan website was used to predict the possible binding sites between miR-379-5 p and AKR1C3 (Fig. 8B). Dual luciferase reporter assays indicated miR-379-5p significantly reduced the relative luciferase activity of wild-type AKR1C3-3 ‘UTR compared to the negative control, while such reduction was not observed in AKR1C3-3’ UTR mutation (Fig. 8B). These results suggest that miR-379-5p targeted binding to AKR1C3-3 ‘UTR caused the mRNA degradation of AKR1C3, resulting in decreased expression of AKR1C3.

miR-379-5p targets AKR1C3 to sustain HCC progression. (A) Western blot detected the protein expression of AKR1C3 in HCC-LM3 and MHCC 97-H transfected with miR-379-5p mimic or inhibitor.(B) Schematic of AKR1C3 3′ UTR wild-type (wt) and mutant (mut) luciferase reporter vectors; Luciferase activity of HEK-293T cell co-transfected with miR-379-5p mimic and AKR1C3 3′-UTR (wt) or AKR1C3 3′-UTR (mut) vectores determined by dual-luciferase reporter assays, Mean ± SD, n = 3, ***p < 0.001. (C,D) Cell viability of HCC cells transfected with miR-379-5p inhibitor alone or co-transfected with si-AKR1C3, measured by CCK8 (Mean ± SD, n = 6, **p < 0.01, ***p < 0.001). (E,F) DNA replication capacity of HCC-LM3 (E) and MHCC 97-H (F) cells co-transfected with miR-379-5p inhibitor and si-AKR1C3, evaluated by EdU assays (Mean ± SD, n = 3, ***p < 0.001). scale bar: 100 μm. (G–I) The cells migration of HCC-LM3 and MHCC 97-H transfected with miR-379-5p inhibitor and si-AKR1C3, assessed by scratch-healing experiments (G,H) and transwell migration assays (I). Mean ± SD, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, scale bar:100 μm. (J). The invasion ability of HCC cells transfected with miR-379-5p inhibitor and si-AKR1C3, measured by transwell invasion assays. Mean ± SD, n = 3, ***p < 0.001, scale bar:100 μm. (K) Western blot analyzed the expression of AKR1C3 in HCC-LM3 and MHCC 97-H co-transfected with sh-circPTK2 and miR-379-5p inhibitor. (L) Schematic diagram shows circPTK2 promoting HCC cell proliferation, migration and invasion via miR-379-5p/AKR1C3 axis.
It was reported that miR-379-5p acts as a tumor suppressor in multiple tumors20,21,22. To further explore the role of miR-379-5p in HCC cells proliferation, migration and invasion, we co-transfected miR-379-5p inhibitor and si-AKR1C3 into HCC-LM3 and MHCC 97-H cells. Functional assay showed miR-379-5p inhibitor significantly increased cells proliferation, but this phenomenon could be reversed by co-transfecting si-AKR1C3 (Fig. 8C–F). In the scratch-healing and transwell assays, we found miR-379-5p knockdown prominently promoted the migration and invasion of HCC cells, which could also be alleviated by AKR1C3 knockdown(Fig. 8G–J). These results manifest that silencing miR-379-5p promotes HCC cells proliferation, migration and invasion via targeting AKR1C3.
Finally, the regulatory relationship among circPTK2, miR-379-5p, and AKR1C3 was confirmed by Western blot, as showed in Fig. 8K, circPTK2 silencing significantly inhibited the expression of AKR1C3, but the inhibition effect could be reversed by miR-379-5p inhibitor. In general, these results indicate that circPTK2 acts as miR-379-5p sponge and up-regulates the expression of AKR1C3, thus promoting the proliferation, migration and invasion of HCC cells(Fig. 8L).
Discussion
circRNAs represent a newly identified class of non-coding RNAs that perform critical biological roles in tumorigenesis and tumor progression23. Despite this, the functions of numous circRNAs in HCC have not been fully elucidated. In our research, we identified a novel circRNA, hsa_circ_0008305(circPTK2), which is markedly up-regulated in HCC tissues. Forethemore, the expression levels of circPTK2 exhibit a positive correlation with tumor diameter, T stage, and vascular invasion. Our findings also demonstrate that circPTK2 enhances the HCC cell proliferation, migration, and invasion both in vitro and vivo. Mechanistically, we elucidated that circPTK2 potentially functions as a molecular sponge for miR-379-5p, thereby alleviating the inhibitory effect of miR-379-5p on AKR1C3, consequently leading to the up-regulation of AKR1C3.
Increasing evidence has indicated that circRNAs play a crucial role in the onset and progression of HCC24.The biological function of circPTK2(circ_0008305) was initially reported in non-small cell lung cancer(NSCLC), where it was found to inhibit TGF-β-induced EMT and tumor cell invasion by regulating TIF-1γ through miR-429/miR-200b-3p axis15. circPTK2 has been shown to be positively associated with cell migration and invasion in ovarian cancer via the miR‑639 and FOXC1 regulatory pathway16. In bladder cancer, circPTK2 significantly enhancerd cell migration, invasion, and resistance to gemcitabine in vitro, as well as promoted lymph node metastasis in vivo17. Despite ongoing research, the biological function and molecular mechanism of circPTK2 in HCC remain inadequately understood. In this study, GO and KEGG enrichment analysis demonstrated that circPTK2 is significantly associated with pathways related to cell proliferation and migration (Figure S2A,B). Functional assays, both in vitro and in vivo, indicated that silencing circPTK2 suppresses cell proliferation, invasion, and migration, whereas its overexpression produces the opposit biological effects. Overall, circPTK2, functions as an oncogene in HCC, playing a crucial role in tumor progression, which is expected to become a potential therapeutic target for patients with HCC.
A growing number of studies suggest that circRNAs frequently exert biological role by modulating target genes and subsequently influencing critical signaling pathways. For instance, circ_0001588 induces malignant progression of hepatocellular carcinoma through regulating miR874/CDK4 axis, in which CDK4 serves as a pivotal regulator of the cell cycle25. Additionally, circPIAS1 inhibits ferroptosis by competitively binding to miR-455-3p, resulting in the upregulation of Nuclear Protein 1 (NUPR1), which is a key regulator of iron metabolism in cancer cells14. In this study, we validated the positive regulatory effect of circPTK2 on AKR1C3 in HCC cells by using RNA-seq and Western blot analysis. AKR1C3 (also known as type 5 17b-hydroxysteroid dehydrogenase), an enzyme belonging to the aldo-ketoreductase family, plays an important biological role in the metabolism of prostaglandins, steroid hormones and retinoids26. AKR1C3 is highly expressed in a variety of cancers and contributes to poor prognosis27,28,29. In HCC, a recent study reported that AKR1C3 promotes HCC cell proliferation and cisplatin resistance by reducing PARP1 ubiquitination30. Zhou et al. revealed that AKR1C3 positively regulates HCC cell proliferation and metastasis via AKR1C3/NF-kB/STAT3 signaling loop26. In this study, we investigated the role of AKR1C3 in HCC progression and invasion from a novel perspective, focusing on the post-transcriptional competitive endogenous RNA (ceRNA) regulatory network. Ourfindings align with previous research, indicating that AKR1C3 expression is elevated in HCC tissues and cells and that its overexpression promotes HCC cells proliferation, migration and invasion.Notably, we discovered that AKR1C3 could reverse the effect of circPTK2 on the malignant progression of HCC.
Recently, the regulatory mechanism of circRNAs in tumorigenesis is gradually being uncovered, including microRNA(miRNA) sponges, binders of proteins or DNA, protein translation, and so forth31. A recent review highlighted that 79.6% of circRNAs in HCC function as miRNA sponge32.These endogenous sponges primarily arise from circRNA interacting with miRNA response elements (MRE) and binding to AGO proteins, which are key crucial components of RNA-induced silencing complex(RISC), in which miRNAs exert gene silencing functions33. According to the competing endogenous RNA (ceRNA) hypothesis, circRNA can competitively bind to miRNAs, thereby acting as sponges to regulate the silencing effect of miRNAs on thire target genes34. In this study, we verified the circular structure and subcellular localization of circPTK2, and found circPTK2 could be significantly enriched on AGO2 protein within cytoplasm, suggesting that circPTK2 may function as an endogenous miRNA sponge. Subsequently, we employed bioinformatics analysis, FISH assay, biotin-labeled miRNA pulldown assay and dual luciferase reporter gene experiments to investigate the potential miRNA/mRNA axis of circPTK2. Our study demonstrates that circPTK2 promotes the proliferation, invasion and migration of HCC cells by binding to miR-379-5p, thereby preventing the miRNA-mediated degradation of AKR1C3. Previous researches have reported that miR-379-5p plays an inhibitory role in various cancers, including glioma35, nasopharyngeal carcinoma36, non-small cell lung cancer37, breast cancer22, and endometrial cancer21. In HCC, the downregulation of miR-379-5p is related to an increased TNM stages and metastasis, miR-379-5p inhibits the migration, invasion, and epithelial-mesenchymal transformation (EMT) by targeting FAK/AKT signal axis38. In the present study, we observed that miR-379-5p is down-regulated in HCC tissues and cells. miR-379-5p acts as a tumor suppressor hindering cell proliferation, migration and invasion in HCC, which is consistent with the results reported in previous study38. Furthermore, miR-379-5p inhibitor can reverse the depressant effect of circPTK2 knockdown on the proliferation and invasion of HCC cells. In addition, miR-379-5p directly binds to the 3’ untranslated region (UTR) of AKR1C3, leading to the degradation of AKR1C3 mRNA. Functional assays indicated silencing AKR1C3 partially mitigates the oncogenic effects of miR-379-5p inhibition in HCC cells proliferation, migration and invasion. In summary, these results fully demonstrated that circPTK2 regulates the proliferation, migration and invasion via miR-379-5p/AKR1C3 axis in HCC.
This study also has some limitations. Firstly, we did not include prognostic factors when discussing the clinical significance of circPTK2. Secondly, this study has elucidated the effect of circPTK2/miR-379-5p/AKR1C3 axis on the HCC progression, but the downstream signal pathway of AKR1C3 needs to be further explored. Besides, we found the expression level of circPTK2 in the HCC plasma was higher than that in healthy populations (Fig. 1D), suggesting that circPTK2 (especially in exosomal circPTK2) in the circulatory system may promote the development and metastasis of HCC. However, the relationship between circPTK2 and exosome, as well as the biological effect of exosomal circPTK2 still need in-depth discussion. Therefore, in future studies, we will collect the survival data and increase HCC plasma samples to enrich our results.
In conclusion, this study demonstrated that circPTK2 significantly contributes to the malignant progression of HCC by acting as a sponge for miR-379-5p and regulating the expression of AKR1C3. The findings from this research provide a novel insights into the molecular mechanism of HCC progression, and may open a way to find a promising molecular-targeted therapies.
Materials and methods
Patient tissue specimens and ethics approval
A total of 72 HCC tissues specimens was collected from the Second Affiliated Hospital of Nanchang University between October 2020 and May 2022. The inclusion criteria is that patients with pathological diagnosis of hepatocellular carcinoma and had undergone radical resection. The exclusion criteria are as follows: (1) patients with preoperative anti-cancer therapy, including thermal ablation, hepatic artery interventional therapy, chemotherapy, targeted drugs, immunotherapy, or traditional Chinese medicine therapy; (2) patients with other malignant tumors or systemic immune diseases; (3) patients without complete clinical information. All tissue specimens collection were immediately frozen in liquid nitrogen within 15 min after removal from the patient and and stored at − 80 °C for long-term storage. The collection of HCC tissues in this study was approved by the Second Affiliated Hospital of Nanchang University Medical Research Ethics Committee, and the written informed consent was provided from each patient in accordance with the Helsinki Declaration. The detailed clinical characteristics, as well as its correlations with hsa_circ_0008305 expression, were described in the Table 1.
In silico analysis
The HCC circRNA microarray datasets were downloaded from Gene Expression Omnibus (GEO, https://www.ncbi.nlm.nih.gov/geo/) database, the circRNA datasets of HCC were as follows: GSE97332 (including 7 HCC tumor tissues and 7 normal liver tissues), GSE94508 (including 5 liver cancer tissues and 5 adjacent tissues), and GSE166678 (including 3 plasma samples of HCC and 3 plasma samples from healthy individuals). Subsequently, the differential circRNAs between HCC and normal liver tissues in GSE97332 were screened out according to the criteria of | log2FC | > 1.5 and p < 0.05. The differential expression of circPTK2 was further verified in HCC tissues and plasma samples based on GSE94508 and GSE166678 datasets.
The mRNA sequencing data of HCC dataset in TPM format was downloaded from UCSC Xena (https://xenabrowser.net/datapages/ ). The clinical information was also obtained from UCSC Xena database, including age, gender, histological grade, pathological staging, vascular invasion status, Overall Survival (OS), Progression Free Interval (PFI), and Disease Specific Survival (DSS). The mRNA differential expression between HCC and normal tissues was analyzed using the “limma” and “ggplot2” packages in R 4.0.5 software. The Wilcoxon rank sum test was used for statistical analysis, and p < 0.05 was considered statistically significant. Kaplan-Meier analysis was applied to explore the relationship between mRNA expression and OS, PFI, and DSS of HCC patients.
Cell lines
Human HCC cell lines HCC-LM3, MHCC97-H, HepG2, SMMC7721, and Huh-7 were purchased from Procell Life Sciences and Technology Co., Ltd. (Wuhan, China), the normal human liver cell L02 was acquired from the cell bank of Chinese Academy of Science. All these cells were cultured in high glucose DMEM (Solaibio, Beijing, China) supplemented with 10% FBS (Gibco, Grand Island, NY, USA) and 1% streptomycin/penicillin sodium (Biotechnology, Beijing, China) under 37 °C with 5% CO2 environment. All the cell lines have been authenticated using STR (or SNP) profiling within the last three years. All the experiments were performed with mycoplasma-free cells.
Cell transfection
The sequence of circPTK2 and AKR1C3 was cloned into pHBLV-CMV-circ-EF1-ZsGreen-T2A-Puro and pcDNA3.1 vectors to construct circPTK2 overexpression (OE-circPTK2) and AKR1C3 overexpression (OE-AKR1C3) plasmids, respectively. Short hairpin RNA (shRNA) targeting circPTK2 (sh-circPTK2) was inserted to pLKO plasmid. All the above plasmids was constructed by Hanheng Biotechnology (Shanghai, China). The specific small interfering RNAs (siRNA) targeting AKR1C3(si-AKR1C3) were synthesized by Hanheng Biotechnology (Shanghai, China). miR-379-5p mimic, inhibitor, and control plasmids (miR-NC) were purchased from RiboBio (Guangzhou, China). According to the manufacturer’s instructions, HCC-LM3 and MHCC97-H(2 × 105 cells) were uniformly inoculated into a 6-well plate, and were transfected with plasmids or siRNA using Lipofectamine 3000 reagent (Invitrogen). After 48 h of transfection, RT‒qPCR was conducted to analyze the knockdown or overexpresion efficiency. The target sequence for sh-circPTK2 is 5’-GCCGTCTCTGTGTCAGAAA AG-3’, and for control is 5’-GGGCGAGGAGCTGTTCACCG-3’. The target sequences for si-AKR1C3#1 is 5’-GGUGAGGAACUUUCACCAATT-3’, for si-AKR1C3#2 is 5’-GGUGAGGAACUUUCACCAATT-3’, for si-AKR1C3#3 is 5’-CCAGAGGUU CCGAGAAGUAAATT-3’, and for control is 5’-UUCUCCGAA CGUGUCACGUTT-3’.
Quantitative real-time PCR and RNA nucleoplasmic separation
Nuclear and cytoplasmic RNAs were isolated from HCC cells using Nuclear and Cytoplasmic Extraction Reagents (Beyotime, Shanghai, China). Total RNA of HCC tissues and cells was extracted using Trizol Reagent (Invitrogen, Carlsbad, CA, USA). Next, RNA was reverse-transcribed into complementary DNA (cDNA) using EasyScript® One-Step gDNA Removal and cDNA Synthesis SuperMix (TransGen Biotech, Beijing, China). Then, qPCR was performed with TB Green® Premix ExTaq™ II (TaKaRa, Dalian, China). GAPDH and U6 was used as endogenous control for mRNAs and miRNAs, respectively. The relative mRNA expression in HCC cells was determined by 2−△△CT method, and that in HCC tissues was reckoned by 2−△CT. The qRT-PCR primers were designed and synthesized by Sangon Biotech Co., Ltd. (Shanghai, China), and the sequences were displayed in Table S1.
Circular structure verification
The circular structure of hsa_circ_0008305 was validated by Dideoxy sequencing, RNase R and actinomycin D treatment. First, the PCR products of circPTK2 were delivered to Geneseed Biotechnology Co, Ltd. (Guangzhou, China) for Dideoxy sequencing. For the RNase R treatment, 4 ug of total RNA isolated from HCC-LM3 and MHCC 97-H was incubated with 10U RNase R (Geneseed Biotechnology Co, Ltd., Guangzhou, China) at 37 ℃ for 30 min to fully digest linear RNA, followed by incubation at 70℃ for 10 min to deactivate RNase R. qRT-PCR was conducted to assess the related mRNA expression of circPTK2 and linear PTK2. For the Actinomycin D treatment, HCC-LM3 and MHCC 97-H was disposed with Actinomycin D (2 ug/mL, AAT Bioquest, USA). Then total RNA of HCC cells was extracted when co-cultured with actinomycin D for 0, 2, 6, 12, and 24 h, respectively. The mRNA levels of circPTK2 and linear PTK2 were detected by qRT-PCR.
Fluorescence in situ hybridization (FISH)
CY3-labeled probe for circPTK2 and FITC-labeled probe for miR-379-5p were designed and synthesized by Geneseed Biotechnology (Guangzhou, China), which were used to labeled the distribution of circPTK2 and miR-379-5p in HCC-LM3 and MHCC 97-H. The FISH assay was conducted by using a Fluorescence in Situ Hybridization Kit (Servicebio, Wuhan, China) according to the manufacturer’s guidelines. Cell nuclei were stained with DAPI. Images were acquired using Olympus FluoView FV1000 confocal microscope (Tokyo, Japan). The probe sequence for circPTK2 was 5’ CY3-AACAACATCTTTTCTGACACAG-3’ CY3, and for miR-379-5p was 5’FITC-CCTACGTTCCATAGTCTACC A-3’.
RNA sequencing (RNA-seq) and enrichment analysis
RNA sequencing (RNA-seq) was conducted in HCC-LM3 transfected with sh-circPTK2 or sh-NC plasmids, and was completed by Lianchuan Biotechnology Co., Ltd. (Hangzhou, China). The differential genes between sh-circPTK2 group and sh-NC group was screened in accordance with the criteria of |log2FC > = 1| and p < 0.05. To illuminate the biological functions and pathways associated with differential genes, we performed enrichment analysis of Gene Ontology (GO, http://www.geneontology.org/) and Kyoto Encyclopedia of Genes and Genomes (KEGG, http://www.genome.jp/kegg/pathway.html). The top 20 pathways were displayed using R “ggplot2” package.
Western blot analysis
The total protein was extracted from HCC cells and HCC tissues by using RIPA lysis buffer (Beyotime, Shanghai, China), separated in 10% SDS-PAGE gel, and then transferred onto PVDF membrane (Millipore, Schwalbach, Germany). The membrane was sealed in TBST containing 5% nonfat milk for 1 h, and then incubated with corresponding diluted primary antibody at 4 °C overnight. The primary antibodies used in this study were anti-AKR1C3(1:1000, Abmart, Shanghai, China), anti-Tublin (1:5000, Proteintech, Wuhan, China), anti-GAPDH(1:5000, Proteintech, Wuhan, China). The membranes were incubated with horseradish peroxidase-conjugated secondary antibody for 1 h at 37 °C. The proteins bands were visualized by ECL chemiluminescence reaction regent(US Everbright Inc., Suzhou, China).
Cell proliferation assays
Cell proliferation was estimated using CCK8, EdU assays, and colony formation assays. For the CCK8 assays, the transfected HCC-LM3 and MHCC 97-H was adjusted to 2 × 104 cells/mL, and 100 µL cell suspension was uniformly seeded in 96-well plates. After cultivation for 0, 24, 48, and 72 h, 10 µL CCK8 reagent was added to each well and incubated in dark for 2 h at 37 ℃. The relative absorbance at 450 nm wavelength was measured using a spectrophotometer (Bio-Rad, Hercules, CA, USA). For the EdU assay, the cell density was adjusted to 2 × 105 cells /mL, and 100µL cell suspension was seeded in a 96-well plate. After 48 h cultivation, EdU assay was performed using YF 555 Click-iT EdU kit (C6016L, US Everbright® Inc., China). Images were acquired using a fluorescence microscope, and ImageJ software was used to analyze the proportion of EdU-positive cells. For the colony formation assays, the transfected cells (500 cells/well) were homogenously inoculated in a 6-well plate and cultured in 37 °C with 5% CO2 incubator for 10–14 days. Then, the cell colonies were fixed with 4% paraformaldehyde for 20 min and stained with 0.1% crystal violet for 10 min. Image J software was used to count the number of colonies.
Cell migration and invasion assays
Cell migration and invasion ability was assessed using scratch-healing assays and Transwell chambers. For the scratch-healing assays, the transfected cells were evenly incubated in 6-well plates (5 × 105 cells/well) until 100% confluence. Next, the cell mono-layer was scratched using a sterile 100 µL pipette tip and washed with PBS buffer to remove the floating cells. The cells migration was recorded by an optical microscope (Olympus, Tokyo, Japan) at 0 h and 36 h, respectively. For the Transwell migration assays, a total of 3 × 104 cells in 200 µL serum-free medium was plated in the upper chamber inserted into a 24-well plate, and the bottom chamber was added 600 µL DMEM containing 20% FBS. After 48 h penetration, the unpenetrated cells were gently wiped with wet cotton swab, and the migrated and invaded cells were fixed with 4% paraformaldehyde for 20 min and stained with 0.1% crystal violet for 10 min at room temperature. The procedure of Transwell invasion assays is similar to the migration assay, except that the upper chamber was pre-covered with 10% Matrigel. Finally, the migrating or invading cells were calculated using an inverted microscope.
Biotin-labeled miRNA pull-down assay
Biotin-labeled miR-379-5p and negative control(miR-NC) probes were synthesized by Generay Biotech Co., Ltd.(Shanghai, China). The sequence of biotin-coupled miR-379-5p was 5’-UGGUAGACUAUGGAACGUAGG-3’, the sequence of miR-NC was 5’-UUCUCCGAACGUGUCACGUTT-3’. Then, the biotin-coupled miRNA was transfected into HCC-LM3 and MHCC 97-H, followed by collecting cell lysates with lysis buffer. The miRNA-binding complex were pulled down by co-incubation with the M280 magnetic beads at 4℃overnight. The binding RNAs was extracted using Trizol Reagent, and qRT-PCR was conducted to measure the related expression of circPTK2 and AKR1C3.
RNA immunoprecipitation (RIP) assay
A total of 2 × 107 cells were collected according to conventional cell culture method. RNA immunoprecipitation Kit(Geneseed Biotechnology, Guangzhou, China) was applied in RIP assay. Briefly, the cells were added with lysis buffer, mixed well, and placed on crushed ice to cleave 10 min. The cell supernatant was obtained after centrifugation at 14,000 g for 10 min. Then, protein A + G breads (B23201, Bimake, USA) was pre-coated with anti-argonaute 2 (anti-AGO2) antibody (ab186733, Abcam, UK) or negative control anti-IgG antibody (#2729, CST, USA). Afterward, 450µL cell lysis was added to the magnetic beads bound with antibody, and the reaction was rotated at 4 ℃, 10 rpm / min for 1 h. The cell supernatant-bread-antibody complexes was eluted by elution buffer. Finally, RNA purification column was used to collect the RNA in the complexes, and qRT-PCR assay was carried out to detect the enrichment of circPTK2.
Double luciferase reporter assay
Targeted genes of miR-379-5p was predicted using bioinformatics websites, including mirDIP(http://ophid.utoronto.ca/mirDIP/index), miRDB(http://www.mirdb.org/ custom.htmL), and Targetscan (https://www.targetscan.org/vert72/). The targeted binding of hsa_circ_0008305 to miRNA was forecasted by miRanda v3.3a based on RNA secondary structure and free energy. Double luciferase reporter assay was performed to validate the above prediction. Firstly, wild-type vectors of circPTK2(circPTK2-WT) and AKR1C3 (AKR1C3-WT) was constructed by cloning their sequences containing corresponding binding sites with miR-379-5p into pSI-Check2 plasmid (Hanheng Biotechnology, Shanghai, China). Binding sites mutated fragments were inserted into pSI-Check2 plasmid forming mutation-type vectors of circPTK2 (circPTK2-MUT) and AKR1C3 (AKR1C3-MUT). The synthetic Luciferase reporter vectors and miR-379-5p mimic or miR-NC were co-transfected into the human embryonic kidney cell lines HEK-293T with lip3000 reagent(Invitrogen). After 48 h transfection, the luciferase activities were determined by Double Luciferase detection kit (TransGen Biotech, Beijing, China). Firefly luciferase activities/Renilla luciferase activities was used to calculate the relative luciferase activity.
In vivo tumor xenograft assay
All animal experiments were approved by the Institutional Animal Care and Use Committee of Nanchang Royo Biotech Co,. Ltd. All the following mice experiments were performed in accordance with the relevant guidelines and regulations. BALB/c nude mice(5-week-old, female, 18–20 g) was purchased from Ziyuan Experimental Animal Science and Technology Co., Ltd.(Hangzhou, China) and was held in Nanchang Royo Biotech CO,.Lt(Nanchang, China). The animal studies were approved by the Institutional Animal Care and Use Committee of Nanchang Royo Biotech Co,. Ltd. The mice were randomly assigned to four groups (n = 5 of each group). The HCC-LM3 cells (5 × 106 cells), which transfected with sh-circPTK2 lentiviruses, OE-circPTK2 lentiviruses, or correspond control lentiviruses, were injected into the right flank of the mice. Tumor sizes and weights were recorded twice a week. Tumors were harvested after 4 weeks of injection. Finally, the tumor tissues were fixed in 4% polyformaldehyde and subjected to immunohistochemistry staining.
Immunohistochemistry (IHC)
The fixed tumor tissues from mice were dehydrated with gradient ethanol, penetrated with xylene, and then were embedded in paraffin. The tissues were cut into 5 μm slices and attached to the slides coated with poly lysine. After dewaxing and hydration, the tissue slices were incubation with 3% H2O2 and soaked in citric acid buffer. 3% BSA was added to cover the tissue evenly for sealing. Next, the sections were incubated with anti-Ki67 (ABclonal, Wuhan, China) overnight at 4 ℃. And then the slices were incubated with HRP-polymer‐conjugated secondary antibodies. After DAB coloration and hematoxylin re-staining, tissue slices were imaged under a light microscope.
Statistical analysis
Statistical analyses were performed using R software (https://www.r-project.org/version 4.0.4) and GraphPad Prism version 9.0(GraphPad Software, CA, USA. The experiment was repeated at least three times, and the results of continuous data were reported as as the Mean ± SD. The Kolmogorov–Smirnov test was used for normality analysis, and unpaired Student’s t test or one-way analysis of variance(ANOVA)(followed by Turkey’s Honest Significant Difference test) was applied in multiple groups comparison. The correlation between the expression of circPTK2 and clinicopathological features were analyzed using Chi-squared or Fisher’s exact tests, as appropriate. The gene expression correlation was assessed using Spearman correlation analysis. All statistical tests were two-sided, and p < 0.05 was taken as statistical significance.
Data availability
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and the project name can be found in The Cancer Genome Atlas database (https://portal.gdc.cancer.gov/repository, Project: TCGA-HCC). The raw data are available from the corresponding author on reasonable request.
Change history
02 September 2025
A Correction to this paper has been published: https://doi.org/10.1038/s41598-025-13503-4
Abbreviations
- HCC:
-
Hepatocellular carcinoma
- circRNA:
-
Circular RNA
- GEO:
-
Gene expression omnibus
- OS:
-
Overall survival
- PFI:
-
Progression free interval
- DSS:
-
Disease specific survival
- RNA-seq:
-
RNA sequencing
- GO:
-
Gene ontology
- KEGG:
-
Kyoto encyclopedia of genes and genomes
- RIP:
-
RNA immunoprecipitation
- AKR1C3:
-
Type 5 17b-hydroxysteroid dehydrogenase
References
Siegel, R. L., Miller, K. D., Fuchs, H. E. & Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 72 (1), 7–33 (2022).
Sung, H. et al. Global Cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71 (3), 209–249 (2021).
Parikh, N. D. & Pillai, A. Recent advances in hepatocellular carcinoma treatment. Clinical gastroenterology and hepatology: the official clinical practice. J. Am. Gastroenterol. Assoc. 19 (10), 2020–2024 (2021).
Lu, Q. et al. Nanoparticles in tumor microenvironment remodeling and cancer immunotherapy. J. Hematol. Oncol. 17 (1), 16 (2024).
Singal, A. G., Kanwal, F. & Llovet, J. M. Global trends in hepatocellular carcinoma epidemiology: implications for screening, prevention and therapy. Nat. Rev. Clin. Oncol. 20 (12), 864–884 (2023).
Tian, Y. et al. Crosstalk between 5-methylcytosine and N(6)-methyladenosine machinery defines disease progression, therapeutic response and pharmacogenomic landscape in hepatocellular carcinoma. Mol. Cancer 22 (1), 5 (2023).
Yang, J. D. et al. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat. Rev. Gastroenterol. Hepatol. 16 (10), 589–604 (2019).
Casadei-Gardini, A., Orsi, G., Caputo, F. & Ercolani, G. Developments in predictive biomarkers for hepatocellular carcinoma therapy. Expert Rev. Anticancer Ther. 20 (1), 63–74 (2020).
Chen, L. L. The biogenesis and emerging roles of circular RNAs. Nat. Rev. Mol. Cell. Biol. 17 (4), 205–211 (2016).
Shen, H. et al. Circular RNAs: characteristics, biogenesis, mechanisms and functions in liver cancer. J. Hematol. Oncol. 14 (1), 134 (2021).
Zhang, Z. et al. Circular RNA: new star, new hope in cancer. BMC Cancer 18 (1), 834 (2018).
Wang, F., Nazarali, A. J. & Ji, S. Circular RNAs as potential biomarkers for cancer diagnosis and therapy. Am. J. Cancer Res. 6 (6), 1167–1176 (2016).
Hu, Z. et al. Exosome-derived circCCAR1 promotes CD8 + T-cell dysfunction and anti-PD1 resistance in hepatocellular carcinoma. Mol. Cancer 22 (1), 55 (2023).
Zhang, X. Y. et al. CircPIAS1 promotes hepatocellular carcinoma progression by inhibiting ferroptosis via the miR-455-3p/NUPR1/FTH1 axis. Mol. Cancer 23 (1), 113 (2024).
Wang, L. et al. Circular RNA hsa_circ_0008305 (circPTK2) inhibits TGF-β-induced epithelial-mesenchymal transition and metastasis by controlling TIF1γ in non-small cell lung cancer. Mol. Cancer 17 (1), 140 (2018).
Wu, S. G. et al. circ-PTK2 (hsa_circ_0008305) regulates the pathogenic processes of ovarian cancer via miR-639 and FOXC1 regulatory cascade. Cancer Cell. Int. 21 (1), 277 (2021).
Meng, X. et al. CircPTK2/PABPC1/SETDB1 axis promotes EMT-mediated tumor metastasis and gemcitabine resistance in bladder cancer. Cancer Lett. 554, 216023 (2023).
Wang, M., Chen, D., Zhang, H., Luo, C. & Circular, R. N. A. circPTK2 modulates migration and invasion via miR-136/NFIB signaling on triple-negative breast cancer cells in vitro. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 71 (4), 409–421 (2022).
Thomson, D. W. & Dinger, M. E. Endogenous microRNA sponges: evidence and controversy. Nat. Rev. Genet. 17 (5), 272–283 (2016).
Wu, D. et al. MicroRNA-379-5p plays a tumor-suppressive role in human bladder cancer growth and metastasis by directly targeting MDM2. Oncol. Rep. 37 (6), 3502–3508 (2017).
Liang, M., Chen, H. & Min, J. Mir-379-5p inhibits proliferation and invasion of the endometrial cancer cells by inhibiting expression of ROR1. Acta Biochim. Pol. 68 (4), 659–665 (2021).
Yang, K. et al. MiR-379-5p inhibits the proliferation, migration, and invasion of breast cancer by targeting KIF4A. Thorac. Cancer (2022).
Zhou, Y., Mao, X., Peng, R. & Bai, D. CircRNAs in hepatocellular carcinoma: characteristic, functions and clinical significance. Int. J. Med. Sci. 19 (14), 2033–2043 (2022).
Liu, Z., Yang, F., Xiao, Z. & Liu, Y. Review of novel functions and implications of circular RNAs in hepatocellular carcinoma. Front. Oncol. 13, 1093063 (2023).
Bin, X. et al. circ_0001588 induces the malignant progression of hepatocellular carcinoma by modulating miR-874/CDK4 signaling. J. Immunol. Res. 2021, 3759879 (2021).
Zhou, Q. et al. A positive feedback loop of AKR1C3-mediated activation of NF-κB and STAT3 facilitates proliferation and metastasis in hepatocellular carcinoma. Cancer Res. 81 (5), 1361–1374 (2021).
Zhong, T., Xu, F., Xu, J., Liu, L. & Chen, Y. Aldo-keto reductase 1C3 (AKR1C3) is associated with the doxorubicin resistance in human breast cancer via PTEN loss. Biomed. Pharmacother. 69, 317–325 (2015).
Jansson, A. K., Gunnarsson, C., Cohen, M., Sivik, T. & Stål, O. 17beta-hydroxysteroid dehydrogenase 14 affects estradiol levels in breast cancer cells and is a prognostic marker in estrogen receptor-positive breast cancer. Cancer Res. 66 (23), 11471–11477 (2006).
Wang, B. et al. AKR1C3, a crucial androgenic enzyme in prostate cancer, promotes epithelial-mesenchymal transition and metastasis through activating ERK signaling. Urol. Oncol. 36 (10) , 472.e11–e20. (2018).
Pan, D. et al. AKR1C3 regulated by NRF2/MAFG complex promotes proliferation via stabilizing PARP1 in hepatocellular carcinoma. Oncogene 41 (31), 3846–3858 (2022).
Niu, Z. S. & Wang, W. H. Circular RNAs in hepatocellular carcinoma: recent advances. World J. Gastrointest. Oncol. 14 (6), 1067–1085 (2022).
Cheng, D., Wang, J., Dong, Z. & Li, X. Cancer-related circular RNA: diverse biological functions. Cancer Cell. Int. 21 (1), 11 (2021).
Conn, V. M., Chinnaiyan, A. M. & Conn, S. J. Circular RNA in cancer. Nat. Rev. Cancer 24 (9), 597–613 (2024).
Liu, C. X. & Chen, L. L. Circular RNAs: characterization, cellular roles, and applications. Cell 185 (12), 2016–2034 (2022).
Yang, B., Xia, S., Ye, X., Jing, W. & Wu, B. MiR-379-5p targets microsomal glutathione transferase 1 (MGST1) to regulate human glioma in cell proliferation, migration and invasion and epithelial-mesenchymal transition (EMT). Biochem. Biophys. Res. Commun. 568, 8–14 (2021).
Zhang, F., Duan, C., Yin, S. & Tian, Y. MicroRNA-379-5p/YBX1 Axis regulates cellular EMT to suppress migration and invasion of Nasopharyngeal carcinoma cells. Cancer Manag. Res. 12, 4335–4346 (2020).
Jiang, Y., Zhu, P., Gao, Y. & Wang, A. miR–379–5p inhibits cell proliferation and promotes cell apoptosis in non–small cell lung cancer by targeting β–arrestin–1. Mol. Med. Rep. 22 (6), 4499–4508 (2020).
Chen, J. S. et al. MicroRNA-379-5p inhibits tumor invasion and metastasis by targeting FAK/AKT signaling in hepatocellular carcinoma. Cancer Lett. 375 (1), 73–83 (2016).
Funding
This research was supported by the National Natural Science Foundation of China (NO. 82060435), the Science and technology project of Jiangxi Provincial Department of Education(No.GJJ2400117), and the National Natural Science Foundation incubation project of The Second Affiliated Hospital of Nanchang University(No. 2023YNFY12031).
Author information
Authors and Affiliations
Contributions
JW contributed to conception and design this study. SH performed development of methodology and writing, review and revision of the paper. KL and YX provided acquisition, analysis and interpretation of data; HW and SF provided technical and material support. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
All experiments with HCC tissues were approved by The Second Affiliated Hospital of Nanchang University Medical Research Ethics Committee, and the written informed consent was provided from each patient in accordance with the Helsinki Declaration. All animal experiments were approved by the Institutional Animal Care and Use Committee of Nanchang Royo Biotech Co,. Ltd. This study is conducted in accordance with ARRIVE guidelines.
Consent for publication
Consent for publication has been obtained from the patients.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this Article was revised: The original version of the Article contained an error in Figure 3E, Figure 7L and Figure 8I, in which the data was duplicated. The original Article has been corrected.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, S., Liu, K., Xu, Y. et al. hsa_circ_0008305 facilitates the malignant progression of hepatocellular carcinoma by regulating AKR1C3 expression and sponging miR-379-5p. Sci Rep 15, 1309 (2025). https://doi.org/10.1038/s41598-025-85737-1
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-85737-1
Keywords
This article is cited by
-
DDX1 facilitates lenvatinib resistance in hepatocellular carcinoma through regulating ephrin-A3 and activating the Wnt/β-catenin signaling pathway
Functional & Integrative Genomics (2025)
