
Abstract
Muscular dystrophies (MD) are a group of hereditary diseases marked by progressive muscle loss, leading to weakness and degeneration of skeletal muscles. These conditions often result from structural defects in the Dystrophin–Glycoprotein Complex (DGC), as seen in Duchenne Muscular Dystrophy (DMD) and Becker Muscular Dystrophy (BMD). Since MDs currently have no cure, research has focused on identifying potential therapeutic targets to improve patients’ quality of life. In this study, skeletal muscle tissue samples from DMD and BMD patients, as well as non-dystrophic controls, were analyzed using label-free mass spectrometry (MS/MS) to characterize the proteomic profile of these conditions and identify biomarkers for differential diagnosis. In-silico analysis revealed that dystrophic muscle tissues are linked to biological processes related to cellular energy metabolism, including oxidation of organic compounds, energy production, and cellular respiration. Enrichment of functions associated with cell structure and RNA binding was also observed, including cytoskeletal protein binding and RNA binding. The human phenotypes most related to the proteomic signature were abnormal circulating metabolites, muscle physiology, and weakness. Quantitative analysis identified significant changes in proteins associated with sarcomere organization and protein ubiquitination, such as myomesin, myozenin, and E3 ubiquitin-protein ligase rififylin, suggesting these as potential therapeutic targets.
Similar content being viewed by others


Introduction
Skeletal muscles (SM) constitute a significant portion of the human muscular mass, comprising about 40% of body weight. SM accounts for approximately 50–75% of the body’s proteins and consists of 75% of water, 20% of proteins, including inorganic salts, minerals, fats, and carbohydrates, which together represent about 5% of the constituents of SMs1. The histological architecture of SM is characterized by the presence of cylindrical, multinucleated cells, composed by myofibrils, which are formed by repeated contractile units responsible for the muscle contraction process called sarcomeres2. The Dystrophin–Glycoprotein Complex (DGC) is the major structural element present in the muscle fibres and is responsible for maintaining the stability and integrity of the fibres through a network that couples the extracellular matrix components to the cytoskeleton3. This complex consists of Dystrophin, which in turn, binds to transmembrane sub-complexes composed by dystroglycans, sarcoglycans, and sarcospan, as well as cytoplasmic proteins such as syntrophin, dystrobrevin, and neuronal nitric oxide synthase (nNOS)4,5. Muscular Dystrophies (MD) represent a heterogeneous group of hereditary diseases characterized by progressive loss of muscle tissue leading to weakness and degeneration of skeletal muscles6. Most muscular dystrophies occur due to structural alterations in DGC proteins, such as in Duchenne Muscular Dystrophy (DMD) and Becker Muscular Dystrophy (BMD).
DMD is a lethal muscular disease with a prevalence of 4.8 individuals/100,0007. Clinically, the first signs of DMD can be observed before the age of five. The main signs include frequent falls, difficulty in climbing stairs, running, getting up from the floor, scoliosis accompanied by hyperlordosis, and calf hypertrophy8. DMD is characterized by symmetrical and progressive loss of muscle mass, starting at the pelvic girdle (hips and legs), later affecting the upper limbs. Fiber degeneration, and subsequent fibrosis, result in gait disturbances in early childhood, followed by loss of ambulation and cardiopulmonary complications9. The severity of the disease progresses with age, and patients become wheelchair-bound, with some cases requiring mechanical ventilation10.
Patients with DMD present a variable phenotype11, which is characterized by progressive and irreversible degeneration of skeletal muscle12, ranging from gait disturbance in early childhood to nearly a small weakness in adulthood13. Typically, DMD onset is juvenile accompanied by muscular weakness, calf hypertrophy, with gradual progression that can lead to loss of motor function and cardiomyopathy14.
BMD is a milder and less lethal form of the disease with a later age of onset, also presenting an indolent clinical progression. BMD presents a global prevalence estimated at 1.6/100,0007. In addition to differences in incidence and clinical manifestations, these two types of dystrophies are genetically distinct. While DMD often presents frame-shift mutations, abolishing dystrophin expression or generating extremely low amounts of dystrophin protein, BMD presents in frame deletions mutations that lead to protein truncations, thus generating an aberrant dystrophin protein with altered functions5,15. These alterations typically lead to a pattern of reduced anti-Dystrophin immunostaining in BMD and absence in DMD. Interestingly, both diseases are caused by different types of mutations in the gene encoding the Dystrophin protein16,17. The absence of dystrophin destabilizes the DGC, leading to increased fragility and permeability of the plasma membrane, resulting in dysregulation of calcium levels and increased oxidative damage18,19. Necrosis is closely associated with increased inflammation and oxidative stress, which in turn leads to regenerative myogenesis. Furthermore, the progressive increase in fibrosis caused by myonecrosis and inflammation impairs myogenesis and muscle regeneration, resulting in severe degeneration of muscle tissues. As patients age, the regenerative capacity of muscle decreases, and associated connective tissue gradually replaces muscle fibers20.
Like all degenerative diseases, MDs have no cure, and for this reason, clinical and experimental studies have focused on characterizing putative therapeutic targets to increase the time and quality of life for patients21. Although advances in genetic sequencing have successfully mapped and identified genetic mutations related to MDs, especially for DMD, the genetic and clinical heterogeneity of this group of diseases makes it indispensable to better characterize the underlying pathophysiological mechanisms of the degenerative process. For instance, genetic sequencing studies for Limb-Girdle Muscular Dystrophy (LGMD) have shown that BMD is often underdiagnosed, with patients being diagnosed as limb-girdle dystrophy22,23.
We have observed a consistent increase in proteomic studies related to muscle diseases. However, many of these studies are conducted using animal genetic models, human plasma, urine or even cell culture24,25. Although different proteomic studies have successfully employed label-free mass spectrometry technique for investigating muscular dystrophies in experimental models26,27, this powerful approach has proven to be limited for application in humans due to evident constraints on sample collections from human tissue biopsies. Therefore, the search for more accurate diagnostic tools, which can highlight unique characteristics of each type of dystrophy, is extremely relevant today. In this context, proteomic analysis stands out as an important strategy for the identification and characterization of protein biomarkers that can significantly contribute both to a more detailed understanding of the physiology and homeostasis of healthy muscle as well, and to a more accurate histopathological diagnosis of MDs. It is important to note that although biopsy is an invasive technique, it is still widely used and economically viable for the histopathological study of MDs using histochemical and immunohistochemical markers. These features may facilitate multi-omics approaches to better characterize the main clinical and physiological aspects of dystrophies.
In this work, skeletal muscle tissue samples from DMD and BMD patients obtained by biopsy, as well as from non-dystrophic patients, were used. The tissues were analysed using a label-free mass spectrometry MS/MS proteomic methodology to characterize the proteomic profile of DMD and BMD both qualitatively and quantitatively and search for putative useful biomarkers for the differential diagnosis of dystrophies. Our in-silico analysis showed that the molecular signature of dystrophic muscle tissue is related to biological processes associated with cellular energy metabolism, such as energy derivation by oxidation of organic compounds, generation of precursor metabolites and energy, and cellular respiration. We also observed an enrichment of molecular functions related to cell structure and RNA binding, such as structural molecule activity, cytoskeletal protein binding, and RNA binding. In fact, the human phenotypes most related to the proteomic signature of DMD were associated with abnormal circulating metabolite concentration, abnormal muscle physiology and muscle weakness. Quantitative analysis showed that Becker and Duchenne muscular dystrophy significantly alter the abundance of proteins related to sarcomere organization and protein ubiquitination as myomezin, myozenin and E3 ubiquitin-protein ligase rififylin suggesting them as putative therapeutic targets.
Methodology
All samples in this study were obtained in the Neuropathology sector of the Clementino Fraga Filho Hospital (HUCFF)at Federal University of Rio de Janeiro, between 2011 and 2021. The protocol (129/2023) was approved by the Ethics Committee of the HUCFF (approval number 6,528,766). Although researchers provided informed consent to all patients, our sample exhibited a high mortality rate, and some patients had outdated contact information, rendering them unreachable. In accordance with the Ethics Committee guidelines and Brazilian legislation, the Ethics Committee of the HUCFF authorized the utilization of stored human biological samples. We ensure the privacy and confidentiality of the research regarding the personal data ensuring that the patients were not identified in the research. All experiments followed the guidelines and regulations in accordance with the Declaration ofHelsinki.
We used 4 samples of human muscle tissue obtained by biopsies from patients diagnosed with DMD and 3 samples from patients diagnosed with BMD, patients’ age ranges from 5 to 11 years. For the Control group, 3 samples of muscles were used from non-dystrophic patients. All the biopsies were collected from biceps or quadriceps femoral muscles. Biopsy samples were removed from liquid nitrogen where they were stored and sectioned in a cryostat into 20 slices measuring 20 μm each. The sections were immediately placed in Eppendorf microtubes and kept in liquid nitrogen to be further processed for mass spectrometry analysis.
Extraction of skeletal muscle proteins
To remove red blood cells, two washes with phosphate buffered saline (PBS) were performed, followed by centrifugation for 5 min at 1,500 rpm at 4 °C, with the supernatant discarded after each wash. The pellets immersed in liquid nitrogen were macerated with a plastic pestle, and then the lysis buffer, adapted from Kolbel et al.28(50 mM Tris-HCl pH 7.8, 150 mM NaCl, 1% SDS, and Halt Protease Inhibitor Cocktail—Thermo Scientific), was added. Subsequently, the samples were sonicated for 5 s on ice, three times, with a 1-minute rest between each sonication. After vortex homogenization for 1 min, with 5-minute intervals, three times, the samples were centrifuged at 20,000 g for 20 min at 4 °C. The resulting supernatant was collected, and proteins were quantified using the BCA Protein Assay colorimetric kit (Thermo Scientific, Ref. 23228) exactly as recommended by the fabricant using a Spectramax 100 reader at a wavelength of 562 nm.
Protein hydrolysis
The protein extracts were hydrolysed according to the “filter-aided sample purification” (FASP) protocol by Wiśniewski et al.29. In brief, 50 µg of total proteins from tissue lysates were incubated for 30 min at 60 °C in the presence of 10 mM Dithiothreitol (DTT), after this the samples were centrifuged at 3,000 rpm for 2 min at 20 °C. Following that, the samples were subjected to two washes in 8 M Urea in 0.1 M Tris/HCl pH 8.5 by centrifugations at 14,000 g for 40 min. Thereafter, the samples were incubated with 50 mM Iodoacetamide for 20 min. After that, the samples were centrifuged for 30 min at 14,000 g, and the supernatant was discarded. The pellet was submitted to two washes with 8 M Urea in 0.1 M Tris/HCl pH 8.0 followed by centrifugation for 30 min at 14,000 g. The pellets were then subjected to enzymatic digestion with Lys-C Mass Spec (Promega) in 8 M Urea and 0.1 M Tris/HCl pH 8.0 at an enzyme/protein ratio of 1:50. The samples were homogenized for 1 min on a thermo-shaker (Nova Instrument) at 600 rpm and incubated for 20 h. After incubation, the filters were transferred to new microtubes, where the second enzymatic digestion step was performed with Trypsin (Promega) in 50mM Ammonium Bicarbonate, at an enzyme/protein ratio of 1:100. The samples were homogenized for 1 min on the thermo-shaker (Nova Instruments) at 600 rpm and incubated at 37 °C for 4 h, followed by centrifugation for 40 min at 14,000 g. Next, 50 µL de NaCl 0,5 M were added and after centrifugation at 14.000 g for 20 min the filtered peptides were collected and acidified with TFA at 10%.
Peptides purification
The peptides were concentrated, desalted, and eluted using 100 µL C18 tips (Thermo Scientific Ref.87784) exactly as recommended by the fabricant. In short, 100 µL of 50% Acetonitrile (ACN) was aspirated and discarded, this process was repeated twice. Next, 100 µL of 0.1% Trifluoroacetic Acid (TFA) were aspirated and discarded, twice. After this step, 100 µL of the protein extract was aspirated, and the solution was homogenized with the pipette for better sample elution. Next for the rinse step, 100µL of 0.1% TFA solution in 5% ACN was used. Then the peptides were eluted with 100 µL of 0.1% Formic Acid in 75% ACN and the samples were completely dehydrated using the Speed vac (Thermo Scientific) and sent to LADETEC at Instituto de Química, da Universidade Federal do Rio de Janeiro for analysis by NANOLC-MS/MS.
Mass spectrometry
The mass spectrometry analysis was done using an Easy1000 nanoLC system (Thermo Fisher) coupled to a Quadrupole Orbitrap mass spectrometer (Q Exactive Plus, Thermo Scientific). The dried peptides were solubilized in 0.1% formic acid and a volume of 4 µL was applied to a Trap column with an internal diameter of 200 μm and a length of 2 cm packed with Reprosil-Pur C18 resin (Dr. Maisch), with pores of 200 Å and particle size of 5 μm (packaged in the laboratory). The analytical column was packed with Reprosil-Gold C18 resin (Dr. Maisch), with 300 Å pores and 3 μm particle size (packaged in the laboratory) with 75 μm in diameter and 25 cm in length. Peptide elution was performed using a gradient from 98% solvent A (5% ACN and 0.1% formic acid) to 20% solvent B (95% ACN and 0.1 formic acid) for 85 min, 20–40% solvent B in 22 min and 40–95% solvent B in 5 min. After that, the column was re-equilibrated with solvent A. The Orbitrap mass spectrometer was controlled by the Xcalibur 2.2 software, which was programmed to operate in the data dependent acquisition (DDA) mode. The MS1 spectrum was acquired with a resolution of 70,000 at 200 m/z (mass/charge). MS1 spectrum reading was performed using 106 ions (AGC) and 50 ms Maximum IT. The reading spectrum comprised ions with 375 to 2000 m/z. The 20 most intense ions were fragmented and then subjected to MS2 acquisition, using collision induced dissociation (HCD) and 200–2000 m/z range. MS2 resolution was 17,500 to 200 m/z, AGC of 105 ions, Maximum IT of 100 ms. Ion isolation window was 1.2 m/z, normalized collision energy (NCE) was 30, dynamic exclusion time was 60s. Peptides with undetermined charges and + 1 were rejected. The samples (3 Controls, 4 DMD and 3 BMD) were injectedin technical duplicates for each biological sample.
Database searches and identification of peptides and proteins were conducted using Proteome Discoverer v2.5.0.400. Searches were performed against a concatenated Homo sapiens (human) database downloaded from UniProt (The UniProt Consortium, 2022) database (reference proteome, 82,485 entries) and contaminants list, utilizing the Sequest HT algorithm. The precursor mass tolerance used was 10 ppm, fragment mass tolerance of 0.1 Da, fixed modification of carbamidomethyl (Cys) and variable modification of N-terminal acetylation, Met-loss (Met), Met-loss + acetyl (Met) and oxidation (Met). Full tryptic cleavage specificity where two missed cleavage sites were allowed. False discovery rates (FDR) were obtained using Percolator and the proteins considered have medium and high confidence.
Experimental design and statistical analysis
The data obtained from mass spectrometry were filtered using the Interactive Venn software (https://www.interactivenn.net/) to identify common proteins among the samples of each experimental group. The proteins common to the experimental groups C1, C2, and C3 formed the Control Group (CG), the proteins common to the experimental groups D1, D2, D3, and D4 made up the Duchenne Muscular Dystrophy (DMD) group, and the proteins common to the experimental groups B1, B2, and B3 were selected as the Becker Muscular Dystrophy (BMD) group. Then using these filtered proteins, the abundance of each biological sample (obtained by the average of the technical duplicate) was considered for the statistical analysis used to identify the increase and decrease in protein abundances. Statistical analysis was carried out in Perseus v 1.6.2.2 and R. Protein area values were converted to log2 scale and normalized by subtracting the median of the sample distribution. Proteins with < 50% valid values in each group (Control, DMD and BMD) were removed from the analysis. The remaining proteins were subjected to missing value imputation using the default parameters (width 0.3, down shift 1.8) of the Perseus Imputation tool.
In silico analysis
All the proteins identified in each experimental group were analysed in the STRING platform (https://string-db.org/) to evaluate the gene ontology distribution in biological processes and in molecular functions. The association with human phenotype was analysed based on the Monarch Initiative platform (https://monarchinitiative.org). In each of these gene ontology annotations, the most enriched biological processes, molecular functions, and human phenotypes were plotted according to the False Discovery Rate (FDR) value. The most enriched pathways in each experimental group were analysed in the reactome platform (https://reactome.org/). The InteractVenn platform was used to show the distribution of the identified proteins between the groups (https://www.interactivenn.net/).
Heatmaps were done with Morpheus (https://software.broadinstitute.org/morpheus/) to show statistically significant proteins with abundance increased or decreased.
Results
Characterization of the molecular signature of muscle tissues
Although the DGC has been well characterized as the major progressive structural alterations responsible for the BMD and DMD the search for new proteins is still necessary to better understand these structural changes allowing the search for mechanisms that lead to reversing or even slowing down disease progression.
In our proteomic analysis a total of 1,649 proteins were identified for all the three experimental groups including control, Becker and Duchene dystrophies (Supplementary Table 1). A total of 1,126 proteins were identified in control 1 (C1), 1,266 proteins were identified in control 2 (C2) and 1,413 proteins were identified in control 3 (C3). Among these, 960 proteins were common to all 3 controls and will be referred to as control group (CG) (supplementary Fig. 1 and Supplementary Table 2). In Becker Muscular Dystrophy 1 (B1) 1,278 proteins were identified, in BMD 2 (B2) 1,303 proteins were identified and in BMD 3 (B3) 1,055 proteins were identified. Among these, 867 proteins were common to all the 3 Becker patient samples and will be referred to as BMD group (BG) (supplemental Fig. 1 and Supplemental Table 2). In Duchenne Muscular Dystrophy 1 (D1) 1,372 proteins were identified, in DMD 2 (D2) 995 proteins were identified, in DMD 3 (D3) 1,278 proteins were identified and in DMD 4 (D4) 1,057 proteins were identified. Among these, 801 proteins were common to all the 4 DMD patients and will be referred to as DMD group (DG) (supplemental Fig. 1 and Supplemental Table 2).
The most enriched biological processes were related to cellular energy metabolism
To characterize the molecular signature of the muscle samples all the proteins identified in each experimental group were examined according to the STRING GO classification. Analysing the biological process, we observed that the three most enriched processes, based on the FDR value, were associated with cellular energy metabolism, such as generation of precursor metabolites and energy (GO:0006091), energy derivation by oxidation of organic compounds (GO:0015980) and cellular respiration (GO:0045333) (Fig. 1; Supplemental Table 3). The biological process of Energy derivation by oxidation of organic compounds (GO:0015980) is represented by 96 proteins identified in CG, 87 proteins identified in DMD, and 88 proteins identified inBMD (Supplemental Table 4). The biological process Generation of precursor metabolites and energy (GO:0006091) is represented by 115 proteins in CG, 105 proteins in DMD, and 108 proteins in BMD (Supplemental Table 4). The biological process Cellular respiration (GO:0045333) is represented by 79 proteins in CG, 71 proteins in DMD, and 74 proteins in BMD (Supplemental Table 4).

GO String classification showing the biological process enrichment for the top fifteen biological processes based on the adjusted (adj) FDR value.
The most enriched molecular functions were related to structural molecule activity, cytoskeletal protein binding, and RNA binding
Next, we used the STRING GO classification system to assess the enrichment of molecular functions for all the proteins identified in each group. We observed that the most enriched molecular functions were related to cell structure and oxidative stress, such as structural molecule activity (GO:0005198), cytoskeletal protein binding (GO:0008092), and RNA binding (GO: GO:0003723) (Fig. 2; Supplemental Table 5). We identified 158 proteins in the molecular function related to structural molecule activity in CG, 149 proteins in DMD, and 142 proteins inBMD (Supplemental Table 6). For the molecular function of cytoskeletal protein binding, 151 proteins were found in GC, 128 proteins in DMD, and 135 proteins in BMD (Supplemental Table 6). For the molecular function of RNA binding, 197 proteins were found in CG, 169 in DMD, and 180 proteins inBMD (Supplemental Table 6).
The human phenotypes most enriched were related to abnormal circulating metabolite concentration, muscle weakness, and abnormal muscle physiology
To further investigate the relationship of the identified proteins in the experimental groups with diseases, we used the Monarch initiative platform, a challenging way to integrate genetic data, phenotype and diseases.

GO String classification showing the molecular function enrichment for the top fifteen molecular functions based on the adjusted (adj) FDR value.
We found that the three most enriched human phenotypes in the experimental groups were associated with abnormal circulating metabolite concentration (HP:0032180), muscle weakness (HP:0001324) and abnormal muscle physiology (HP:0011804) (Fig. 3; Supplemental Table 7). We observed 161 proteins in abnormal circulating metabolite concentration CG, 144 proteins for DMD, and 154 proteins forBMD (Supplemental Table 8). For abnormal muscle physiology, 238 proteins were found in CG, 203 proteins in DMD, and 222 proteins inBMD (Supplemental Table 8). In the classification of muscle weakness, 146 proteins were found for CG, 134 proteins in DMD, and 140 proteins inBMD (Supplemental Table 8).
Signalling pathway analysis showed a decrease in metabolism of proteins
To analyse the alteration of signalling pathways between CG, BG and DG, we performed a Reactome analysis with all proteins identified in each group. The results showed an overall decrease in protein metabolism from CG to BG and DG, respectively (Fig. 4A).Specially processes involved in post translational protein modification as protein methylation and protein deubiquitination as Ub specific processing proteases that were less enriched in BG and DG compared to CG.
Enrichment analysis of the main pathways showed that most of them decreased compared to the control and that mitochondrial protein degradation decreased as dystrophy worsened (Fig. 4B).In the CG a total of 41 proteins were identified, in the BG 38 proteins were identified and in the DG 33 proteins were identified in the mitochondrial protein degradation pathway. Analyzing these proteins we found that 3 proteins were identified only in the CG (ADP/ATP translocase 2, NADH-ubiquinone oxidoreductase chain 5, NADH-ubiquinone oxidoreductase chain 1), and 5 proteins were communs to CG and BG (Fumarate hydratase, mitochondrial, Transcription factor A, mitochondrial, ATP synthase-coupling factor 6 mitochondrial, Short/branched chain specific acyl-CoA dehydrogenase mitochondrial, NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 2) and these 8 proteins were not identified in DG.

Monarch Initiative classification showing the human phenotype enrichment for the top fifteen human phenotypes based on the adjusted (adj) FDR value.

Reactome Pathway Database analysis with all the proteins identified and quantified in each group (CG, BG and DG). (A) Reacfoam showing p value coverage for all proteins identified and quantified in control, Becker and Duchenne groups. (B) Graphical representation of the top fifteen reactome pathways based on adjusted p value enrichment.
Becker and Duchenne muscular dystrophy quantitatively alter the molecular signature of muscle tissues
Although the dystrophin abundance alteration in Becker and its severe absence in Duchene has been well established, the possibility to identify and quantify more proteins differentially expressed in these dystrophies could be important to better understand the molecular mechanism involved in these diseases.
Our mass spectrometry analysis revealed that the mean protein abundances of the dystrophin, sarcoglycans and dystroglycan were decreased in BG and DG in comparison to CG (Supplemental Table 9, Supplemental Fig. 2). Although dystrophin was identified and quantified in all groups, including the Duchene group, its mean abundance in Becker and Duchenne was less than 6% compared to the control group which is in accordance with the dystrophin severe absence established for duchenne dystrophy ranging from 0.7 to 7%30.
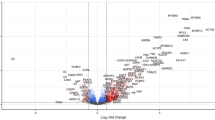
Comparing the protein distribution between BG and CG (Fig. 5A), we could observe that 755 proteins were common to BG and CG, 112 proteins were identified only in BG and 205 proteins were identified only in CG. The 755 common proteins showed 17 proteins whose differences in abundances were statistically significantly decreased when compared to the control group (Fig. 5B, C; Table 1).
On the other hand, we observed 16 proteins whose differences in abundances were statistically significantly increased in BG when compared to CG (Fig. 5B, C; Table 2).
To analyse the PPI between these proteins with abundance differences statistically significant we used the STRING platform (Fig. 5D). We found a PPI enrichment p value of 0.000622 for protein interactions within proteins classified in actin binding, muscle protein and sarcomere organization (Fig. 5D). The halo colour in the protein network represents the abundance of the proteins, we could observe the high abundant protein with the strong halo colour (rififylin) and the low abundance proteins with the weak halo colour.

(A) Interact Venn Diagram distribution between the 960 proteins identified and quantified in CG and the 867 proteins identified and quantified in BG by our label free proteomic MS/MS data. (B) Volcano plots of all proteins from CG and BG. Proteins with increased fold change ratio are indicated by red circles whereas the blue circles denote those proteins presenting a decreased fold change ratio. (C) Heat Map of the proteins with a relative fold change higher than 2 (0.5 < FC > 2) for the ratio BG/CG. (D) String protein-protein interaction network of the proteins with a relative fold-change higherthan 2 (0.5 < FC > 2) for the ratio BG/CG. The halo colour is based on the FC of the protein.
Comparing all the identified and quantified proteins between DG and CG (Fig. 6A), we found 696 proteins common to DG and CG, 105 proteins were identified only in DG and 264 proteins were identified only in CG. We observed 8 proteins whose differences in abundances between DG compared to CG were statisticallysignificantlydecreased (Fig. 6B, C; Table 3).
Conversely, we observed 16 proteins whose differences in abundance was statistically significantly increased (Fig. 6B, C; Table 4).
Analysing the PPI between the proteins with abundance differences statistically significant in the STRING platform we found a PPI enrichment p value of 0.000376 forprotein interactions within proteins classified in sarcomere organization, myofibril assembly and striated muscle cell differentiation. The halo colour in the protein network represents the abundance of the proteins, we could observe the high abundant protein with the strong halo colour (rififylin) and the low abundance proteins with the weak halo colour (Fig. 6D).

(A) Interact Venn Diagram distribution between the 960 proteins identified and quantified in CG and the 801 proteins identified and quantified in DG by our label free proteomic MS/MS data. (B) Volcano plots of all proteins from CG and BG. Proteins with increased fold change ratio are indicated by red circles whereas the blue circles denote those proteins presenting a decreased fold change ratio. (C) Heat Map of the proteins with a relative fold change higher than 2 (0.5 < FC > 2) for the ratio DG/CG. (D) String protein-protein interaction network of the proteins with a relative fold change higher than 2 (0.5 < FC > 2) for the ratio DG/CG. The halo colour is based on the FC of the protein.
Finally, we compared all the identified and quantified protein distribution between DG and BG (Fig. 7A), we could observe that 649 proteins were common to DG and BG, 152 proteins were identified only in DG and 218 proteins were identified only in BG. When comparing the DMD and BMD groups, we observed 10 proteins whose differences in abundance were statistically significantly decreased in DMD compared to BMD (Table 5).
In contrast, we observed 17 proteins whose differences in abundance were statistically significantly increased when comparing DG vs. BG (Fig. 7B, C; Table 6).
The PPI between the proteins with abundance differences statistically significant in the STRING platform showed a PPI enrichment p value of 4.07 × 107 forprotein interactions within proteins classified in collagen trimer and collagen-containing extracellular matrix. The halo colour in the protein network represents the abundance of the proteins, we could observe the high abundant protein with the strong halo colour (Profilin-1) and the low abundance proteins with the weak halo colour (Fig. 7D).

(A) Interact Venn Diagram distribution between the 867 proteins identified and quantified in BG and the 801 proteins identified and quantified in DG by our label free proteomic MS/MS data. (B) Volcano plots of all proteins from DG and BG. Proteins with increased fold change ratio are indicated by red circles whereas the blue circles denote those proteins presenting a decreased fold change ratio. (C) Heat Map of the proteins with a relative fold change higher than 2 (0.5 < FC > 2) for the ratio DG/BG. (D) String protein-protein interaction network of the proteins with a relative fold change higher than 2 (0.5 < FC > 2) for the ratio DG/BG. The halo colour is based on the FC of the protein.
Of the proteins mentioned above, we highlight 2 proteins with decreased abundance in both DMD and BMD, when compared to control: Myozenin-2 (MYOZ2; Q9NPC6; fold change: 0.49) and Myomesin-2 (MYOM2; P54296). Four proteins were significantly increased to both dystrophies compared to control: T-complex protein 1 subunit theta (CCT8; P50990); X-ray repair cross-complementing protein 6 (XRCC6; P12956); 60 S ribosomal protein L12 (RPL12; P30050) and E3 ubiquitin-protein ligase rififylin (RFFL; Q8WZ73). We also observed 2 proteins upregulated in DMD compared with both CG and BMD: Calreticulin (A0A7P0T861), Collagen alpha-1(XIV) chain (COL14A1; Q05707). And finally, we observed 2 other proteins whose abundances were decreased in DMD compared with both CG and BMD: Exportin-1 (A0A7I2V2S3) and Eukaryotic translation initiation factor 4 gamma 1 (EIF4G1; E7EX73).
We also find one protein whose abundance was increased in BG, usually associated to Limb Girdle Muscular Dystrophy (LGMD), Delta-sarcoglycan (SGCD; Q92629) and two other proteins whose abundance was decreased, associated to muscular disorders in Congenital Myopathy: Tropomyosin alpha-3 chain (TPM3; P06753-5) and Leiomodin-3 (LMOD3; Q0VAK6).
Discussion
Although Duchenne and Becker Muscular Dystrophies are directly related to the dystrophin protein, it is very important to find other proteins that contribute to the clinical heterogeneity among patients with this disease. Two proteins stand out for being less abundant in both types of dystrophies compared to the control, suggesting the loss or silencing of these proteins during the degenerative process caused by the dystrophy. One of them, Myozenin-2, is a Z-disc protein exclusively observed in striated muscles, which is strongly associated with studies of human hypertrophic cardiomyopathy31. Ruggiero et al. concluded that alterations in the Z disc may favour the induction of cardiac hypertrophy caused by mutations in the MYOZ2 gene32. Murphy et al. found a reduced abundance of the isoform 1 of myozenin comparing the dystrophic mdx-4cv mouse model versus age-matched leg muscle microsomes during the investigation of the differential protein expression patterns between dystrophic and normal skeletal muscle33.
The other protein, myomesin-2, with reduced abundance in both BMD and DMD is a M-line sarcomere protein important in the monitoring of sarcomere assembly and integrity as it is the last protein to be incorporated into the sarcomere. Myomesin assays have been described as a more specific method for detecting sarcomere damage than muscle creatine kinase in striated muscle disease34.
Our Gene Ontology analyses using STRING emphasize the function of this protein, since it was detected in Molecular Functions related to the structural activity of the cell. The importance of proteins from sarcomere in maintaining the integrity of skeletal striated muscle cells is well understood, which raises the hypothesis of a probable loss of these proteins in dystrophic tissue.
One protein was highlighted, the protein E3 ubiquitin-protein ligase rififylin, that had the highest increased in both BMD (fold change 14.72) and in DMD (fold change 39.44). This protein has been described as a risk factor for cardiac repolarization via ubiquitination of multiple proteins that differently affect different potassium channels and cardiac action potential duration35,36. Also known as an apoptosis inhibitor, rififylin protein regulates several biological processes via ubiquitination leading to proteasomal degradation of several target proteins as caspase-8 and caspase-10 as well as p5337,38.
Of the proteins found with abundance decreased only in DMD, suggesting a more aggressive loss in this type of dystrophy compared to its milder variant BMD and the control group, Exportin-1 plays an essential role in the nucleocytoplasmic trafficking of key transcription factor proteins39. Suárez-Sánchez et al. showed that Dp71d, a shortened form of dystrophin in Duchene muscular dystrophy, is transported between the nucleus and cytoplasm by the conventional nuclear transporters, importin (IMP) α/β and the exportin-1 CRM140. Exportin-1 have been described to interact with polyglutamine (polyQ) proteins that are produced in patients with amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). In these patients the defective nuclear export activity regulated by exportin-1 was observed41,42. Some selective inhibitors of nuclear export (SINEs) have been developed initially for cancer therapies, however, recent discoveries have demonstrated that exportin-1 could serve as a target in neurological and neuromuscular disorders containing pathogenic expansion repeats affecting export in protein activity39,43.
The Eukaryotic translation initiation factor 4 gamma 1 (EIF4G1) has been related to breast cancer and Parkinson disease44,45. Sanson et al. described the involvement of EIF4G2 in the mitochondrial metabolism of glucocorticoid used in Duchenne patients’ treatment, through its target by miR37946.
Of the proteins found increased only in DMD, which suggest an overexpression in this type of dystrophy in relation to CG and BMD, Calreticulin is a Ca2 + binding protein in the endoplasmic reticulum of muscle and nonmuscle cells47. Morales, et al., 2024 found an increase of calreticulin at a transcriptional level in a canine DMD model. The authors concluded that increase in the cytosolic calcium levels were an important mechanism in the regulation of muscle degradation in DMD. High levels of Ca2 + that also accumulates in mitochondria contributes to mitochondrial dysfunction and muscle damage in DMD48,49,50.
The protein Collagen alpha-1(XIV) chain plays a role in integrating collagen fibrils51. It is known that patients with DMD express higher levels of collagen and extracellular matrix factors52,53. Capitanio et al. using an in gel label free proteomic strategy found an increase in extracellular matrix proteins specially collagens in DMD compared to BMD18. In our proteomic analysis we also found collagen classified as contained in extracellular matrix increased in DMD compared to BMD patients. Comparing DMD and BMD, the profilin-1 protein was highlighted with a FC of 7.19 times higher in DMD compared to BMD. Profilin-1 is a protein that binds to actin and affects the organization of the cytoskeleton, however, in a higher concentration it is capable of preventing actin polymerization54,55.
The Delta-sarcoglycan protein (SGCD), which is usually associated with Limb Girdle Muscular Dystrophy (LGMD), was found to be more abundant in BMD compared to control. This transmembrane protein is a member of DGC which binds to dystroglycans, which in turn, were associated with the third domain of Dystrophin. This increase in expression was also reported by Zanotti et al. who demonstrated a significant increase in delta-sarcoglycan in patients with Becker, compared to other types of sarcoglycans56. Our findings may contribute with information that may justify reports of under diagnosis of BMD that are sometimes classified as LGMD22,23. Our analysis on the Monarch initiative revealed that this protein is associated with the Human Phenotypes revealing participation in muscle weakness, physiology and abnormal concentration of metabolites. We raised the hypothesis of SGCD overexpression as a cellular strategy for correction in the face of damage, although further studies are necessary to evaluate this hypothesis.
Of the proteins that were less abundant in BMDwe highlight Leiomodin-3 (LMOD3), which is a slow-growing end-binding protein of filamentous actin, and Isoform 5 of Tropomyosin alpha-3 chain (TPM3), which is a troponin-actin complex-binding protein both proteins have been associated with congenital myopathies57,58. These proteins were classified in our gene ontology analysis using STRING in the most enriched molecular functions and human phenotypes demonstrating their participation have been related to structural activities and abnormal functions of muscle.
The data found in our study demonstrate that we have taken an important step in the investigation of biomarkers and molecular signatures of DMD and BMD. Although the diagnosis of these diseases is currently largely linked to genetic investigation, in inconclusive cases, muscle biopsy is necessary to determine the presence of Dystrophin. It is important to emphasize that diagnosis through genetic tests is not accessible to the entire population, as they are more expensive59. Therefore, our studies have contributed to the knowledge of more proteins involved in DMD and BMD pathologies.
Conclusion
This study shows a LC-MS/MS label free proteomic approach analysis of muscle tissues from patients with Becker and Duchenne muscular dystrophies. Among the proteins identified and quantified, most were associated with structural activities of the muscle cell and physiological abnormalities of muscle or neurodegenerative diseases. Due to the challenges of obtaining more biopsy samples from rare pediatric diseases, new studies are necessary to validate our results. However, our findings were corroborated by the literature, which confirms a clear path in the search for new targets in DMD and BMD diseases.
Data availability
Data Availability: The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD050694, at [http://proteomecentral.proteomexchange.org/cgi/GetDataset].
References
Frontera, W. R. & Ochala, J. Skeletal muscle: a brief review of structure and function. Calcif. Tissue Int. 96, 183–195 (2015).
Boyle, J. et al. Molecular biology of the cell, 5th edition by,and Biochem. Mol. Bio Educ. 36, 317–318 (2008).
Hoffman, E. P., Brown, R. H. & Kunkel, L. M. Dystrophin: The protein product of the duchenne muscular dystrophy locus. Cell 51, 919–928 (1987).
Ervasti, J. M. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim. Biophys. Acta Mol. Basis Dis. 1772, 108–117 (2007).
McGreevy, J. W., Hakim, C. H., McIntosh, M. A. & Duan, D. Animal models of Duchenne muscular dystrophy: from basic mechanisms to gene therapy. Dis. Models Mech. 8, 195–213 (2015).
Mercuri, E. & Muntoni, F. Muscular dystrophies. Lancet 381, 845–860 (2013).
Salari, N. et al. Global prevalence of Duchenne and Becker muscular dystrophy: a systematic review and meta-analysis. J. Orthop. Surg. Res. 17, 96 (2022).
Connolly, A. M. et al. Motor and cognitive assessment of infants and young boys with Duchenne muscular dystrophy: results from the muscular dystrophy Association DMD Clinical Research Network. Neuromuscul. Disord. 23, 529–539 (2013).
Carberry, S., Brinkmeier, H., Zhang, Y., Winkler, C. K. & Ohlendieck, K. Comparative proteomic profiling of soleus, extensor digitorum longus, flexor digitorum brevis and interosseus muscles from the mdx mouse model of Duchenne muscular dystrophy. Int. J. Mol. Med. 32, 544–556 (2013).
Messina, S. & Vita, G. L. Clinical management of Duchenne muscular dystrophy: the state of the art. Neurol. Sci. 39, 1837–1845 (2018).
Flanigan, K. M. Duchenne and Becker muscular dystrophies. Neurol. Clin. 32, 671–688 (2014).
Godoy, V. C. W. P. D. & Lanzillotta, P. Treinamento muscular respiratório na distrofia muscular de Becker. Rev. Neurocienc 20, 138–143 (2001).
Momma, K. et al. Rimmed vacuoles in becker muscular dystrophy have similar features with inclusion myopathies. PLoS ONE 7, e52002 (2012).
Bello, L. et al. Functional changes in Becker muscular dystrophy: implications for clinical trials in dystrophinopathies. Sci. Rep. 6, 32439 (2016).
Sheikh, O. & Yokota, T. Advances in genetic characterization and genotype–phenotype correlation of duchenne and becker muscular dystrophy in the personalized medicine era. JPM 10, 111 (2020).
Monaco, A. P., Bertelson, C. J., Liechti-Gallati, S., Moser, H. & Kunkel, L. M. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 2, 90–95 (1988).
de Albuquerque, M. A. V. Distrofia Muscular de Cinturas em crianças: Caracterização Clínica, Histológia E Molecular. https://doi.org/10.11606/T.5.2013.tde-03012014-154533 (2013).
Capitanio, D. et al. Comparative proteomic analyses of Duchenne muscular dystrophy and Becker muscular dystrophy muscles: changes contributing to preserve muscle function in Becker muscular dystrophy patients. J. Cachexia Sarcopenia Muscle 11, 547–563 (2020).
Law, M. L., Cohen, H., Martin, A. A., Angulski, A. B. B. & Metzger, J. M. Dysregulation of calcium handling in duchenne muscular dystrophy-associated dilated cardiomyopathy: mechanisms and experimental therapeutic strategies. JCM 9, 520 (2020).
Portela, M. V. M., Weksler, F. L., Vallim, Y. V. & Siqueira, E. C. D. Distrofia muscular de Duchenne. Acervo Saude 23, e12912 (2023).
Luna-Angulo, A. et al. Pharmacological treatments and therapeutic targets in muscle dystrophies generated by alterations in dystrophin-associated proteins. Medicina 60, 1060 (2024).
Töpf, A. et al. Exome reanalysis and proteomic profiling identified TRIP4 as a novel cause of cerebellar hypoplasia and spinal muscular atrophy (PCH1). Eur. J. Hum. Genet. 29, 1348–1353 (2021).
Nallamilli, B. R. R. et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann. Clin. Transl Neurol. 5, 1574–1587 (2018).
Wilson, K. et al. Duchenne and Becker muscular dystrophies: a review of animal models, clinical end points, and biomarker quantification. Toxicol. Pathol. 45, 961–976 (2017).
Dowling, P. et al. Emerging proteomic biomarkers of X-linked muscular dystrophy. Expert Rev. Mol. Diagn. 19, 739–755 (2019).
Holland, A. et al. Comparative label-free mass spectrometric analysis of mildly versus severely affected mdx mouse skeletal muscles identifies annexin, lamin, and vimentin as universal dystrophic markers. Molecules 20, 11317–11344 (2015).
Carr, S. J., Zahedi, R. P., Lochmüller, H. & Roos, A. Mass spectrometry-based protein analysis to unravel the tissue pathophysiology in Duchenne muscular dystrophy. Prot. Clin. Appl. 12, 1700071 (2018).
Kolbel, H. et al. Identification of candidate protein markers in skeletal muscle of laminin-211-deficient CMD type 1A-patients. Front. Neurol. 10, 470 (2019).
Wiśniewski, J. R., Zougman, A., Nagaraj, N. & Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009).
Beekman, C., Janson, A. A., Baghat, A., Van Deutekom, J. C. & Datson, N. A. Use of capillary western immunoassay (Wes) for quantification of dystrophin levels in skeletal muscle of healthy controls and individuals with Becker and Duchenne muscular dystrophy. PLoS ONE 13, e0195850 (2018).
Osio, A. et al. Myozenin 2 is a novel gene for human hypertrophic cardiomyopathy. Circ. Res. 100, 766–768 (2007).
Ruggiero, A., Chen, S. N., Lombardi, R., Rodriguez, G. & Marian, A. J. Pathogenesis of hypertrophic cardiomyopathy caused by myozenin 2 mutations is independent of calcineurin activity. Cardiovasc. Res. 97, 44–54 (2013).
Murphy, S. et al. Simultaneous pathoproteomic evaluation of the dystrophin-glycoprotein complex and secondary changes in the mdx-4cv mouse model of duchenne muscular dystrophy. Biology 4, 397–423 (2015).
Prill, K., Carlisle, C., Stannard, M., Reid, W., Pilgrim, D. B. & P. J. & Myomesin is part of an integrity pathway that responds to sarcomere damage and disease. PLoS ONE 14, e0224206 (2019).
Gopalakrishnan, K. et al. Augmented rififylin is a risk factor linked to aberrant cardiomyocyte function, short-QT interval and hypertension. Hypertension 57, 764–771 (2011).
Kabakov, A. Y. et al. E3 ubiquitin ligase rififylin has Yin and Yang effects on rabbit cardiac transient outward potassium currents (Ito) and corresponding channel proteins. J. Biol. Chem. 300, 105759 (2024).
Yang, W., Dicker, D. T., Chen, J. & El-Deiry, W. S. CARPs enhance p53 turnover by degrading 14-3-3σ and stabilizing MDM2. Cell. Cycle 7, 670–682 (2008).
McDonald, E. R. & El-Deiry, W. S. Suppression of caspase-8- and -10-associated RING proteins results in sensitization to death ligands and inhibition of tumor cell growth. Proc. Natl. Acad. Sci. U.S.A. 101, 6170–6175 (2004).
Hightower, R. M. et al. The SINE compound KPT-350 blocks dystrophic pathologies in DMD zebrafish and mice. Mol. Ther. 28, 189–201 (2020).
Suárez-Sánchez, R. et al. Nucleocytoplasmic shuttling of the Duchenne muscular dystrophy gene product dystrophin Dp71d is dependent on the importin α/β and CRM1 nuclear transporters and microtubule motor dynein. Biochim. Biophys. Acta Mol. Cell. Res. 1843, 985–1001 (2014).
Chan, W. M. et al. Y. E. expanded polyglutamine domain possesses nuclear export activity which modulates subcellular localization and toxicity of polyQ disease protein via exportin-1. Hum. Mol. Genet. 20, 1738–1750 (2011).
Lattante, S. et al. Contribution of ATXN2 intermediary polyQ expansions in a spectrum of neurodegenerative disorders. Neurology 83, 990–995 (2014).
Mendonca, J. et al. Selective inhibitors of nuclear export (SINE) as novel therapeutics for prostate cancer. Oncotarget 5, 6102–6112 (2014).
Li, K. et al. EIF4G1 is a potential prognostic biomarker of breast cancer. Biomolecules 12, 1756 (2022).
Deng, H., Wu, Y. & Jankovic, J. The EIF4G1 gene and Parkinson’s disease. Acta Neurol. Scand. 132, 73–78 (2015).
Sanson, M. et al. miR-379 links glucocorticoid treatment with mitochondrial response in Duchenne muscular dystrophy. Sci. Rep. 10, 9139 (2020).
Fliegel, L. & Michalak, M. Fast-twitch and slow-twitch skeletal muscles express the same isoform of calreticulin. Biochem. Biophys. Res. Commun. 177, 979–984 (1991).
Morales, E. D. et al. Transcriptional changes of genes encoding sarcoplasmic reticulum calcium binding and up-taking proteins in normal and duchenne muscular dystrophy dogs. BMC Musculoskelet. Disord. 25, 811 (2024).
Dubinin, M. V. et al. Reduction of mitochondrial calcium overload via MKT077-induced inhibition of glucose-regulated protein 75 alleviates skeletal muscle pathology in dystrophin-deficient mdx mice. IJMS 25, 9892 (2024).
Bonato, A., Raparelli, G. & Caruso, M. Molecular pathways involved in the control of contractile and metabolic properties of skeletal muscle fibers as potential therapeutic targets for Duchenne muscular dystrophy. Front. Physiol. 15, 1496870 (2024).
Schuppan, D. et al. Undulin, an extracellular matrix glycoprotein associated with collagen fibrils. J. Biol. Chem. 265, 8823–8832 (1990).
Kharraz, Y., Guerra, J., Pessina, P. & Serrano, A. L. Muñoz-Cánoves, P. understanding the process of fibrosis in duchenne muscular dystrophy. Biomed. Res. Int. 2014, 1–11 (2014).
Suárez-Calvet, X. et al. Decoding the transcriptome of Duchenne muscular dystrophy to the single nuclei level reveals clinical-genetic correlations. Cell. Death Dis. 14, 596 (2023).
Shao, J., Welch, W. J., DiProspero, N. A. & Diamond M. I. Phosphorylation of profilin by ROCK1 regulates polyglutamine aggregation. Mol. Cell. Biol. 28, 5196–5208 (2008).
Liu, X., Pimm, M. L., Haarer, B., Brawner, A. T. & Henty-Ridilla, J. L. Biochemical characterization of actin assembly mechanisms with ALS-associated profilin variants. Eur. J. Cell Biol. 101, 151212 (2022).
Zanotti, S. et al. Immunofluorescence signal intensity measurements as a semi-quantitative tool to assess sarcoglycan complex expression in muscle biopsy. Eur. J. Histochem. 66, 1 (2022).
Yuen, M. et al. Leiomodin-3 dysfunction results in thin filament disorganization and nemaline myopathy. J. Clin. Investig. 124, 4693–4708 (2014).
Moraczewska, J. et al. Congenital myopathy-related mutations in tropomyosin disrupt regulatory function through altered actin affinity and tropomodulin binding. FEBS J. 286, 1877–1893 (2019).
Srivastava, P. et al. Diagnosing muscular dystrophies: comparison of techniques and their cost effectiveness: a multi-institutional study. JNRP 11, 420–429 (2020).
Acknowledgements
We thank the Unidade de Espectrometria de Massas e Proteômica (UEMP) from the Universidade Federal do Rio de Janeiro for the use of the facilities for sample preparations.
Funding
The authors acknowledge Fundação de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ 260003/009300/2021) for research funding.
Author information
Authors and Affiliations
Contributions
J.C.T.S. and M.R.A.N. and conducted the proteins extraction and hydrolysis experiments, the purification peptides experiments, the data analysis and wrote the manuscript. F.C.S.N. and YMS conducted the mass spectrometry experiments. N.H.S.C conceived and designed the study. K.C. conceived and designed the study, contributed to the data analysis, the in silico analysis and wrote the manuscript. D.A.P. conceived and designed the study contributed to the experiments, the data and the in silico analysis, the statistical analysis, and wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
da Silva, J.C.T., Nogueira, M.R.A., da Silva, Y.M. et al. Label-free proteomic analysis of Duchenne and Becker muscular dystrophy showed decreased sarcomere proteins and increased ubiquitination-related proteins. Sci Rep 15, 3293 (2025). https://doi.org/10.1038/s41598-025-87995-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-87995-5
