
Abstract
Traditional virtual screening methods need to explore expanse and vast chemical spaces and need to be based on existing chemical libraries. With the development of deep learning techniques for the de novo generation of molecules, also known as inverse molecular design, the increasingly widespread application of various types of deep learning algorithms has led to revolutionary changes in de novo molecular generation research. In particular, the emergence of a novel natural language processing (NLP) architecture called the transformer has improved the state-of-the-art performance of existing AI technologies. In this study, we modified one top-performing molecular generation model on the basis of the generative pretraining transformer (GPT) architecture in three directions. Moreover, we propose an integrated end-to-end neural network learning framework based on one complete encoder-decoder architecture transformer model: Transfer Text-to-Text Transformer (T5), by learning the embedding vector representation space of conditional molecular properties to encode and guide the vector representation of SMILES sequences, resulting in the output of the final decoder block with a softmax output (maximum likelihood objective). Moreover, we evaluated the performance of these NLP-based generation models and another new model architecture based on a selective state space and selected the best approach jointing a transfer learning strategy for de novo drug discovery to target L858R/T790M/C797S-mutant EGFR in non-small cell lung cancer.
Similar content being viewed by others
Introduction
De novo drug design, also known as molecular generation, is the process of producing novel chemical structures with desirable pharmacological and physicochemical properties that meet the desired molecular profile1. Chemical space is an expanse and vast space with up to 1023–1060 drug-like compounds2. The screening of large chemical libraries by brute force is computationally intractable, and traditional screening methods are still based on existing chemical libraries. In contrast, molecular generation aims to generate novel molecules with desirable properties by tuning compounds from chemical space. With the development of deep learning algorithms, especially the great stride of deep generative models in natural language processing (NLP), a novel NLP architecture called the transformer, the incorporation of the new paradigm of artificial intelligence has changed the mode of molecular generation. Especially as the development of large language models, Many fascinating discoveries have been established by many variants of the transformer model, such as BERT3, GPT4, and T55, in organic synthesis prediction6,35,36 and molecular property prediction7,8. These results provide encouraging evidence that language models can capture sufficient chemical and structural information because of their ability to process large-scale character data. In particular, Deva Priyakumar et al.9 provided evidence of the excellent performance of the generative pretraining (GPT) model MolGPT, a transformer-decoder model for the generation of drug-like molecules, in comparison with previously proposed modern machine learning frameworks, including CharRNN10, variational autoencoder (VAE)11, junction tree-VAE12, AAE13, and LatentGAN14.
When GPT works in dealing with sequence problems, the position information of each word in the sequence is very important. For the attention mechanism to distinguish words in different positions, it is necessary to introduce certain positional information to each word. The traditional GPT model uses alternating sine and cosine functions to create position codes. However, the original encoding was designed for fixed-length sequences, and beyond the maximum length model could not generate encoding. Second, the original position encoding could not handle relative positions, the original position encoding used sine and cosine functions to generate encoding, and the periodicity of these functions could make it difficult for the model to handle long-distance dependencies. Therefore, in this study, we first modified the rotary position embedding (RoPE) method proposed by Su et al.15, which encodes the absolute position with a rotation matrix and meanwhile incorporates the explicit relative position dependency in the self-attention formulation, named GPT-RoPE.
Additionally, deep neural networks involve the stacking of many layers, and the parameter updates in each layer can cause changes in the input data distribution of the upper layers. Through layer-by-layer stacking, the input distribution changes in the higher layers will be very dramatic, which means that the higher layers need to constantly adapt to the parameter updates in the lower layers, leading to training instability of the large language model16. The conventional module of the transformer framework Add&Norm adopts Post-LN17 for layer normalization, but its stability needs to be improved. Second, we modified the layer normalization function of the GPT via DeepNorm18 to modify the residual connection in the transformer, which combines the good performance of the Post-LN and stable training of the Pre-LN, named GPT-Deep.
Furthermore, in deep learning, activation functions determine the output of neurons and affect the learning ability and performance of the network. Therefore, here we introduce one novel activation function for GPT, proposed by the Google company19, which combines the properties of the activation functions GELU (Gaussian error linear unit) and GLU (gated linear unit) to improve the expressiveness and flexibility of the model by dynamically adjusting the degree of activation of the neurons, named GPT-GEGLU.
On the other hand, despite the dominance of the transformer in the realm of large-scale models, as the model size increases and the length of sequences to be processed increases, the computational cost of the self-attention mechanism increases quadratically with increasing context length. To address these limitations, researchers have explored various approaches. One such method, called Mamba, is currently receiving much attention from researchers20. Studies have shown that it can match or even beat transformers in language modeling21,22. Mamba is based on a new model architecture called the selective state space model, which is a simple generalization of structured state spaces for sequence modeling23. It determines the output variables of the system by using system state variables and input variables. This model is able to capture the system’s internal state or hidden state and can be used to predict the system’s future behavior. Consequently, to evaluate the performance of Mamba in de novo drug design, we also evaluated Mamba performance in molecular generation tasks.
However, although Deva Privakumar et al. reported that GPT-based molecular generation displays excellent performance and good representation of chemical space, MolGPT adopted only the decoder module of the transformer architecture to predict a sequence of SMILES tokens for molecular generation by predicting tokens as a result of masked self-attention applied to all previously generated tokens. In fact, it is an unconditional molecular generation architecture. Although the authors trained MolGPT conditionally to explicitly learn certain molecular properties, the molecular scaffold representation was concatenated at the start of the molecular sequence of the embeddings and distinguished from the SMILES tokens by segment tokens. However, this approach does not consider or learn the mapping relationship between the conditional properties and SMILES sequences, which may lead to a loss of control of specified molecular properties; therefore, it may not be enough for certain conditional drug-like molecular generation tasks.
Actually, many biological and chemical processes require molecules with certain properties. For example, due to L858R and T790M/C797S-EGFR mutations, drug generation in non-small cell lung cancer patients requires overcoming acquired resistance. In 2015, the third-generation EGFR tyrosine kinase inhibitor (TKI) osimertinib, which is potent for the EGFR T790M mutation and other activating EGFR mutations, was shown to be acquired resistance caused by the C797S mutation. Therefore, fourth-generation drugs overcoming the EGFR C797S mutation are currently extremely urgent24,25. On the basis of this goal, drug generation from immense and vast chemical space is needed to be limited for tyrosine kinase inhibitors. On this basis, we developed one complete encoder-decoder transformer implementation from the T5 model for the conditional molecular generation task named T5MolGe, which is leveraged to learn the conditional properties’ internal relationships over a property-controlled encoder model. Second, the original deformed SMILES, which, after multihead attention and layer normalization, received the first part output to employ further nonlinear transformations, finally obtained the prediction (maximum likelihood) through decoding of the transformed Softmax vector (more details can be found in Methods).
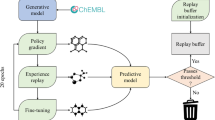
Therefore, in this study, on the one hand, we modified the former MolGPT model in three main directions, GPT-RoPE, GPT-Deep, and GPT-GEGLU; on the other hand, we proposed one T5-based model for the conditional molecular generation task named T5MolGe and considered another selective state space model, Mamba. By comparing the performance of these models on non-conditional and conditional generation tasks, the optimal de novo drug generation model combined with a transfer learning strategy (to overcome the bottleneck of small datasets in AI-aided novel drug discovery) for targeting L858R/T790M/C797S-mutant EGFR in non-small cell lung cancer was selected. The entire framework of this study is shown in Fig. 1.

The entire framework of this study.
Theory and methods
Modification of position encoding
GPT-RoPE. RoPE15 implements relative position encoding via absolute position encoding, which combines the advantages of absolute position encoding and relative position encoding to capture relative position information and handle complex structures and long-distance dependencies. The absolute position encoding function of vector \(q,k\) is defined as \(f(q,m) = R_{f} (q,m)e^{{i\Theta f(q,m)}} \quad f(k,n) = R_{f} (k,n)e^{{i\Theta f(k,n)}}.\) Where, \(R_{f} (q,m) = ||q||,{\kern 1pt} {\kern 1pt} {\kern 1pt} R_{f} (k,n) = ||k||\), the magnitude of the vector, \(\Theta = \left\{ {\theta _{i} = 10000^{{ - 2(i - 1)/d}} ,} \right.\) \(\left. {i \in [1,2,...,d/2]} \right\}\) .
Modification of layer normalization
To further improve the stability of transformers, many efforts to improve the optimization by means of better initialization26 or better architecture27 have been established, yet none of them have succeeded when the model is scaled to 1000 layers. H. Wang et al.18 proposed one new normalization method, which successfully scales transformers up to 2,500 attention and feedforward network sublayers, where the formulation of DeepNorm is written as: \(x_{l + 1} = LN(\alpha x_{l} + G_{l} (x_{l} ,\theta_{l} ))\), where LN is Layer normalization, \(\alpha\) is a constant and \(G_{l} (x_{l} ,\theta_{l} )\) is a function of l-th transformer attention or a feedforward network with parameters. DeepNorm expands the residual with parameters before performing the layer norm, thereby avoiding the instability caused by disappearance of the gradient18.
Modification of the activation function
GEGLU19 works by performing two parallel inputs: one is a pure linear transformation, and the other is a linear transformation through the GELU activation function. The result of these two transformations is then multiplied element-by-element: \(GEGLU(x,W,V,b,c) = GELU(xW + b) \otimes (xV + c)\), where vector \(x\) is the input of the hidden representation at a particular position in the sequence, \(W,V\) represents linear transformation matrices, and \(b\) represents the bias vector.
Mamba model
It is capable of dynamically adjusting its parameters in response to changes in the input variables, thus enabling the model to selectively pass or ignore information23. Mamba is constructed on the basis of a selective state space model, which defines a sequence-to-sequence transformation in two stages with four parameters \((X,A,B,C)\), mapping the sequence or function \(x(t) \in R \mapsto y(t) \in R\) through an implicit latent state \(h(t) \in R^{N}\). The state space is usually composed of two basic differential equations:
where \(A,B,C\) are coefficient matrixes. The change of state is described by matrix A. And how does the input affect the state described by matrix B. Meanwhile, the output equation of matrix C describes how the state is converted to the output. These three matrices are parameters that would be learned through model iteration.
Since the data inputs obtained in practical applications are usually discrete, it is necessary to transform the continuous state space model into a discrete state space model. Through zero-order hold, defined as \(\overline{A} = \exp (\Delta A),{\kern 1pt} {\kern 1pt} {\kern 1pt} {\kern 1pt} \overline{B} = (\Delta A)^{ - 1} (\exp (\Delta A) - I)\Delta B\), a discrete state space model can be obtained as:
Therefore, the mamba model has greater flexibility and adaptability by introducing selective parameters into the discrete state space.
T5-based conditional molecular generation model T5MolGe
T5MolGe (the framework is shown in Fig. 2 is an integrated transformer model with an encoder module for embedding conditional representations and a decoder module, which is a GPT architecture that works on a masked self-attention mechanism as a generator by predicting a sequence of SMILES tokens. T5MolGe adopted a similar SMILES tokenizer as that in9 to break SMILES strings into a set of relevant tokens, corresponding to individual atoms and bonds. For the conditional properties of molecules, we extract the specific scaffolds from molecules in ChEMBL28 datasets via RDkit29. The whole model overview is described below. First, the tokens of the input sequence of the molecular scaffold are mapped to a sequence of embeddings and then passed into the encoder module, which consists of a stack of “blocks”. Each of these blocks is composed of a self-attention layer and a small feed-forward network. Here, six identical blocks were adopted. Through a self-attention mechanism, three sets of vectors, query (Q), key (K), and value vectors (V), are calculated, and a Softmax function is subsequently applied to acquire corresponding weights by \(soft\max \left( {\frac{{QK^{T} }}{{\sqrt {d_{k} } }}} \right)V\) where \({d}_{k}\) is the dimension of the query and key vectors, and attention is provided to all the tokens of a sequence. Moreover, combined with the assistance of a feedforward neural network, the molecular scaffold information was extracted by this neural network, which would be fed to another stack as conditional information.

(A) The molecular design using T5MolGe model, (B) Sampling and property extraction from molecular chemical space for virtual screening by Encoder and Decoder of T5MolGe model.
Next, the molecular SMILESs from the training database are tokenized and fed into the decoder module with a start token, which is obtained via weighted random sampling from a list of tokens that appear first in the SMILES string in the training set. Then, with the provided starting label, the model predicts the next label in turn by training with a maximum likelihood of the output probabilities of generated molecules, thus generating a new molecule. Moreover, the decoder module works on a masked self-attention mechanism to mask attention to all sequence tokens that are important in future time steps. Moreover, in each of all blocks of the decoder module, multihead attention was also adopted to provide better representations in different subspaces. Therefore, the T5MolGe model can achieve the ability of molecule generation and optimization under specific conditions.
Transfer learning
To generate anticancer agents targeted to the notorious pathway, the EGFR L858R/T790M/C797S mutation, which involves the substitution of the Cys797 residue with serine 797, leads to the loss of covalent interactions and significantly reduces drug efficacy. The artificial neural networks need to search in one specific dataset related to the EGFR pathway, which would be too small to achieve efficient learning results. However, the bottleneck in AI training caused by a small dataset could be overcome by transfer learning (TL) by retraining a small dataset on the basis of a pretrained model of integrated data.
All of the models were built via PyTorch, and the parameters of the models are consistent, where complete hyperparameter settings can be found in Table 1. The initial learning rate is set to 10−4 and adjusted by the learning rate warm-up and decay strategies so that the learning rate varies between 10−3 and 10−4. The cross-entropy loss is the loss function of the model. To avoid gradient disappearance or gradient explosion in the process of backpropagation, PyTorch’s automatic gradient scaling mechanism is adopted in the training process. Among them, the parameter of the number of self-attention heads is only applicable to the GPT model but not to the Mamba model.
Datasets
In this work, we used the benchmark dataset GuacaMol, which is a subset of the ChEMBL 2428 database and contains 1.6 million molecules, published by Benevolent AI for training and evaluation of our model. This benchmark dataset is specifically developed for molecular design, the primary goal of which is to advance machine learning research in the field of drug discovery. The dataset contains goal-directed molecular generation tasks and molecular optimization tasks, covering all aspects of generating molecules with specific properties to optimize molecular properties. The GuacaMol dataset also defines a series of evaluation metrics, such as the reasonableness, uniqueness, and novelty of the generated molecules, providing a detailed benchmark for evaluating model performance in this study. In addition, the GuacaMol dataset contains a training dataset and a test dataset for different training and test model performances.
Moreover, we downloaded 2,431,025 compounds from ChEMBL and selected those named ‘-tinib’, which is an already known tyrosine kinase inhibitor that has certain pharmacological effects on various tyrosine kinases, including EGFR, also known as teninids, a type of molecularly targeted therapy that inhibits the growth and division of cancer cells by inhibiting key tyrosine kinases in signal transduction pathways, resulting in 171 compounds. To support the training of molecular generative models, the SMILES molecular randomization strategy was subsequently used to enrich the rain dataset by expanding the original size by ten times.
Experimental setup
In this work, all the experiments were performed on a Linux ubuntu20.04 operating system with 24 CPU cores, an AMD EPYC 7642 CPU, 80 GB of memory, and a GPU driver with 24 GB of memory. The implemented procedure codes were all based on the open source framework “Pytorch” (https://pytorch.org). The transformer models adopted the open source deep learning platform “Hugging Face library” (https://huggingface.co/t5-base). Subsequently, Schrödinger Maestro 12.8 (https: //www.schrodinger.com, accessed on 20 January 2024) was used for the precise docking of their ligands and receptors.
Training procedure and evaluation metrics
Each model in our study was trained for 10 epochs via the Adam optimizer. The number of layers of the model is set to 8, and the number of heads of the self-attention mechanism is also set to 8. The embedding dimension is set to 256; that is, each unit of the sequence is converted to a 256-dimensional vector. In addition, to prevent overfitting and improve the generalization ability of the model, a discard rate of 0.1 is used in the model training process. Moreover, the initial learning rate is set to \({10}^{-4}\) and adjusted by learning rate warm-up and decay strategies so that the learning rate varies between \({10}^{-3}\) and \({10}^{-4}\). The cross-entropy loss is implemented as a loss function for the model through PyTorch. To avoid gradient disappearance or gradient explosion in the process of backpropagation, PyTorch’s automatic gradient scaling mechanism is adopted in the training process. A total of 10 epochs during each period need 1 day, and the entire training and evaluation procedure includes five models in the nonconditional generational pretraining task and six models in the conditional generational pretraining tasks, which last nearly 20 days.
The evaluation metrics for the assessment of the molecule generation models adopted here include validity, uniqueness, novelty, Frechet ChemNet distance (FCD)30, and KL diversity31 for nonconditioned generation model evaluation, whereas validity, uniqueness, novelty, and the ratio of Tanimoto similarity for conditioned generation model evaluation are described in detail below:
-
Validity the proportion of generated sequences that conform to SMILES syntax to the total number of generated sequences. In this study, RDKit was used to test the validity of the molecules. Measurements of validity demonstrate the model’s ability to learn and master SMILES syntax as well as valency rules.
-
Uniqueness the proportion of nonrepeating molecules in the valid molecules produced.
-
Novelty the proportion of valid molecules generated that are not included in the training molecule set.
-
FCD a measure of the distribution similarity between the generated molecule set and the target molecule set. The calculation of FCD requires feature extraction via ChemNet, a pretrained neural network used to extract useful chemical properties from molecular structures. The smaller the FCD is, the closer the distribution of the generated molecule is to that of the target molecule. Calculation of the FCD between the generated data distribution (\(G\)) and training data distribution (\(D\)):
$$FCD(G,D) = ||\mu _{G} - \mu _{D} ||^{2} + Tr\left( {\sum\limits_{G} - \sum\limits_{D} - 2\left( {\sum\limits_{G} {\sum\limits_{D} {^{{\frac{1}{2}}} } } } \right)} \right)$$where \(\mu_{G}\) is the average of distribution G, \(\sum_{G}\) represents the covariance matrix of distribution G, and \(Tr\) is the trace of the matrix. The final FCD score is reported as \(S = \exp ( - 0.2FCD)\). Therefore, a higher \(S\) value is considered to be better.
-
Similarity The similarity between the generated molecules and the conditional scaffold, Tanimoto (T) similarity, was used as the metric, which is widely used in cheminformatics to compare the similarity of molecular fingerprints. The molecular fingerprints needed can be obtained via the RDkit package, which converts the SMILES sequence into a fixed length of 01 sequence, to assess the accuracy and consistency of the model-generated molecules. Here, we computed the proportion of Tanimoto similarity greater than 0.8 and recorded it as the similarity proportion to describe the performance of different models in maintaining the characteristics of conditional scaffolds.
-
KL divergence the measurement of the difference between two probabilities distributions. In molecular generation tasks, it can be used to measure the difference between the property distribution of the generated molecule (e.g., molecular weight, polarity, etc.) and the property distribution of the target molecule. The KL divergence between the generated dataset \(Q\) and the training dataset \(P\) can be calculated as follows: \(D_{KL} (P||Q) = \sum\limits_{i} {P(i)\log (\frac{P(i)}{{Q(i)}})}\), where \(P(i)\) is the probability of dataset \(P\) at data point \(i\), and and \(Q(i)\) are the probabilities of dataset \(Q\) at data point i. Finally, the KL divergence values for each attribute of the two datasets are used to calculate the score: \(S = \frac{1}{k}\sum\limits_{i = 1}^{k} { - D_{KL,i} }\).
-
QED Quantitative estimates of drug likeness, which quantifies drug-likeness by calculating some predefined rules or models to take into account the main molecular properties. It ranges from 0 (all properties unfavorable) to 1 (all properties favorable).
-
SAS Synthetic Accessibility score, which is the measurement of difficulty of synthesizing a compound, ranging from 1 (easy to make) to 10 (difficult to make).
Results
In this section, we first present the evaluation results of five unconditional generation models, including three kinds of improved GPT-based models, the archetype GPT model, mamba and six conditional generation models, including our proposed model T5MolGe, and the remaining five unconditional models are applied in conditional scenarios. As mentioned by Priyakumar et al.9, the chemical space is too infinte to explore entirely. Here, we also adopted these metrics, which measure how well the models learn the molecular grammar, including the internal diversity scores, validity, uniqueness, and novelty, to measure the extent of chemical space traversed by the models and FCD and KL divergence to measure the statistics and distributions of features of the dataset captured by different models. U. Priyakumar et al. demonstrated the exceptional performance of the GPT model over other state-of-the-art approaches, such as CharRNN32, VAE11, AAE13, LatentGAN14, and JT-VAE12. Therefore, in this study, we evaluated the performance of the GPT model compared with three improvements of the GPT model, one new star selective-state-space-based model Mamba, and the transformer-based T5MolGe model in unconditional and conditional generation tasks separately.
Unconditional molecular generation
Table 1 exhibited the loss value changes of different models in test dataset within 20 epochs, showing that in performance of loss values Mamba model is obviously superior to GPT and its improved model. Moreover, among the GPT models, GPT-ROPE and GPT-GEGLU models showed the most significant decline in losses. When evaluating the molecular generation model, besides the model’s performance on the test set, it is also necessary to consider the model’s ability to generate molecules and the indicators of generating molecular sets. Table 2 shows the performance of the molecular sets generated by each model in terms of validity, uniqueness, novelty, FCD and KL divergence, which were trained on the benchmark dataset GuacaMol for non-conditional generation. From the perspective of validity, GPT-RoPE achieves the highest score of 0.98, indicating that rotational position coding can promote SMILES sequence position information to improve the model’s ability to learn SMILES sequence syntax and that it is necessary to prompt location information when training molecular generation models to handle long-range dependencies very well. However, GPT-RoPE has the lowest FCD score, showing that it is more difficult to simulate the molecular properties and data distributions in target datasets than other models are. On the other hand, all the models achieved consistent scores for uniqueness and novelty, which were all close to or equal to 1, suggesting that all the models generate molecules novelly and randomly without falling into fixed patterns. For the FCD score and KL divergence, the Mamba model yields the best results, indicating that the distribution of the molecule set generated by Mamba is the closest to the distribution of the target molecule set. Therefore, for non-conditional generation task, Mamba achieved the most optimal efficiency.
Generation based on a conditional molecular scaffold
The performance of generation models must be evaluated when certain properties are required for many specific biological and chemical processes. We test the model’s ability to control specific molecular scaffolds/scaffolds by training on the GuacaMol dataset. The main purpose of selecting a molecular scaffold as a condition is to generate molecules with a specific molecular scaffold, which helps to ensure that the generated new molecules are structurally consistent with the given scaffold while also exploring and exploiting the possible chemical changes and functions of this scaffold. For the conditional molecular generation task, we compared the performance of six models trained and evaluated under the same conditions, including T5MolGe, Mamba, GPT, GPT-RoPE, GPT-Deep, and GPT-GEGLU.
Firstly, the loss variation of each model under conditional molecule generation is reported in Table 3. The final loss values were significantly lower than that of non-conditional generation models, which shew the input of molecular scaffold effectively guides the formation of corresponding molecules. Concretely, the original GPT model has a high loss, while the GPT-RoPE model with improved position coding shows a significant decrease in loss, which indicates that the optimization of position coding has a positive effect on conditional molecule generation based on molecular scaffold. The Transformer model shows even more outstanding performance, with a loss value of as low as 0.082, indicating that a model with a full Encoder-Decoder architecture is more suitable for handling molecular scaffold-based conditional generation tasks.
Next, we randomly selected 100 molecular scaffolds that were not included in the training set, generated 100 molecules for each scaffold, and then calculated the metrics of Validity, Uniqueness, Novelty and Similarity ratio. The average performance was shown in Table 4, and the distributions of these evaluation metrics for all 100 scaffolds in terms of the box plots are shown in Fig. 3. The box plots show the performance of different models in generating molecular sets on each scaffold, with the middle box representing the range from the first quartile to the third quartile, the thick line representing the median, and the thin line extending to the upper and lower marginal values of the data, which is 1.5 times the upper quartile and lower quartile seperately, while the black dots represent the outliers. Therefore, from Fig. 3, we can see that except for GPT-RoPE and T5MolGe, all the other models encountered cases where the molecular scaffold could not be recognized. That is, there are points with a similarity ratio value of 0, which suggests that these models fail to generate molecules with a high degree of similarity to the target scaffold for some molecular scaffolds. In contrast, the T5MolGe model performed well in terms of similarity ratios, suggesting that it can control almost all molecular scaffolds while achieving the best performance in terms of validity and novelty. The performance of uniqueness is only slightly lower.

The boxplot of distributions of Validity, Uniqueness, Novelty, Similarity of different approaches based on these molecular skeletons.
Compared with those of the former GPT model, the average value of validity and similarity ratios of the GPT-RoPE model improved from 0.945 to 0.984 (p-value = 0.001) and from 0.862 to 0.941 (p-value < 0.001), respectively, when rotational position coding was introduced. This finding shows that the rotational position coding enhances the ability of the model to capture the molecular scaffold features, thus improving the quality of the generated molecules and the matching degree of the target scaffold. While sacrificing in terms of uniqueness, this suggests that models may be learning with a greater emphasis on structures that are similar to the molecular scaffold, to some extent at the expense of molecular diversity. Moreover, GPT-GEGLU outperforms the basic GPT model in terms of validity and similarity ratio, which indicates that the activation function with the introduction of a gating mechanism is more competent for the conditional molecular generation task than the traditional activation function is.
However, for this conditional generation task, the Mamba model is inferior to the basic GPT model, which may indicate that deep learning models based on attention mechanisms can process conditional inputs more efficiently than state-transition models can, thus extracting information from conditions to effectively improve molecule generation. In summary, T5MolGe and GPT-RoPE achieved better performances in this conditional generation task and therefore were selected as pretraining models in the next transfer learning conditional generation task.
Transfer learning assists in AI-based drug discovery for EGFR TKIs
On the basis of the above pretraining models, transfer learning was carried out to generate specific L858R/T790M/C797S-mutation EGFR TKIs related to NSCLC. The transfer learning strategy was applied both in nonconditioned situations and conditional situations. Those five models were operated on the ‘-tinib’ small dataset after pretraining on the large-scale molecular dataset GuacaMol. Since the number of such compounds is not enough to be used in transfer learning, randomization was adopted to expand the 171 ligands to 1710 SMILEs sequences. One randomization effect example was shown in Supplementary Figure S1 and Table S1.
Nonconditioned TL generation
The loss values of all the models on this small dataset shown in Fig. 4, exhibited gradually decreasing and converging at approximately 20 epochs, except for GPT-GEGLU, which showed an increasing trend after 20 epochs, indicating overfitting during the training process. Moreover, GPT-deep and Mamba performed poorly on losses compared with the other models, which illustrates that these two models have relatively difficulty learning the grammatical rules of SMILES sequences. The GPT-RoPE model performed best in both training and testing and showed the fastest decline in loss values, demonstrating the excellent properties of rotational position coding in generating molecular compounds.

Loss value curves of different models integrated TL algorithm in non-conditional generation task.
In terms of the properties of the generated molecular sets, the performance of each model is shown in Table 5. To prevent overfitting, the model is saved when the loss of all the models on the test set is no longer reduced. In terms of validity, the GPT-RoPE model performed best at 0.794. The validity of the Mamba model is slightly greater than that of the GPT model, but its uniqueness is slightly lower. In fact, the test loss of the Mamba model no longer decreases after approximately 14 rounds of training, indicating that its generalizability needs to be improved.
Conditional TL generation
In this conditional generation task, transfer learning is carried out on the PGT-ROPE model, which performs well during pretraining, and the T5MolGe model, whose loss curves are shown in Fig. 5. By comparing Figs. 4 and 5, in terms of transfer learning based on the ‘-tinib’ specific dataset, conditional molecular generation based on a scaffold can achieve a lower loss value than nonconditioned generation models can achieve. Moreover, Fig. 5 clearly shows that the test loss value of the T5MolGe model is significantly lower than that of the GPT, which indicates that the transformer model with the complete encoder-decoder architecture performs better in transfer learning scenarios than the GPT-based model does. Finally, the properties of generated molecular sets by conditional generaion models are shown in Table 6, which illustrates that compared with the GPT-RoPE model, T5MolGe can reproduce the input molecular scaffold structure more effectively and maintain high efficiency.

Loss value curves of T5MolGe and GPT-Rope integrated TL algorithm in conditional generation task.
‘-tinib’ drug generation
In this study, we finally selected the T5MolGe model to learn ‘-tinib’ drug molecules for scaffold-based conditional ‘-tinib’ drug generation. The resulting AI-generated molecules were filtered and screened to obtain desirable ligands by filtering on the basis of molecular weight (MW), lipid water partition coefficient of molecules (LogP), topologically polar surface area (TPSA), number of hydrogen bond donors (HBAs), number of hydrogen bond receptors (HBDs), HBA + HBDs, and the number of rotatable bonds (NumRotBonds) to meet the encoding rules. The parameters of the screening filters used here were as follows: \(300 \le MW \le 700,{\kern 1pt} {\kern 1pt} {\kern 1pt} 2.0 \le L{\text{og}}P \le 6.0,{\kern 1pt} {\kern 1pt} {\kern 1pt} 2.0 \le HBD \le 6.0,{\kern 1pt} 0 \le {\kern 1pt} HBV \le 12.0,{\kern 1pt} {\kern 1pt} {\kern 1pt} HBA + HBD \le 14.0,{\kern 1pt} {\kern 1pt}\)\(60.0 \le TPSA \le 140.0\) and a rotational bond \(\le 12.0\).
After the filtering process, we obtained 7059 standard and nonrepeated SMILES sequences from the more than 20,000 AI-generated SMILES sequences. Furthermore, to investigate the similarity between the generated ‘-tinib’ drugs and the original ‘-tinib’ drugs, we determined the distributions of these two molecular sets with different properties. As shown in Fig. 6, each subgraph represents the distribution of an attribute, including MW (molecular weight), LogP, HBA, HBD, TPSA, NumRotBonds, QED, and SAS.

Comparison of the feature distribution between the generated molecular set and the original Tinib dataset.
Next, to explore the affinity of the generated candidate drugs to the target, virtual screening of the generated molecular sets was conducted. The L858R/T790M/C797S mutant EGFR (PDB: 6LUD) was selected as the target for affinity calculations of the candidate compounds. DeepPurpose33 is a deep learning library based on a convolutional neural network (CNN) and multilayer perceptron (MLP) for encoding and downstream prediction of proteins and compounds. Affinity is evaluated by the pKd score, where the Kd value is a standard for measuring the binding strength of a ligand and protein, and the smaller the value is, the closer the binding and the stronger the affinity. The pKd value is the negative pair of Kd values and is used to more intuitively represent the magnitude of affinity. As shown in Fig. 7, compared with the nonconditional generation strategy, although there was no significant difference in the overall distribution (Fig. 7A,B), conditional molecule generation produced more molecules with high pKd values, as explained by the local distribution diagram (Fig. 7C,D). These findings indicate that the molecular scaffold can be used to guide the model to generate more molecules with high binding strengths between ligands and proteins. Meanwhile, the virtual screening workflow module was used to perform virtual screening. Three generated molecules with Tanimoto Similarity ratio 0.95, 0.85, and 0.75, was randomly sampled to simulate the docking results targeting to EGFR mutant T790M/C797S (Fig. 8), where the 2D structures of original molecules and the generated ones were exhibited in the Supplementary Figure S2.

Distribution of the predicted pKd value of generated molecular set.

The sample molecular docking results of generated molecules targeting to EGFR-T790M/C797S. (A) The generated molecule with Similarity ratio 0.95; (B) the generated molecule with Similarity ratio 0.85; (C) the generated molecule with Similarity ratio 0.75.
Discussion and conclusion
Since 2000, the average cost associated with drug development from the research stage to approval has increased, reaching $2.6 billion in the first half of the twenty-first century34. Additionally, the number of drug-like compounds in the chemical space may be as high as 1023–10602. The diversity and complexity of compounds, as well as the interactions between drugs and targets, make drug design complex and time-consuming. Therefore, computer-based simulations have become a new tool for drug development, but the search range through screening of known compounds (such as high-throughput virtual screening) is still much smaller than the chemical space (approximately 109). Generative models have been proposed for generating novel molecules with desirable properties from scratch, and these models are expected to more intelligently explore vast chemical spaces rather than just relying on sifting through existing libraries. As the development of deep learning algorithms, especially natural language processing, the natural language description of molecules can be input into algorithm system to abstract more accurate molecular features and functions, which greatly accelerated the process of molecular design and drug discovery. For example, research has achieved the ability to generate molecules from proposing a GPT-based framework, MolGPT, by comparing with the previously proposed modern machine learning frameworks. However, there are still many mechanisms could be improved in this GPT-based generation model, moreover some new generation architectures emerging continuously. Furthermore, generating drug-like ligands for prospective tyrosine kinase inhibitors of specific proteins or pathways are needed to train models based on some specific dataset, which would violate big data request of deep learning algorithms. Therefore, the integration of one natural language processing model and transfer learning ideology is one important and practical design method for de novo molecular generation targeting to some specific protein targets.
Therefore, in this study, firstly we improved the former GPT-based generation models and compared their performance with the former one and another selective state space model. Then we proposed one end-to-end language based model and evaluated its performance on both non-conditional and conditional generation tasks. Finally, through integrating transfer learning ideology, the T5MolGe model was demonstrated to be able to generated the specific TKIs targeting to L858R/T790M/C797S-mutation EGFR in NSCLC. We extracted 171 ligands associated with TKIs from approximately 2.4 million compounds in ChEMBL database, next performed the end-to-end neural language model T5MolGe, which has been trained by large enough GuacaMol dataset to generated 2,4700 SMILES associated with TKIs. Through parameter-filtering such as molecular weight and lipid water partition coefficient of molecules, 7059 drug-like ligands were narrowed down. Then we used the DeepPurpose for fast virtual screening, to predict the binding affinities by resulting in higher pKd score, indicating higher infinity of the generated drug-like ligands with the L858R/T790M/C797S mutant EGFR.
However, there are still plenty of exploratory space after our study, such as the experiments of efficacy, toxicity, pharmacodynamics, etc. of the candidates. And there is still a long road to drug discovery and successful drug approval from the AI-assist molecular generation. But our study gave one feasible scheme by integrating the end-to-end transformer algorithm and transfer learning ideology. Meanwhile comparing the generation performances of the existing GPT-based methodologies and one new-style challenging model Mamba. And demonstrated that Mamba is not proper to molecular generation by comparing the neural language models. Furthermore, the full model of transformer composed by Encoder and Decoder is superior in the conditional molecular generation task, also in transfer learning scene.
Data availability
Molecular sequence and ligands data, along with Python code that support the findings of this study have been all deposited to GitHub: https://github.com/Yswangustb/T5MolGe-drug-generation.
References
Kutchukian, P. & Shakhnovich, E. De novo design: Balancing novelty and confined chemical space. Expert Opin. Drug Discov. 5, 789–812 (2010).
Polishchuk, P. G., Madzhidov, T. I. & Varnek, A. Estimation of the size of drug-like chemical space based on GDB-17 data. J. Comput. Aided Mol. Des. 27, 675–679 (2013).
Devlin, J., Chang, M. W., Lee, K. & Toutanova, K. BERT: Pretraining of deep bidirectional transformers for language understanding. In NAACL; Association for Computational Linguistics. pp 4171−4186 (2019).
Radford, A., Narasimhan, K., Salimans, T. & Sutskever, I. Improving language understanding by generative pre-training. Technical report, OpenAI, (2018).
Raffel, C. et al. Exploring the limits of transfer learning with a unified text-to-text transformer. J. Mach. Learn. Res. 21, 140 (2020).
Schwaller, P. et al. Molecular transformer: A model for uncertainty-calibrated chemical reaction prediction. ACS Cent. Sci. 5(9), 1572–1583 (2019).
Ross, J. et al. Large-scale chemical language representations capture molecular structure and properties. Nat. Mach. Intell. 4, 1256–1264 (2022).
Chen, D. et al. Algebraic graph-assisted bidirectional transformers for molecular property prediction. Nat. Commun. 12, 3521 (2021).
Bagal, V., Aggarwal, R., Vinod, P. K. & Priyakumar, U. MolGPT: Molecular generation using a transformer-decoder model. J. Chem. Inf. Model. 62, 2064 (2021).
Kotsias, P.-C. et al. Direct steering of de novo molecular generation with descriptor conditional recurrent neural networks. Nat. Mach. Intell. 2, 254–265 (2020).
Lim, J., Ryu, S., Kim, J. W. & Kim, W. Y. Molecular generative model based on conditional variational autoencoder for de novo molecular design. J. Cheminf. 10, 1–9 (2018).
Jin, W., Barzilay, R., Jaakkola, T. Junction tree variational autoencoder for molecular graph generation. Preprint at arXiv:1802.04364. (2018).
Hong, S. H., Ryu, S., Lim, J. & Kim, W. Y. Molecular generative model based on an adversarially regularized autoencoder. J. Chem. Inf. Model. 60, 29–36 (2020).
Prykhodko, O. et al. A de novo molecular generation method using latent vector based generative adversarial network. J. Cheminf. 11, 74 (2019).
Su, J., Lu, Y., Pan, S., Wen, B. & Liu, Y. RoFormer: Enhanced transformer with rotary position embedding (2021).
Brown, T. B., et al. Language models arefew-shot learners. In NeurIPS (2020).
Nguyen, T. Q. & Salazar, J. Transformers without tears: Improving the normalization of self-attention. Preprint at arXiv:1910.05895 (2019).
Wang, H., Ma, S., Dong, L., Huang, S., Zhang, D., & Wei, F. DeepNet: Scaling transformers to 1000 layers. Preprint at arXiv:2203.00555 (2022).
Shazeer N. Glu variants improve transformer. Preprint at arXiv:2002.05202 (2020).
Gu, A., Dao, T. Mamba: Linear-time sequence modeling with selective state spaces. Preprint at arXiv:2312.00752 (2023).
Liu, M., et al. CM-UNet: Hybrid CNN-Mamba UNet for remote sensing image semantic segmentation. Preprint at arXiv:2405.10530 (2024): n. pag.
Ali, B., Farnoosh, H., Graph Mamba: Towards learning on graphs with state space models. Preprint at arXiv:2402.08678.
Leo, F., Joseph, B., Maria, V., Paul-Henry, C. & Stergios, C. Structured state space models for multiple instance learning in digital pathology. Preprint at arXiv:2306.15789.
Ghosh, A. K., Samanta, I., Mondal, A. & Liu, W. R. Covalent inhibition in drug discovery. ChemMedChem 14, 889–906. https://doi.org/10.1002/cmdc.201900107 (2019).
Grabe, T., Lategahn, J. & Rauh, D. C797S resistance: The Undruggable EGFR mutation in non-small cell lung cancer?. ACS Med. Chem. Lett. 9, 779–782. https://doi.org/10.1021/acsmedchemlett.8b00314 (2018).
Huang, X. S., Perez, F., Ba, J. & Volkovs, M. Improving trans-former optimization through better initialization. In ICML, ser. Proceedings of Machine Learning Research, vol. 119, pp.4475–4483 (2020).
Liu, L., Liu, X., Gao, J., Chen, W. & Han, J. Understanding the difficulty of training transformers. In Proceedings of the 2020 Conference on Empirical Methods in Natural Language Processing (EMNLP), pp. 5747–5763 (2020).
Gaulton, A. et al. The ChEMBL database in 2017. Nucleic Acids Res. 45, D945–D954 (2017).
Landrum, G. RDKit: A software suite for cheminformatics, computational chemistry, and predictive modeling (2013).
Preuer, K., Renz, P., Unterthiner, T., Hochreiter, S. & Klambauer, G. Frechet ChemNet distance: A metric for generative models for molecules in drug discovery. J. Chem. Inf. Model. 58, 1736–1741 (2018).
Brown, N., Fiscato, M., Segler, M. H. & Vaucher, A. C. GuacaMol: Benchmarking models for de novo molecular design. J. Chem. Inf. Model. 59, 1096–1108 (2019).
Santana, M. V. S. & Silva-Jr, F. P. De novo design and bioactivity prediction of SARS-CoV-2 main protease inhibitors using recurrent neural network-based transfer learning. Bmc Chem. 15, 8 (2021).
Huang, K. et al. DeepPurpose: A deep learning library for drug-target interaction prediction. Bioinformatics 36(22–23), 5545–5547 (2021).
Takebe, T., Imai, R. & Ono, S. The current status of drug discovery and development as originated in United States Academia: The influence of industrial and academic collaboration on drug discovery and development. Clin. Transl. sci. 11(6), 597–606 (2018).
Jablonka, K. M. et al. Leveraging large language models for predictive chemistry. Nat. Mach. Intell. 6, 161–169 (2024).
Ai, C. et al. MTMol-GPT: De novo multi-target molecular generation with transformer-based generative adversarial imitation learning. PLOS Comput. Biol. 20(6), e1012229 (2024).
Funding
This research was funded by the National Natural Science Foundation of China (NO. 3161194 and No. 72271028).
Author information
Authors and Affiliations
Contributions
Y.W. and D.A. conceived and designed the graph network model and implemented the simulated study and real data set analysis; X.C. and M.G. wrote the codes for experiments; Y.W. wrote the whole manuscript. All authors have participated sufficiently in the work to take responsibility for it. All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wang, Y., Guo, M., Chen, X. et al. Screening of multi deep learning-based de novo molecular generation models and their application for specific target molecular generation. Sci Rep 15, 4419 (2025). https://doi.org/10.1038/s41598-025-86840-z
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-86840-z
Keywords
This article is cited by
-
Current AI technologies in cancer diagnostics and treatment
Molecular Cancer (2025)
