
Abstract
The intratumoral microbiota, fatty acid metabolism (FAM), and tumor microenvironment (TME) all provide insights into the management of colon adenocarcinoma (COAD). But the biological link among the three remains unclear. Here, we analyzed intratumoral microbiome samples and matched host transcriptome samples from 420 patients with COAD in The Cancer Genome Atlas (TCGA). All patients were divided into two subtypes (FAM_high and FAM_low) based on the Gene set variation analysis (GSVA) score of FAM pathway. Furthermore, we found significant difference in the intratumoral microbiota signatures between the two subtypes. In-depth analysis suggested that specific microbes in tumors may indirectly modify the TME, particularly stromal cell populations, by modulating the FAM process. More importantly, the crosstalk between the three can have a significant impact on prognosis, response to immunotherapy, and drug sensitivity of patients. Pathological image profiling showed that changes in the TME originating from intratumoral microbiota disturbance could be reflected in pathological image features. In summary, our study provides novel insights into the biological links among the intratumoral microbiota, FAM, and the TME in COAD, and offer guidance for the therapeutic opportunities that target intratumoral microbes.
Similar content being viewed by others


Introduction
Colorectal cancer (CRC) is a common malignant tumor of the digestive system, and the global incidence of CRC is increasing year by year1. Colon adenocarcinoma (COAD), the most common histological subtype of CRC, is a highly heterogeneous solid tumor that is highly aggressive2. Because there are no obvious early symptoms and most patients have metastasized by the time of diagnosis. The five-year overall survival (OS) for patients with COAD remains low despite the availability of various targeted drugs and immunotherapies in recent years3,4. Therefore, preventive measures such as exploring the pathogenesis of patients with COAD, screening and finding new therapeutic targets are essential to improve the survival and prognosis of patients with COAD.
Tumor development is closely related to the physiological state of the tumor microenvironment (TME)5, and therapeutic targeting of cells, processes, and signaling pathways in TME is considered to be a promising strategy. TME includes a variety of immune cells, cancer-associated fibroblasts (CAFs), endothelial cells and extracellular matrix (ECM). These cells were once considered bystanders to tumorigenesis, but are now known to play a key role in the pathogenesis of cancer6. For example, CAFs, as a type of stromal cells, can help tumors evade immune control through multiple mechanisms7. ECM facilitates intercellular communication by acting as a substrate for secretory molecules and a substrate for cell adhesion and migration, thus forming a complex signaling network in cancer8. Immune cells can support anti-tumor immune response and protect the human body from tumor cells. However, they can also act as pro-tumor cells, thus promoting tumor progression and survival9. The study of controlling the TME has become a key link in the field of tumor therapy and has a pivotal position. In recent years, there have been numerous studies on the characterization of TME and molecular correlation by deep learning based on pathological images. One study provided mappings of tumor-infiltrating lymphocytes (TILs) utilizing H&E images obtained from 13 tumor types in The Cancer Genome Atlas (TCGA)10. Another study developed a deep learning system that uses histopathology images to predict the spatial expression levels of high-dimensional genes in the corresponding regions, the TME of a large cancer cohort with only pathological image data and no spatial transcriptome data was characterized11. Jin et al. established a large-scale multi-omics cohort of breast cancer and identified four molecular subtypes12. In addition, the authors developed a convolutional neural network model to distinguish these subtypes based on digital pathology for potential clinical translation.
In recent years, with the development of detection technology and in-depth understanding of TME, more and more evidence has confirmed the existence of intratumoral microbiota13,14,15, which provides new therapeutic strategies and targets for tumor treatment16. Intratumoral microbiota can not only enhance anti-tumor immunity through STING signal activation and other mechanisms, but also reduce anti-tumor immune response through up-regulation of ROS and promotion of anti-inflammatory environment, thus promoting cancer progression17. The composition of a particular microbe can indicate the stage of cancer or disease progression. Tissue/organ microbiome signatures were identified from biological samples of major gastrointestinal cancer types such as esophageal cancer (ESCA), esophageal adenocarcinoma (EAD), and esophageal squamous cell carcinoma (ESCC), gastric adenocarcinoma (STAD), colon adenocarcinoma (COAD), and rectal adenocarcinoma (READ) by Wang et al. based on the public databases of The Cancer Microbiome Atlas (TCMA) and TCGA. It has been found that the composition of Alisteria, Brautiella, pasteurella and porphyromonas is correlated with the clinical features of gastrointestinal cancers, especially patients with COAD18. Galeano Nino et al. revealed the host-microbial interactions in space, cells and molecules, and confirmed the influence of intratumoral microbiota on tumor heterogeneity through the application of in situ spatial analysis technology and single-cell RNA sequencing technology in oral squamous cell carcinoma and colorectal cancer19.
Dysregulation of lipid metabolism has been shown to be a key metabolic feature of cancer cells and has important therapeutic implications20. In a variety of cancers including COAD, the regulation of key enzymes involved in fatty acid (FA) uptake, synthesis and oxidation (FAO) is abnormal, which leads to the reprogramming of fatty acid metabolism (FAM) and further aggravates the malignant characteristics of cancer, indicating the importance of lipid metabolism imbalance in the development of cancer21,22,23. FAM is closely related to cell activity in TME. For example, regulatory T cells (Tregs) drive immunosuppression in TME, while lipid signaling forces functional specialization of Tregs within tumors24.
In recent years, the pairwise relationship between intratumoral microbiota, FAM and TME of patients with COAD has been confirmed, but the serial progression relationship among the three remains unclear. In this study, we obtained matched intratumoral microbiome and host transcriptome data from 420 patients with COAD in TCGA. Based on this, we discussed the interplay among the intratumoral microbiome, FAM and TME in COAD, and their relationship with drug sensitivity. Moreover, to enhance the clinical utility, we correlated the FAM-based COAD subtyping with pathological images and investigate the possibility of determining the COAD subtyping based on pathological images from patients.
Materials and methods
Data acquisition
The intratumoral microbiome abundance data and metadata used in this study were downloaded at https://github.com/knightlab-analyses/mycobiome25. Narunsky-Haziza et al. systematically analyzed the tumor microbiome from the WIS cohort, TCGA, Hopkins, and UCSD cohorts25. The data on bacterial abundance in tumor tissue from patients with COAD in TCGA were used in this study. The TCGA data were filtered through computational software and compared with results from WIS, HMP projects, and more than 100 other papers. More details on decontamination are in the original article. The total survival time and survival status of samples were acquired from UCSC Xena (http://xena.ucsc.edu/). The RNA-Seq data for host gene expression was downloaded from https://portal.gdc.cancer.gov/. The gene list of FAM was obtained from the MSigDB (https://www.gsea-msigdb.org/gsea/msigdb).
GSVA, GO and KEGG enrichment analysis
Gene Set Variation Analysis (GSVA) is a non-parametric, non-supervised algorithm that calculates the degree to which each sample in a gene expression profile is associated with a specified set of genes, converting it into a continuous score26. The GSVA score of FAM was calculated to represent the relationship between the COAD sample and the set of genes associated with FAM.
Gene Ontology (GO)27 and Kyoto Encyclopedia of Genes and Genomes (KEGG)28 were used to explore the biological pathways of FAM-related Differentially expressed genes (DEGs), both of which were implemented by the R package“clusterProfiler”.
Intratumoral microbiota analysis
Microbial alpha diversity is represented by the Shannon and Simpson indices and calculated by the “vegdist” function in the R package “vegan”. LDA Effect Size analysis (LEfSe) was used to identify microorganisms with significantly different relative abundances29. In this analysis, we used LDA score greater than 3 as a threshold.
t-SNE and NMDS dimension reduction methods
t-Distributed Stochastic Neighbor Embedding (t-SNE) is a machine learning algorithm used for dimension reduction, widely used in bioinformatics, image analysis, and other fields30. t-SNE was used to reduce and visualize the microbial abundance dimensions of COAD samples.
Non-Metric Multidimensional Scaling (NMDS) is a data analysis method that simplifies multi-dimensional control research objects (samples or variables) into low-dimensional space for positioning and classifying, while preserving the original relationship of objects31. NMDS was used here to visualize the immune infiltration levels.
Immune characteristics and drug sensitivity analysis
The Tumor Immune Dysfunction and Exclusion (TIDE) score was used to calculate the immune escape capacity of patients with COAD32. In general, higher TIDE scores are associated with poorer immune checkpoint suppression therapy. The immune infiltration levels of FAM_high and FAM_low groups were calculated and compared by six methods: TIMER33, CIBERSORT34, QUANTISEQ35, MCPCOUNTER36, XCELL37 and EPIC38. The 50% inhibiting concentration (IC50) value of the 195 drugs was inferred using the R package “oncoPredict” and the value was normally transformed. A higher IC50 value usually means that the drug is less effective on cells, that is, a higher concentration is needed to achieve the same inhibitory effect.
Statistical analysis
All statistical calculations were performed using R (version 4.3.2). The Wilcoxon test was used to compare the differences between the two groups. The survival curve was plotted using Kaplan-Meier (KM) method, and the significance was determined by logarithmic rank test. Spearman correlation analysis was used to calculate the correlation between expression of FAM-related genes and abundance of intratumoral microbiota, GSVA score of FAM, immune score, stromal score, and drug IC50 value. Cox univariate analysis was used to calculate the significance of the correlation between FAM-related genes and the prognosis of patients with COAD, and Cox multivariate analysis was used to determine the FAM-related genes coefficient and risk score. p < 0.05 for all tests were considered statistically significant.
Results
Intratumoral Microbiota signatures are closely related to FAM
First, we calculated the GSVA score of each patient. All patients were divided into two groups, FAM_high and FAM_low, based on the median GSVA score (Fig. 1a). Then, we studied the distribution of intratumoral microbiota in these two groups. We did not detect significant difference in microbial alpha diversity between FAM_high and FAM_low (Fig. 1b and c). However, we found that there was a clear separation between these two groups based on the microbial community composition at the genus level (Fig. 1d; PERMANOVA test, p = 0.001), indicating that there was significant difference in the microbial signatures between FAM_high and FAM_low. Based on the LEfSe algorithm, Desulfovibrio, Desulfococcus, Mesorhizobium, Streptococcus, and Mycobacterium were detected to be enriched in the FAM_high group (Fig. 1e). The gene set related to FAM was obtained in MSigDB. Procrustes analysis showed that the microbial biomarkers were significantly correlated with FAM-related genes (Fig. 1f). Interestingly, the microbes enriched in the two groups exhibited distinct correlation pattern with individual FAM-related genes, respectively (Fig. 1g). We observed a significantly positive correlation among the microbes enriched in the FAM_high group (Fig. 1h). In addition, the expression of FAM-related genes was significantly different between the FAM_high and FAM_low groups (Fig. 1i). Our results indicate that the FAM is closely related to intratumoral microbiota in COAD.

The intratumoral microbiome profile is related to FAM. (a) Patients were divided into two groups based on the median of GSVA score of FAM. (b,c) Boxplots showing the difference in alpha diversity between FAM_high and FAM_low groups. Wilcoxon test was used to generate the p value. (d) t-SNE showing the difference in microbiota community between FAM_high and FAM_low groups. PERMANOVA test was used to conduct the statistical test. (e) LEfSe showing the microbial biomarkers in the two groups. (f) Procrustes analysis showing the correlation between intratumoral microbiome and FAM-related genes. (g) Heatmap showing the Spearman correlation between microbial biomarkers and FAM-related genes. (h) Heatmap showing the correlation among microbes enriched in the FAM_high group. (i) Heatmap showing the expression level of FAM-related genes in FAM_high and FAM_low groups. *** indicate p < 0.001.
The biological functions of DEGs between the FAM_high and FAM_low groups
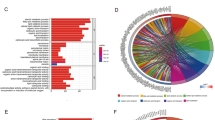
DEGs may reflect the differences in biological characteristics, disease status, drug response and other aspects between groups39,40. Thus, we identified the DEGs between the FAM_high and FAM_low groups (Fig. 2a). In order to further reveal the biological functions and pathways involved in these DEGs, we performed GO and KEGG enrichment analysis for these genes.

The biological functions of DEGs between FAM_high and FAM_low. (a) Volcano plot showing the DEGs of the FAM_high and FAM_low groups. (b–d) GO enrichment analysis results of the DEGs, including Biological Process (BP), Cellular Component (CC) and Molecular Function (MF). (e) KEGG enrichment results of the DEGs.
GO enrichment analysis categorises functions in terms of Biological Process (BP), Cellular Component (CC) and Molecular Function (MF). We have enriched 472, 93 and 61 GO terms in these three aspects, respectively. Figure 2b and d showed the most significantly enriched GO terms in each category. In BP, for example, the term “Cell-cell adhesion via plasma-membrane adhesion molecules” was enriched. This function is essential for maintaining tissue structure and function, as loss of cell adhesion is associated with increased tumor metastasis and aggressive41,42, and FAM may affect the composition and fluidity of cell membranes, thereby indirectly affecting cell adhesion43,44. The term “External encapsulating structure organization” was enriched. This function may involve a variety of extracellular matrix (ECM) composition and structure of the organization, these structures play an important role in tumor development45,46.
KEGG classifies functional pathways into seven broad categories, and here we mainly concentrated on four (Fig. 2e). Among them, ECM-Receptor interaction pathway involves signal transduction between cells and ECM. This interaction plays an important role in the migration, invasion and metastasis of tumor cells47,48, and FAM can influence the composition of cell membranes and ECM49,50. The Calcium signaling pathway plays a key role in multiple cellular processes, including cell proliferation, differentiation, and apoptosis51. The PI3K-Akt signaling pathway is an important intracellular signal transduction pathway, which is related to cell proliferation, survival and metabolism52.
FAM-related genes are associated with prognosis
Next, we sought to investigate the potential role of FAM in prognosis. The FAM_high and FAM_low groups showed significant differences in overall survival (OS) between the two groups, indicating a relationship between FAM levels and patient survival (Fig. 3a). We did not observe significant difference in disease specific survival (DSS) and progression free interval (PFI) between the two groups (Fig. 3b and c). Then, we identified the genes significantly correlated with OS using univariate Cox analysis (Fig. 3d). Subsequently, multivariate Cox regression was performed (Fig. 3e). Based on these four genes, including ADIPOR2, HAO2, ALAD and HPGD, we established a risk score and verified that the risk score could be a powerful survival indicator (Fig. 3f). Besides, we did not observe strong correlation among these genes, indicting the independent prognostic implication of these genes (Fig. 3g). We downloaded the immunohistochemical maps of these gene markers from the HPA database (HAO2-related proteins were not recorded in HPA), and found that the protein expression level of ADIPOR2, ALAD, and HPGD was distinct between tumor tissues and adjacent tissues (Fig. 3h). We have further explored the relationship between microbial markers and risk score, and we found that the microbes enriched in FAM_high were negatively correlated with the risk score, especially the genus Desulfococcus (Fig. 3i).

FAM-related genes are implicated in prognosis. (a–c) Survival curves of the FAM of OS, DSS, PFI. Log-rank test was used for statistical test. (d) Barplot showing the genes significantly associated with OS, conducted by univariate Cox regression. (e) Forest plot of multivariate Cox regression results. (f) Survival curves of the risk score. (g) Heatmap showing the Spearman correlation among the genes in prognostic model. (h) Representative immunohistochemical images of the protein level of ADIPOR2, ALAD, and HPGD in tumors and normal tissues. (i) Correlation between microbial markers and risk score. * indicate p < 0.05, ** indicate p < 0.01, *** indicate p < 0.001.
FAM is implicated in the TME
Studies have shown that FAM can affect tumor tissues from the aspects of tumor-related inflammatory response and TME53,54,55. We used the ESTIMATE algorithm to calculate the stromal score and immune score for each patient, and found that there was significant difference in stromal score between the FAM_high and FAM_low groups, but no significant differences in immune score (Fig. 4a). Next, we calculated the abundance of immune cell populations based on the gene expression, and observed clear separation between the two groups (Fig. 4b). Furthermore, the TIDE score of FAM_high group was significantly higher than that of FAM_low group (Fig. 4c). Specifically, we found that the FAM_high group harbored a lower level of immune infiltration (Fig. 4d and e). In both the two groups, the stromal score was significantly negatively correlated with the GSVA score of FAM (Fig. 4f and g). Next, we identified the top ten genes with the most significant differences between the FAM_high and FAM_low groups (Fig. 4h). We found a significant positive correlation between most of these genes, suggesting a synergistic effect of these genes on the activity level of FAM. In addition, there was no strong association between the expression level of these genes and stromal score and immune sore (Fig. 4i).

FAM is closely related the TME profile. (a) Boxplots showing the difference in immune score and stromal score between the FAM_high and FAM_low groups. Wilcoxon test was used to perform the statistical test. (b) NMDS plot showing the difference in the abundance of immune cell populations between the FAM_high and FAM_low groups. PERMANOVA test was used to perform the statistical test. (c) Boxplot showing the difference in the TIDE score between the FAM_high and FAM_low groups. (d) Heatmap showing the abundance of immune cells in the FAM_high and FAM_low groups. (e) Density plot showing the difference in the abundance of specific immune cells between the two groups. Wilcoxon test was used to perform the statistical test. Correlation between the GSVA score of FAM and stromal score in the (f) FAM_low and (g) FAM_high groups. (h) Barplot showing the top 10 genes with the most significant differences in expression between the two groups. Statistical tests were performed using Wilcoxon test. (i) Mantel test showing the correlation between the 10 genes and stromal score and immune score. Line in red indicates positive correlation, and line in blue indicates negative correlation. **** indicate p < 0.0001.
Drug sensitivity analysis in patients with COAD
Next, we calculated the IC50 value of potential drugs for all patients. We observed significant differences in drug sensitivity between the FAM_high and FAM_low groups (Fig. 5a). Of 195 drug candidates, the patients in the two groups exhibited significantly different sensitivities to 120 drugs (Fig. 5b). Then, we correlated the GSVA score of FAM, TME characteristics, and microbes enriched in the FAM_high group with the IC50 value of the 120 drugs (Fig. 5c). Based on the Spearman correlation analysis, we found that the drugs positively correlated with the GSVA score of FAM and microbes were basically enriched in the FAM_high group. Meanwhile, the trend of correlation between TME characteristics and drug sensitivity was opposite to that of FAM and microbes. In addition, we showed six drug candidates with the most significant differences between the two groups (Fig. 5d and i). Specifically, the IC50 values of JQ1, WEHI-539, and ZM447439 were significantly higher in the FAM_high group, suggesting that patients in the FAM_low group could benefit from these drugs. Another three drugs, including 5-Fluorouracil, Erlotinib, and GDC0810, could be more appropriate for patients in the FAM_high group. We also showed the chemical structure of these drug candidates.

Drug sensitivity analysis and its correlation with FAM. (a) NMDS plot showing difference in IC50 value of drugs between the FAM_high and FAM_low groups. PERMANOVA test was used to perform the statistical test. (b) The statistical significance of the difference in IC50 value of all drugs between the two groups. Wilcoxon test was used to perform the test. (c) Heatmap showing the correlation between the IC50 value of drugs significantly different between the two groups and the GSVA score of FAM, TME, and intratumoral microbes. (d–i) Boxplots showing the difference in the IC50 value of six drugs between the two groups. (j) Chemical structure maps of the six drug candidates. * indicate p < 0.05, ** indicate p < 0.01, *** indicate p < 0.001, **** indicate p < 0.0001.
Pathological image profiling identifying FAM subtypes
Due to the challenges of high costs, long lead times and complex technical processes in clinical implementation of multi-omics analysis, therefore, a cost-effective, rapid and convenient method is needed to infer the FAM-based COAD classification in our study. Therefore, we then explored the heterogeneity at the histopathology image level between the two groups. First, we extracted 11 texture features from each patient’s histopathology images, including Contrast, GLA, Regulation, SGBDA, DLA, Correlation, DLMSE, GLMSE, Entropy, SGSDA, and IDM (Fig. 6a). Then, we correlated these 11 texture features with the TME characteristics (stromal and immune score), intratumoral microbial signatures (alpha and beta diversity), and GSVA score of FAM (Fig. 6b). The results showed that stromal score, intratumoral microbiota, and FAM were all significantly correlated with most of texture features. Intriguingly, we observed that texture features negatively correlated with FAM and microbial signatures were all significantly higher in FAM_low than that in FAM_high (Fig. 6c; Wilcoxon test, p < 0.0001). Besides, texture features positively correlated with FAM and microbial signatures were all significantly higher in FAM_high than that in FAM_low (Wilcoxon test, p < 0.01). Consequently, these results demonstrate that the divergence between the FAM-based COAD classification can be reflected in the histopathology image. In the future, histopathology image-based deep learning models can be developed to predict this classification system.

Pathological images can identify the FAM subtypes. (a) Pipeline of the texture features extraction from pathological images. (b) Heatmap showing the Spearman correlation of 11 texture features with the TME characteristics, intratumoral microbial signatures, and GSVA score of FAM. MDS1 and MDS2 represent the two principal components of the intratumoral microbiota after dimensionality reduction. The color of the font of a texture feature indicates its correlation with other features. (c) Boxplot showing the distribution of 11 texture features in FAM_high and FAM_low. Ns, not significant. ** indicate p < 0.01, *** indicate p < 0.001, **** indicate p < 0.0001.
Discussion
The intratumoral microbiome, FAM and TME are all implicated in the pathogenesis and progression of COAD. In this study, we explored the biological links among the intratumoral microbiota, FAM and TME in patients with COAD. Through drug sensitivity analysis, the concentration direction of GSVA score of FAM, We proposed a novel COAD subtyping based on the GSVA score of FAM, and observed substantial difference in drug sensitivity between the two subtypes. Moreover, we observed that the two FAM-based subtypes could be distinguished by pathological images, highlighting the clinical utility of our COAD subtyping.
In the FAM_high and FAM_low groups based on GSVA score, the distribution characteristics of intratumoral microorganisms were significantly different. We selected microorganisms enriched in FAM_high group and significantly clustered into one class as markers, including Desulfovibrio, Desulfococcus, Streptococcus, Mycobacterium and Desulfococcus. Studies of these COAD-associated microbial markers have also been reported. Li et al. divided patients with CRC into clinical benefit responders (CBR) and no clinical benefit responders (NCB) based on chemotherapy efficacy, found that Desulfovibrio was more abundant in the NCB group56. In cultured colonic epithelial cells and in 2 murine models of intestinal tumorigenesis, found Streptococcus thermophilus is a kind of new prophylactic for CRC prevention in mice57. Our study shows that our microbial markers are associated with FAM in patients with COAD, which is consistent with previous studies. Mycobacterium, especially Mycobacterium tuberculosis, its genome contains a large number of genes related to FAM, these genes encode enzymes involved in FA and protein synthesis, degradation and transport process58. FAM is closely related to the occurrence and development of many diseases, including COAD. By exploring the relationship between microorganisms and FAM, we reveal the role of microorganisms in the process of COAD occurrence.
Previous reports have highlighted the importance of FAM for cancer prognosis. Tang et al. collected and retrieved 269 reliable FAM-related genes (FMGs), and identified 11 candidates OS-related FMGs in the TCGA-BRCA cohort. Based on this, an independent prognostic index FMI was established59. Li et al. found that in fatty acid anabolism, SREBP1, ACLY, ACSs, ACC, FASN, and SCD1 enhanced the occurrence, progression, and metastasis of gastric cancer. CD36, FABPs, and CPT1, which are fatty acid absorption-related molecules, also contributed to the proliferation of gastric cancer cells60. Yang et al. identified genes associated with FAM and differentially expressed in normal and gastric cancer tissues in the TCGA-STAD cohort. Based on this gene set, patients were grouped into two gastric cancer types, one of which was found to have a poor prognosis61. In this study on COAD, we found that the OS rate of the FAM_low group was lower than that of the FAM_low group. We identified four genes, ADIPOR2, HAO2, ALAD and HPGD, and established an independent prognostic factor risk score, indicating their potential significance in COAD prognosis.
Through the enrichment analysis of FAM-related DEGs, we found that most of the enriched biological pathways were related to tumor progression or TME. FAM_low group and FAM_high group can significantly separate TME characteristics and immune infiltration value, which emphasizes the important relationship between FAM and TME of COAD. TME plays an important role in COAD research. Lin et al. analyzed mutation data of immune microenvironment and immunogenicity in different MSI states, and found that compared with CRC patients with MSS/MSI-L tumors, those with MSI-H tumors significantly benefited from ICI treatment. MSI-H CRC had more immune cell infiltration, higher expression of immune-related genes, and higher immunogenicity than MSS/MSI-L CRC62. Zhang et al. established a angiogenesis potential index (API), and divided patients with COAD into high API group and low API group. They found that immune cells and stromal cells were more abundant in tumor tissues with high API scores, and different types of immune cells and immune function scores were higher in the high API group63. Our results suggest that it is possible to influence the TME by regulating FAM, thus achieving the purpose of cancer treatment.
Based on IC50 values, we identified 120 COAD-related drug candidates, including JQ1_2172, 5-Fluorouracil_1073, Erlotinib_1168, GDC0810_1925, etc. The pharmacological uses and activities of these compounds have been reported for a long time. Through the analysis of CRC cells, Wu et al. found that BET-Bromodomain inhibitor JQ1 combined with ABT-263 could synergistically reduce the level of Mcl-1 protein, induce cell apoptosis, and reduce cell vitality64. 5-Fluorouracil inhibits cell growth and division by interfering with DNA synthesis, is an essential component of systemic chemotherapy for colorectal cancer (CRC) in the palliative and adjuvant settings65. We found that drugs positively related to GSVA score of FAM and microbial markers were basically enriched in FAM_high group. Meanwhile, the direction of stromal score, immune score and drug enrichment was basically opposite. This provides an important clue for finding new personalized treatment options for COAD.
There are some limitations in our study. First, microbial data at the generic level have low resolution, and a deeper understanding of microbial diversity and function may require further in-depth data analysis at the species level or even subspecies level. Second, our study on the interaction between microbiota, FAM and the TME is based on a single TCGA-COAD dataset and lacks external independent validation. Finally, further functional experiments and studies are needed to confirm these relationships. For example, the influence of drugs on fatty acids and their metabolic pathways, as well as how such influence interacts with the presence and activity of microorganisms, can be verified through in vitro experiments or animal models.
In conclusion, this study comprehensively explore the trinity interaction among intratumoral microbiota, FAM and TME in patients with COAD. These findings not only provide new insights into the biology of COAD, but also lay a theoretical foundation for developing new therapeutic strategies. Future studies should delve into the mechanistic insight of the FAM-related activities in COAD.
Data availability
The data supporting the findings of this study are available in Materials and methods. Further inquiries can be directed to the corresponding author.
References
Argilés, G. et al. Localised colon cancer: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 31(10), 1291–1305 (2020).
Li, C. et al. Based on cuproptosis-related lncRNAs, a novel prognostic signature for colon adenocarcinoma prognosis, immunotherapy, and chemotherapy response. Front. Pharmacol. 14, 1200054 (2023).
Baldi, S. et al. Novel characterization discoveries of ferroptosis-associated molecules in COAD microenvironment based TCGA data. Front. Mol. Biosci. 9, 1102735 (2022).
Siegel, R. L., Giaquinto, A. N. & Jemal, A. Cancer statistics, 2024. Cancer J. Clin. 74(1), 12–49 (2024).
Roma-Rodrigues, C., Mendes, R., Baptista, P. V. & Fernandes, A. R. Targeting tumor microenvironment for cancer therapy. Int. J. Mol. Sci. 20(4). (2019).
de Visser, K. E. & Joyce, J. A. The evolving tumor microenvironment: From cancer initiation to metastatic outgrowth. Cancer Cell 41(3), 374–403 (2023).
Saw, P. E., Chen, J. & Song, E. Targeting CAFs to overcome anticancer therapeutic resistance. Trends Cancer 8(7), 527–555 (2022).
Huang, J. et al. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct. Target. Therapy 6(1), 153 (2021).
Peña-Romero, A. C. & Orenes-Piñero, E. Dual effect of immune cells within tumour microenvironment: pro- and anti-tumour effects and their triggers. Cancers 14(7) (2022).
Saltz, J. et al. Spatial organization and molecular correlation of tumor-infiltrating lymphocytes using deep learning on pathology images. Cell. Rep. 23(1), 181–193e187 (2018).
Gao, R. et al. Harnessing TME depicted by histological images to improve cancer prognosis through a deep learning system. Cell. Rep. Med. 5(5), 101536 (2024).
Jin, X. et al. Molecular classification of hormone receptor-positive HER2-negative breast cancer. Nat. Genet. 55(10), 1696–1708 (2023).
Azevedo, M. M., Pina-Vaz, C. & Baltazar, F. Microbes and Cancer: friends or Faux? Int. J. Mol. Sci. 21(9). (2020).
Cogdill, A. P., Gaudreau, P. O., Arora, R., Gopalakrishnan, V. & Wargo, J. A. The impact of intratumoral and gastrointestinal microbiota on systemic cancer therapy. Trends Immunol. 39(11), 900–920 (2018).
Wong-Rolle, A., Wei, H. K., Zhao, C. & Jin, C. Unexpected guests in the tumor microenvironment: Microbiome in cancer. Protein Cell 12(5), 426–435 (2021).
Yang, L., Li, A., Wang, Y. & Zhang, Y. Intratumoral microbiota: Roles in cancer initiation, development and therapeutic efficacy. Signal. Transduct. Target. Therapy 8(1), 35 (2023).
Nejman, D. et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Sci. (New York NY) 368(6494), 973–980 (2020).
Wang, J., Wang, Y., Li, Z., Gao, X. & Huang, D. Global analysis of microbiota signatures in four major types of gastrointestinal cancer. Front. Oncol. 11, 685641 (2021).
Galeano Niño, J. L. et al. Effect of the intratumoral microbiota on spatial and cellular heterogeneity in cancer. Nature 611(7937), 810–817 (2022).
Dai, W. et al. PTPRO represses colorectal cancer tumorigenesis and progression by reprogramming fatty acid metabolism. Cancer Commun. (London England) 42(9), 848–867 (2022).
Bacci, M., Lorito, N., Smiriglia, A. & Morandi, A. Fat and furious: Lipid metabolism in antitumoral therapy response and resistance. Trends cancer 7(3), 198–213 (2021).
Munir, R., Lisec, J., Swinnen, J. V. & Zaidi, N. Lipid metabolism in cancer cells under metabolic stress. Br. J. Cancer 120(12), 1090–1098 (2019).
Snaebjornsson, M. T., Janaki-Raman, S. & Schulze, A. Greasing the wheels of the cancer machine: The role of lipid metabolism in cancer. Cell Metabol. 31(1), 62–76 (2020).
Lim, S. A. et al. Lipid signalling enforces functional specialization of T(reg) cells in tumours. Nature 591(7849), 306–311 (2021).
Narunsky-Haziza, L. et al. Pan-cancer analyses reveal cancer-type-specific fungal ecologies and bacteriome interactions. Cell 185(20), 3789–3806e3717 (2022).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7 (2013).
Gene Ontology Consortium. Going forward. Nucleic Acids Res. 43(Database issue), D1049–1056 (2015).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44(D1), D457–462 (2016).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12(6), R60 (2011).
Van der Maaten, L. & Hinton, G. J. J. Visualizing data using t-SNE. 9(11) (2008).
Cox, T. F. & Cox, M. A. Multidimensional Scaling, CRC (2000).
Jiang, P. et al. Signatures of T cell dysfunction and exclusion predict cancer immunotherapy response. Nat. Med. 24(10), 1550–1558 (2018).
Li, B. et al. Comprehensive analyses of tumor immunity: Implications for cancer immunotherapy. Genome Biol. 17(1), 174 (2016).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 12(5), 453–457 (2015).
Plattner, C., Finotello, F. & Rieder, D. Deconvoluting tumor-infiltrating immune cells from RNA-seq data using quanTIseq. Methods Enzymol. 636, 261–285 (2020).
Becht, E. et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol. 17(1), 218 (2016).
Aran, D., Hu, Z. & Butte, A. J. xCell: Digitally portraying the tissue cellular heterogeneity landscape. Genome Biol. 18(1), 220 (2017).
Racle, J. & Gfeller, D. EPIC: A tool to estimate the proportions of different cell types from bulk gene expression data. Methods Mol. Biol. (Clifton NJ) 2120, 233–248 (2020).
Dai, W. et al. Identification of hub genes and pathways in lung metastatic colorectal cancer. BMC Cancer 23(1), 323 (2023).
Fang, L., Liu, Q., Cui, H., Zheng, Y. & Wu, C. Bioinformatics analysis highlight differentially expressed CCNB1 and PLK1 genes as potential anti-breast cancer drug targets and prognostic markers. Genes 13(4). (2022).
Peng, J. M., Lin, S. H., Yu, M. C. & Hsieh, S. Y. CLIC1 recruits PIP5K1A/C to induce cell-matrix adhesions for tumor metastasis. J. Clin. Invest. 131(1) (2021).
Tong, Y. et al. Escape from cell-cell and cell-matrix adhesion dependence underscores disease progression in gastric cancer organoid models. Gut 72(2), 242–255 (2023).
Raza, H. et al. Effects of ultrasound-induced V-type rice starch-tannic acid interactions on starch in vitro digestion and multiscale structural properties. Int. J. Biol. Macromol. 246, 125619 (2023).
Wang, Y. et al. ACSL4 and polyunsaturated lipids support metastatic extravasation and colonization. Cell(2024).
Bansaccal, N. et al. The extracellular matrix dictates regional competence for tumour initiation. Nature 623(7988), 828–835 (2023).
Winkler, J., Abisoye-Ogunniyan, A., Metcalf, K. J. & Werb, Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat. Commun. 11(1), 5120 (2020).
Bao, Y. et al. Transcriptome profiling revealed multiple genes and ECM-receptor interaction pathways that may be associated with breast cancer. Cell. Mol. Biol. Lett. 24, 38 (2019).
Huang, Z. et al. Low NT5DC2 expression predicts favorable prognosis and suppresses soft tissue sarcoma progression via ECM-receptor interaction pathway. Transl Oncol. 44, 101937 (2024).
Mumby, S. et al. Extracellular matrix degradation pathways and fatty acid metabolism regulate distinct pulmonary vascular cell types in pulmonary arterial hypertension. Pulm Circ. 11(1), 2045894021996190 (2021).
Tian, G. et al. Voltage-dependent anion channel 1 (VDAC1) overexpression alleviates cardiac fibroblast activation in cardiac fibrosis via regulating fatty acid metabolism. Redox Biol. 67, 102907 (2023).
Martinez-Reyes, I. & Chandel, N. S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 21(10), 669–680 (2021).
Yu, J. S. & Cui, W. Proliferation, survival and metabolism: the role of PI3K/AKT/mTOR signalling in pluripotency and cell fate determination. Development 143(17), 3050–3060 (2016).
Pacella, I. et al. Fatty acid metabolism complements glycolysis in the selective regulatory T cell expansion during tumor growth. Proc. Natl. Acad. Sci. U.S.A. 115(28), E6546–e6555 (2018).
Yoon, H. & Lee, S. Fatty acid metabolism in ovarian cancer: Therapeutic implications. Int. J. Mol. Sci. 23(4), 2170 (2022).
Zhang, Y. et al. Enhancing CD8(+) T cell fatty acid catabolism within a metabolically challenging tumor microenvironment increases the efficacy of melanoma immunotherapy. Cancer Cell 32(3), 377–391e379 (2017).
Li, G. et al. Desulfovibrio desulfuricans and its derived metabolites confer resistance to FOLFOX through METTL3. EBioMedicine 102, 105041 (2024).
Li, Q. et al. Streptococcus thermophilus inhibits colorectal tumorigenesis through secreting β-galactosidase. Gastroenterology 160(4), 1179–1193e1114 (2021).
Laval, T., Chaumont, L. & Demangel, C. Not too fat to fight: The emerging role of macrophage fatty acid metabolism in immunity to Mycobacterium tuberculosis. Immunol. Rev. 301(1), 84–97 (2021).
Tang, Y. et al. Prognosis and dissection of immunosuppressive microenvironment in breast cancer based on fatty acid metabolism-related signature. Front. Immunol. 13, 843515 (2022).
Li, C., Zhang, L., Qiu, Z., Deng, W. & Wang, W. Key molecules of fatty acid metabolism in gastric cancer. Biomolecules 12(5), 706 (2022).
Yang, S. et al. Fatty acid metabolism is related to the immune microenvironment changes of gastric cancer and RGS2 is a new tumor biomarker. Front. Immunol. 13, 1065927 (2022).
Lin, A., Zhang, J. & Luo, P. Crosstalk between the MSI status and tumor microenvironment in colorectal cancer. Front. Immunol. 11, 2039 (2020).
Zhang, D., Zhao, Y., Wang, S., Wang, X. & Sun, Y. A prognostic model of angiogenesis and neutrophil extracellular traps related genes manipulating tumor microenvironment in colon cancer. J. Cancer 14(11), 2109–2127 (2023).
Wu, Z. et al. The BET-Bromodomain inhibitor JQ1 synergized ABT-263 against colorectal cancer cells through suppressing c-Myc-induced mir-1271-5p expression. Biomed. Pharmacotherapy = Biomedecine Pharmacotherapie. 95, 1574–1579 (2017).
Vodenkova, S. et al. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: past, present and future. Pharmacol. Ther. 206, 107447 (2020).
Author information
Authors and Affiliations
Contributions
G L (Guangyi Liu): Formal analysis, Writing—original draft, Writing—review & editing, Validation. K L (Kun Liu): Software, Methodology, Formal analysis. L J (Lei Ji): Data curation, Writing—review & editing. Software. Y L (Yang Li): Conceptualization, Writing-review & editing, Supervision, Project administration. All authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, G., Liu, K., Ji, L. et al. Intratumoral microbiota, fatty acid metabolism, and tumor microenvironment constitute an unresolved trinity in colon adenocarcinoma. Sci Rep 15, 2568 (2025). https://doi.org/10.1038/s41598-025-87194-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-87194-2
