
Abstract
Copy number variation (CNV) is an important part of human genetic variations, which is associated with various kinds of diseases. To tackle the limitations of traditional CNV detection methods, such as restricted detection types, high error rates, and challenges in precisely identifying the location of variant breakpoints, a new method called MSCNV (copy number variations detection method for multi-strategies integration based on a one-class support vector machine model) is proposed. MSCNV establishes a multi-signal channel that integrates three strategies: read depth, split read, and read pair. First, a one-class support vector machine algorithm is used to detect abnormal signals in read depth and mapping quality values to determine the rough CNV region. Then, the rough CNV region is filtered by using paired read signals to improve the precision of MSCNV method. Finally, MSCNV explores and recognizes tandem duplication regions, interspersed duplication regions, and loss regions. It uses split read signals to determine the precise location of mutation points and to determine the type of variation. Compared with Manta, FREEC, GROM-RD, Rsicnv, and CNVkit, MSCNV significantly improves the sensitivity, precision, F1-score, and overlap density score of CNV detection while reducing the boundary bias of the detection results.
Similar content being viewed by others
Introduction
Next-generation sequencing (NGS) technology has significantly increased the throughput of sample sequencing. Copy number variation (CNV) is a significant form of genetic variation in humans1. CNV can be defined as the losses and duplications of DNA segments ranging from one Kbp to several Mbps2. Moreover, it constitutes approximately 9.5% of the human genome3. According to studies4,5,6, CNVs are closely associated with a variety of genetic diseases, neurological disorders, and tumors, such as congenital heart disease, autism, and cancer. Therefore, the accurate detection of CNVs is crucial for identifying disease-causing genes, gaining a comprehensive understanding of disease pathogenesis, and facilitating the development of appropriate therapeutic strategies.
NGS provides researchers with a vast amount of affordable sequencing data. Currently, the read depth (RD)-based strategy has emerged as the primary approach for CNV detection7, 8. The RD-based method detects CNVs based on the positive correlation between the copy number in the genome and its RD value. ReadDepth9 predicts CNVs by fitting a negative binomial distribution and circular binary segmentation using the RD information obtained from preprocessing. GROM-RD10 models genomic bias and employs statistical models and machine learning methods to estimate expected RD values for each genomic location. FREEC11 preprocesses RD using GC bias correction to generate segmented smoothing profiles for assessing CNVs. iCopyDAV3 uses GC and mapping bias to correct RD values and detects CNVs via divisive or agglomerative segmentation methods. RKDOSCNV12 assigns a relative kernel density outlier score to each RD segment. Based on the relative kernel density outlier score profiles, CNVs are predicted by choosing a reasonable threshold. CNVnator13 identifies potential copy number change signals and estimates CNVs by analyzing the coverage depth of the genome. Rsicnv14 identifies potential CNVs by comparing the sequencing depth distributions of normal and to-be-detected samples using a parametric model with a Poisson distribution. These methods all use RD signals. However, these methods mentioned above cannot distinguish the types of duplication regions, failing to identify interspersed duplication or achieve nucleotide-level breakpoint detection.
In addition, three other strategies, namely read pair (RP), split read (SR), and de novo assembly (AS), can be employed to detect structural variants (SVs) from NGS data15,16,17. GenomeSTRiP18 uses sequence comparison analysis to capture structural differences in the genome. Delly19 analyzes the alignment positions of connecting ends in sequencing data and uses the alignment position, and relative direction of SR segments to detect different types of SVs. LUMPY20 uses a probabilistic model to model the pattern of different SVs, extract SRs, paired reads, and sequencing depth information from sequencing data, and determine the location and type of SVs. Manta21 depends on information such as SR segments, inconsistencies in comparison, and segment sizes to detect SVs. These methods employ multiple strategies simultaneously, enabling the detection of precise variant breakpoints and complex variant types. Currently, there are few tools available for implementing SV detection strategies in CNV detection3, 22, 23.
To address these issues and effectively utilize existing SV strategies, this study proposes a method called MSCNV (Multi-Strategies-Integration Copy Number Variations Detection Method). By integrating three strategies, MSCNV enhances the reliability and accuracy of CNV detection. In the first step, the one-class support vector machine (OCSVM)24 is used to perform nonlinear kernel function mapping on RD and mapping quality (MQ) signals to obtain rough CNVs. This enables the detection of many CNVs that cannot be accurately fitted using linear methods. Moreover, the OCSVM separates positive samples by identifying the optimal hyperplane, making the detection of abnormal samples easier. This effectively addresses the issue of significantly different proportions of normal and abnormal genes in the sample. In the second step, the RP signals are employed to filter out false-positive regions and obtain final CNV regions. Lastly, SR signals are used to explore and identify tandem duplication, interspersed duplication, and lost regions, after which mutation breakpoints are located accurately. When applied to real data, the MSCNV has the ability to detect tandem duplication, interspersed duplication, and loss. Unfortunately, the methods based on the RD strategy are unable to detect interspersed duplication.
In this work, our main contributions are as follows: (1) Employing a multi-strategy integration approach for CNV detection. This approach filters the CNV detected by RD and MQ signals and uses the RP signals to improve the accuracy of CNV detection. SR signals are utilized to search near the variation boundary, update the boundaries of the variant, reduce the deviation of the CNV area boundary, and ultimately ascertain the precise location of the variant point. (2) Expanding the types of CNV detection, achieving the application of tandem duplication variants and interspersed duplication variants in CNV. The tandem duplication region and interspersed duplication region are explored by analyzing the matching information between the testing sample and the reference sequence. (3) Applying machine learning method (OCSVM) to CNV detection, enhancing the efficiency of CNV detection and expanding the scope of its application.
Materials and methods
Overview of MSCNV
MSCNV uses RD, RP, and SR signals to detect CNVs from a single sample, without the need for control-matched samples25, 26. The workflow of the MSCNV method is illustrated in Fig. 1. Initially, the input data consists of the sequenced samples (a Fastq file) and the reference genome (a Fasta file). The short reads obtained from the sequenced sample are aligned to the reference genome using the BWA27 software, resulting in a BAM file. SAMtools28 software is used to sort the BAM files and extract RP and SR segments from the BAM files. Subsequently, read counts and mapping quality profiles are preprocessed, including correction of GC bias, noise reduction, and standardization of RD and MQ signals. Finally, rough CNV regions are identified employing the OCSVM model.

Workflow of the MSCNV method. It consists of three main parts: input, preprocessing, and detection.
To achieve more accurate CNV regions, false-positive regions are filtered out using discordant reads. The breakpoint locations of the CNVs are inferred using the location information of the SR, which helps to refine the boundaries of the CNV regions. In the following subsections, the principles and implementation of each step are described in detail.
Preprocessing
Data preprocessing mainly includes four parts: (1) calculation of RD and MQ; (2) correction of GC bias; (3) denoise; and (4) standardization.
Obtaining RD and MQ
The reference genome is generally incomplete in any version29. “N” is used to replace unidentified bases in the reference genome30, 31. Further processing is required for the “N” positions32, 33. The read count (RC) at position “N” of the reference genome can be set to zero. As all sequencing reads are traversed, the RC value of the alignment position is increased by 1. The RC value for each location of the reference genome is shown in Eq. (1).
where RCl represents the read count at the l-th position of the reference genome, Nrs represents the number of reads covering the l-th position, and Asd represents the average sequencing depth at the l-th position.
To reduce random fluctuations in characteristic signals caused by noisy signals, the read count profile is divided into consecutive and nonoverlapping bins. The value of RD can be calculated by Eq. (2).
where RDm represents the RD value at the m-th bin, RCm represents the sum of RC values at each position within the m-th bin, and binlenm represents the size of the m-th bin.
The value of MQ can be calculated by Eq. (3).
where MQm represents the MQ value at the m-th bin, and MQl represents the MQ value at the l-th position within the m-th bin.
Correction of GC bias
The GC content bias is caused by PCR amplification34. The GC content may affect sequence coverage on some platforms35. Therefore, RD in each bin needs to be corrected. It can be calculated by Eq. (4).
where\(\small RD_{m}^{\prime }\) represents the RD value of the m-th bin after correction, \(\it {\overline {{{\text{sum}}}} _{{\text{rd}}}}\) represents the mean RD value of all bin, and rdgc represents the mean RD value bins with similar GC content to the m-th bin. Bins with similar GC content are defined as windows with a difference in GC content from the m-th bin less than or equal to 0.002.
Denoise
The original sequencing data are noisy due to sample contamination and limitations of the sequencing technology. Incorrectly identifying the noise as CNVs may generate false-positive results. At the same time, CNVs may be masked by noise, failing to correctly detect the true variation. To avoid these consequences, noise reduction from sequencing data should be performed for CNV detection. MSCNV uses total variation (TV) regularization to denoise RD signals36. The TV algorithm is sensitive to noisy data. The algorithm is expressed as the solution to a minimization problem. The regularized TV and least squares are used to process one-dimensional data, as shown in Eq. (5)37.
where RDRm represents the RD value after the noise reduction of the m-th bin, and t represents the total number of bins. Currently, choosing the appropriate value for λ remains a challenge33, 38. When λ is small, the denoising effect of the TV algorithm is mild. When λ is large, the denoising effect of the TV algorithm becomes more significant, leading to reduced discriminability between denoising scores across different bins. Users are allowed to specify the value of λ themselves. Based on relevant literature and experience, it is recommended to choose a value for λ within the range of 0.15 to 0.30.
Standardization
The RD values of the same bin may differ significantly. Therefore, to eliminate dimensional differences between different features, it is necessary to standardize the feature signals. Then, the data can be compared and analyzed at the same scale.
Detecting CNVs
Three algorithms are designed for the detection phase. The first algorithm models the OCSVM algorithm. Then, the outlier score for each bin is calculated to determine whether the sample belongs to the abnormal category. The second algorithm utilizes the discordant reads to filter false-positive regions. The third algorithm employs the SR signal to explore the variation type and the location of breakpoints in the variant region.
Declaration of rough CNVs
After preprocessing, the values of RD and MQ for each bin can be obtained. OCSVM39, 40 algorithm is used to evaluate the degree of abnormality of each genomic segment. An OCSVM model is constructed to identify rough CNVs.
OCSVM is a variant of the support vector machine (SVM) algorithm. SVM is a supervised learning algorithm commonly employed for classification problems. OCSVM is an unsupervised learning algorithm primarily utilized for detecting abnormal or outlier samples. Detecting CNV in genomic sequences is an anomaly test. However, only a small portion of the real genome sequence is mutated. Additionally, the region and type of CNV cannot be obtained in advance. Consequently, OCSVM, as a variant of SVM, is chosen to discern the specific information of CNV in the human genome. This method aims to enhance aggregation by mapping the sample data to a high-dimensional feature space using a kernel function. Subsequently, the optimal hyperplane is determined in the feature space to maximize the separation between the target data and the coordinate origin. The variant regions exhibit larger geometric margins than the normal regions. This approach applies to our target of detecting CNV regions, which can be considered variant regions with a large geometric margin. The process of recognizing rough CNVs is shown in Algorithm 1. Steps 2 to 5 of Algorithm 1 provide a mathematical description of the OCSVM model, demonstrating how to utilize this model to compute the abnormal scores S from the dataset D. The computational complexity of Algorithm 1 is O(n3), where n is the number of samples.

Recognizing rough CNVs based on OCSVM.
In the first step, dataset D consists of two-dimensional data, including RD and MQ, as shown in Eq. (6).
where n represents the number of samples in dataset D and di = [rdi, mqi] represents the i-th data sample point.
In the second step, dataset D is mapped into a high-dimensional space using the radial basis function (RBF). Because the dataset D is not assumed to be linearly distributed, RBF is chosen as the kernel function of OCSVM. Making the originally indivisible data divisible thus improves the classification accuracy of the OCSVM. A kernel function is a mathematical way of representing the result of a dot product in the upscaled space using vector computations in the original space of the data. The exact calculation of the radial basis function kernel is shown in Eq. (7) and Eq. (8).
where \(\small \parallel D - D^\prime \parallel\) represents the Euclidean distance between each pair of samples in the dataset D, β represents the reciprocal of the number of features24, 40, and X is the mapped feature vector. In this study, the value of β was set to 1/2.
In the third step, each sample point is mapped as a feature vector in the feature space. Each feature vector is constructed into a feature matrix, as shown in Eq. (9).
where F represents the feature matrix and Xi represents the feature vector that maps the sample point di to the high-dimensional space.
In the fourth step, an optimal hyperplane HP is obtained by training the feature matrix F. This plane is the “decision boundary” of the data, and all the data will be within the plane as much as possible, as shown in Eq. (10).
where ω represents the normal vector of hyperplane HP and b represents the bias term of the hyperplane.
In the fifth step, when a sample point is correctly classified by the hyperplane, the distance of the point from the hyperplane is defined as the geometric spacing. A larger geometric spacing from the sample point to the hyperplane means that the probability of the point’s abnormality is larger, and vice versa. The abnormality score for each sample point is represented by the geometric spacing from that sample point to the hyperplane, as shown in Eq. (11).
where \(\left\| \omega \right\|\) represents the length of the vector.
In the sixth step, a threshold is set. If the abnormal score of a sample point is greater than the threshold, then the sample point is an abnormal sample, and vice versa for a normal sample. The sample category T is represented by 0 and 1. A value of 1 means that the sample is abnormal, and a value of 0 means that the sample is normal.
In the seventh step, bins with neighboring positions and bins with category T of 1 are merged.
Precision CNVs
Due to the effects of tumor purity, the difference in feature signals between normal samples and tumor samples is small41. Therefore, the discordant reads are utilized to filter the false-positive regions to improve the detection accuracy. The POS and TLEN features are extracted from the file. POS is used to record the position of the short-read alignment to the reference gene. TLEN, which represents the length of the inserted fragment of the short read, is used to filter out false-positive regions. Here, a “short read” refers to a sequence fragment in gene sequencing data, and a “short-read alignment” is the process of aligning a short read with a reference genome to determine the position of the sequence fragment in the reference genome. Additionally, an “inserted fragment” denotes an additional piece of DNA that appears at a genomic location, resulting in an increased DNA sequence length compared to the reference genome. The process for determining the precise CNV region is shown in Algorithm 2. First, the CNV region is initialized. Then, the rough CNV region is traversed. If the length of the inserted fragment significantly differs from the length of the variant region, the region is considered a true-positive region. Conversely, if the lengths are similar, the region is regarded as a false-positive region. Finally, the true positive region is saved to obtain the final CNVs. Algorithm 2 consists of two loops and its computational complexity is O(ij), where i represents the number of rough CNV regions and j represents the length of the i-th rough CNV region.

Using discordant reads to filter false-positive regions.
where rl represents the starting position of the rough CNV region R, and rr represents the ending position of the rough CNV region R.
Refinement of CNV boundaries
The SR signal is utilized to refine the boundaries of the CNV regions after detecting CNVs. POS and CIGAR are the two features extracted from the BAM file. CIGAR provides information about the comparison between short sequence reads and the reference sequence.
CIGAR is a string consisting of numbers and letters. MSCNV is used to detect tandem duplications, interspersed duplications, and losses of CNVs. Therefore, only the marker characteristics “M” and “S” require consideration. CIGAR can be expressed by Eq. (12).
where “C” represents the CIGAR, “M” represents match, “S” represents mismatch, a represents the number of bases where the SR matches the reference genome, b represents the number of bases where the SR does not match the reference genome, and len represents the length of the SR.
The presence of the character “S” in the CIGAR feature indicates that the short reads do not match the reference sequence. One type is called pre-alignment, denoted as “MS”, while the other is called post-alignment, denoted as “SM”. The details of refining the boundaries of the CNV regions are described in Algorithm 3. Algorithm 3 has a computational complexity of O(nl), where n represents the number of CNV regions and l represents the length of the n-th CNV region.

Refinement of the boundaries of CNVs.
While exploring breakpoints, not all SR signals are meaningful to explore. The final CNV regions are scanned, denoted as [m, n]. Here, m and n respectively denote the starting point and ending point of a CNV region.
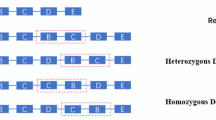
In the third step, the boundaries of the regions of tandem duplication, interspersed duplication, and loss are further explored. Figure 2 depicts in detail the exploration process of the boundaries of the CNV region, where R1 ~ R8 are short reads.

Boundary exploration of variant regions, where (a) is an example of tandem duplication, (b) is an example of interspersed duplication, and (c) is an example of loss.
Figure 2(a) illustrates the exploration process of the boundaries of the CNV tandem duplication region. The position of x is the start of the tandem duplication region, and y is the end position. The matching type of R1 at the x position is post-alignment. The matching type of R2 at the y position is front pre-alignment. The boundaries of the tandem duplication region can be expressed by [TDPx, TDPy+a−1]. Here TDPx represents the position of the reference genome matched with R1, TDPy represents the position of the reference genome matched with R2, and a represents the number of bases matched with R2.
Figure 2(b) provides the exploration process of the boundaries of the CNV interspersed duplication region. The o position is the start position of the interspersed duplication region, and p is the end position. The matching type of R3 from the interspersed duplication at the o position is post-alignment. The matching type of R6 at the p position is pre-alignment. The position q is the position where interspersed duplication occurs. Unlike the tandem duplication region, the R4 is matched in a pre-alignment manner at the q position, and the R5 is matched in a post-alignment manner at the q position. The boundaries of the interspersed duplication region can be expressed by [IDPo, IDPP+b−1]. Here IDPo represents the position of the reference genome matched with R3, IDPp represents the position of the reference genome matched with R6, and b represents the number of bases matched with R6.
Figure 2(c) depicts the exploration process of the boundaries of the CNV loss region. The w position is the start position of the loss region, and z is the end position. The matching type of R8 at the w position is pre-alignment. The matching type of R7 at the z position is post-alignment. The boundary of the loss region can be expressed by [LPw+c, LPz−1]. Here LPw represents the position of the reference genome matched with R8, LPz represents the position of the reference genome matched with R7, and c represents the number of bases matched with R8.
Experiment environment and tools
In this study, we conduct our experiments on the Ubuntu 16.04, which can provide a stable and reliable platform. For algorithm implementation, we utilize Python3.8. To further explore and validate our experimental results, we use MATLAB and Excel for statistical analysis.
Results
Simulated and real datasets were separately tested to evaluate and validate the effectiveness of MSCNV. In the simulated datasets, MSCNV was compared with five other methods: Manta, FREEC, GROM-RD, Rsicnv, and CNVkit. Metrics such as sensitivity, precision, F1-score, and boundary bias were used to evaluate the effectiveness of these six methods. In addition, the run time consumption of these methods was analyzed and the comparison results are shown in Supplementary Table S1. MSCNV method was tested on seven real samples obtained from the 1000 Genomes Project and NCBI database. The effectiveness and usability of MSCNV for the seven real samples were validated using the F1-score and the overlap density score (ODS)12, 33, 42. During the experiments, different values of parameters bin and kernel will impact the results. A detailed discussion is provided in Supplementary Figs. F1 and F2.
Simulated data experiments
The simulated datasets were generated by SInC41 and seqtk (https://github.com/lh3/seqtk) software. The SInC software determines the sequencing coverage (SC) of the simulated data, while the seqtk software determines the tumor purity (TP). The chromosome 21 sequence from the hg38 version was chosen as the reference genome, with SC set at 20X, 30X, and 40X, respectively. To reduce experimental variability, 30 simulation samples were generated for each group. Within each simulation sample, there were 20 CNV regions, comprising 7 tandem duplications, 7 interspersed duplications, and 6 losses. The size of CNV regions ranged from 1 kb to 9 kb for each CNV region.
This study compared MSCNV with five other methods, Manta, FREEC, GROM-RD, Rsicnv, and CNVkit. The comparison includes sensitivity, precision, F1-score, and boundary bias. Sensitivity is the ratio of the number of correctly detected CNVs to the number of CNVs in the ground truth file, as shown in Eq. (13). The precision is the ratio of the number of correctly detected CNVs to the number of CNVs detected by a method, as shown in Eq. (14). The F1-score is a harmonized average of sensitivity and precision, defined as the ratio of twice the product of sensitivity and precision to the sum of sensitivity and precision, as shown in Eq. (15). The boundary bias is defined as the deviation of the boundary of CNVs detected at the nucleotide level from the boundary of actual CNVs, as shown in Eq. (16).
where Sen represents the sensitivity detected by the method, TP represents the number of CNVs correctly detected by the method, and GN represents the number of CNVs in the ground truth file.
where Pre represents the precision detected by the method, and MN represents the number of CNVs detected by a method.
where FS represents the F1-score detected by the method.
where BB represents the boundary bias detected by the method, DR represents the boundary of CNVs detected by the method, TR represents the boundary where CNVs truly occur, and n represents the number of consecutive CNVs detected by the algorithm.
To validate the effectiveness, reliability, and stability of MSCNV, a comparison between MSCNV and five other methods is shown in Figs. 3 and 4. Figure 3 shows that MSCNV demonstrates higher efficiency and reliability across all 12 simulated configurations. Regarding precision, MSCNV ranks first in 11 out of the 12 simulated configurations. For sensitivity, MSCNV ranks first in nine simulated configurations, with FREEC ranking first in the remaining cases. Under certain circumstances, a trade-off between precision and sensitivity may arise, wherein enhancing one metric could result in a decline in the other. Therefore, the trade-off between precision and sensitivity can provide a more comprehensive evaluation of the method’s performance. Concerning the F1-score, MSCNV attains the highest value across all simulation configurations and delivers the optimal performance, followed by FREEC. Figure 4 demonstrates the strong stability of MSCNV. The F1-score distribution of the MSCNV method is unimodal, with the majority of scores concentrated around 0.94. In contrast, the F1-score distributions of the other five methods are multi-modal, and the peak values are all lower than MSCNV. In addition, by looking at the middle line of the violin plot, it is clear that the MSCNV method has a significantly higher median than the other five methods. This suggests that MSCNV has stronger stability and better detection ability.

The effectiveness of MSCNV is evaluated against that of the other five methods on 12 sets of simulated data. Gray curves represent the F1-score. The equations to the left and right of the comma indicate the sequencing coverage (SC) and the tumor purity (TP), respectively.

Simulation data F1-score violin plots of different algorithms under different configurations.
To further evaluate the performance of MSCNV method. Non-parametric tests were conducted on the F1-scores, and the tests were two-tailed. The F1-scores were not normally distributed. The results are shown in Tables 1, 2, 3, 4 and 5. Where, M represents the median of F1-socres and QR represents the quartile distance of F1-scores, with a P-value < 0.05 being considered statistically significant. Based on the data presented in Tables 1, 2, 3, 4 and 5, the F1-scores of the MSCNV consistently outperform the comparison methods (Manta, FREEC, GROM-RD, Rsicnv, and CNVkit) in all configurations. Furthermore, a significant difference in the F1-score between the MSCNV and the comparison method was observed in all configurations (P < 0.05). These findings demonstrate the clear advantages of the MSCNV in CNV detection tasks. Moreover, the MSCNV exhibits superior and more robust detection performance.
The detection of nucleotide-level breakpoints is particularly challenging in CNV studies due to the presence of multiple breakpoints in repeat and complex regions of the genome. Figure 5 shows the boundary bias for all four methods (the boundary bias of Rsicnv and CNVkit is large, which significantly differs from the other four methods). Studies have shown a positive correlation between a smaller boundary bias and higher accuracy in CNV detection methods. In various simulation configurations, Manta demonstrates high accuracy with minimal boundary bias. In 8 out of 12 simulated configurations, MSCNV ranks second, while GROM-RD obtains the second position in the remaining configurations. This phenomenon may be attributed to the involvement of complex genomic structural changes in SV methods, which enable them to capture breakpoint locations more accurately. On the contrary, CNV methods primarily focus on detecting copy number loss or duplication, which may result in relatively crude boundary breakpoint detection.

Comparison of the boundary biases of MSCNV and three other methods on 12 sets of simulation datasets.
To demonstrate the accuracy of MSCNV in exploring tandem duplication, interspersed duplication, and loss, the distribution of MSCNV exploration under 12 simulation configurations is shown in Fig. 6. ID_ALL represents the true number of interspersed duplications. The ID represents the number of interspersed duplications correctly detected by MSCNV. TD_ALL represents the true number of tandem duplications. The TD represents the number of tandem duplications correctly detected by MSCNV. loss_ALL represents the true number of losses. The loss represents the number of losses correctly detected by MSCNV. Increasing SC and TP leads to improved accuracy of the MSCNV method in detecting different variant types. MSCNV consistently identifies all variations and their types accurately on data with medium to high tumor purity (≥ 0.6). In terms of interspersed duplication, MSCNV achieved almost 100% accuracy in all the simulated configurations (except for a sequencing coverage of 20X and a tumor purity of 0.2). In terms of tandem duplication, MSCNV achieved almost 100% accuracy in all the simulated configurations (except for a sequencing coverage of 20X with a tumor purity of 0.2 and a sequencing coverage of 30X with a tumor purity of 0.2). Regarding losses, MSCNV achieves an average detection accuracy of 100% when TC is greater than or equal to 0.6. For loss, the average detection accuracy of MSCNV is 79.17%. This may be because the difference between the loss area signal and the normal area signal is so small that it can easily be missed.

Detection results of MSCNV under different configurations.
In terms of F1-score, MSCNV demonstrates outstanding performance across all datasets. This means that MSCNV achieves a good balance between sensitivity and precision, with high effectiveness, reliability, and stability. In terms of boundary bias, MSCNV achieves excellent performance in detecting CNVs. Moreover, MSCNV proves adept at accurately identifying tandem duplications, interspersed duplications, and deletions. Based on the simulated data, MSCNV outperforms the other five methods in terms of performance and effectiveness.
Applications to real datasets
To verify the efficacy of MSCNV, two families from the 1000 Genomes Project and a real sample from the NCBI database were analyzed. The data can be downloaded from the respective websites: http://www.1000genomes.org and https://ftp.ncbi.nlm.nih.gov. Specifically, the two families in the 1000 Genomes Project consisted of the YRI trio of the Yoruba Nigerian ethnicity (NA19238, NA19239, and NA19240) and the CEU trio of European ancestry (NA12891, NA12892, and NA12878). The real sample (HG002) in the NCBI database originated from a Jewish family.
Analysis of samples from the 1000 genomes project
This study utilized six real samples from the 1000 Genomes Project to conduct ten tests on chromosome 21, and their average performance was analyzed. The results were compared with three commonly used single-sample-based methods (FREEC, GROM-RD, and CNVkit). The ground truth CNVs for these samples, available in the DGV database (http://dgv.tcag.ca/), were utilized to calculate the sensitivity, precision, and F1-score of each method on real data, as depicted in Fig. 7. Among the six real samples, MSCNV method performs best in terms of F1-score. Regarding precision, MSCNV achieves the highest precision among these six real samples. Concerning sensitivity, CNVkit exhibits the highest sensitivity for NA19238, NA19239, NA19240, and NA12891, while FREEC demonstrates the highest sensitivity for the remaining two real samples. However, GROM-RD exhibits moderate performance in NA19238 and NA19239, while performing poorly in the remaining four samples.

The effectiveness of MSCNV is evaluated against that of the other three methods on six sets of real data.
Moreover, the CNVs of the samples reported in the DGV database may not be complete. Consequently, we employed the ODS value to assess the degree of overlap among different methods. The ODS can be expressed by Eq. (17)12, 33, 42, and the corresponding comparison results are presented in Table 6. MSCNV achieved the highest ODS value. However, GROM-RD showed poor performance in some samples. This is likely due to the presence of complex genomic structures or variation profiles in these samples, which surpass the analytical capabilities of the GROM-RD method. Take Na19240 and NA12892 as examples, the GROM-RD could not accurately detect known variants. This may have resulted in the risk of genetic disorders or drug reactions being incorrectly assessed or missed in patients in the clinical setting. In contrast, the MSCNV method demonstrates superior performance in detecting variants in real data samples. Therefore, the MSCNV method was proposed for more accurate variant detection, ensuring that patients receive precise diagnoses and appropriate treatment.
where Meancov represents the average number of overlaps between one method and the other methods, and Ratiocov represents the ratio of the average number of overlaps between one method and the other methods to the total number of variants it detects.
Analysis of samples from the NCBI database
The son (HG002) from the Ashkenazim Jewish (AJ) trio in the NCBI database was selected for analysis. We analyzed 22 autosome chromosomes. The ground truth for HG002 can be obtained from Genome in a Bottle. MSCNV was compared with four other methods: Rsicnv, FREEC, GROM-RD, and CNVkit, as illustrated in Fig. 8. MSCNV exhibited the best performance in terms of the F1-score, achieving the highest value. This shows that MSCNV can provide relatively accurate results. Specifically, MSCNV achieved the highest F1-score, 27% higher (36% versus 9%), 10% higher (36% versus 26%), 1% higher (36% versus 35%), and 12% higher (36% versus 24%) than Rsicnv, FREEC, CNVkit, and GROM-RD, respectively. In addition, the sensitivity of MSCNV was 23% greater than that of the second-ranked method (49% versus 26%). This shows that MSCNV can effectively detect CNVs. Nevertheless, the precision and sensitivity of each method are not exceptionally high for the whole-genome data. This is because there are a significant number of gene families, repetitive sequences, and types of variations in the whole-genome data. These complexities can make it challenging for methods to accurately identify and distinguish CNVs, thereby reducing the accuracy of detection43.

The effectiveness of MSCNV is evaluated against that of the other four methods on HG002.
In conclusion, MSCNV exhibits a stable detection ability in real datasets. MSCNV not only had the best F1-score and ODS in the real samples from the 1000 Genomes Project but also had the highest sensitivity and F1-score in the real samples from the NCBI database. The effectiveness of MSCNV can be attributed to the integration of three strategies, namely RD, SR, and RP, for CNV detection. The combination of these strategies leads to a significant improvement in both the sensitivity and precision of CNV detection. Additionally, MSCNV incorporates the OCSVM model, well-suited to identify anomalies in nonlinear distributions, further enhancing the sensitivity and stability of CNV detection. MSCNV applies machine learning algorithms to CNV detection while combining multiple strategies to make it more suitable for detecting high-coverage data. These findings affirm the reliability and efficacy of MSCNV in detecting CNV in real datasets, making it an invaluable tool for research and practical applications in related fields.
Discussion
To expand the detection types of CNV variation and improve the accuracy of CNV detection, this study proposes a multi-strategies hybrid CNV detection method based on OCSVM. The method achieves rough CNV by using OCSVM; improves the accuracy of CNV detection by using discordant reads; and explores tandem duplication regions, interspersed duplication regions, and loss regions by using SR signals, which predicts CNVs and accurately locates variant locations.
Compared with traditional methods, MSCNV exhibits two novel characteristics: (1) Considering the uniqueness of CNV features and establishing a multi-signals channel, the CNV regions detected by RD and MQ signals based on OCSVM model are filtered employing the RP signals, improving the accuracy of CNV detection. SR signals are also used to search near the boundary of the variation region, update the variant boundary, reduce the boundary deviation of the CNV region, and finally determine the precise location of the variant point. (2) Expanding the types of CNV detection and achieving the application of tandem duplication variants and interspersed duplication variants in CNV. The tandem duplication region and interspersed duplication region are explored by analyzing the matching information between the testing sample and the reference sequence. As a result, tandem duplication regions, interspersed duplication regions, and loss regions are identified.
In this study, MSCNV was compared with five other methods in terms of sensitivity, precision, F1-score, boundary bias, and overlap density score by testing 360 simulated datasets and seven real datasets. The experimental results demonstrate that MSCNV achieves a favorable balance between precision and sensitivity, significantly enhancing the sensitivity, precision, F1-score, and ODS of CNV detection, while reducing the boundary bias of the results.
In the future, we will continue to extend the types of variation detection of MSCNV method. We will consider the effects of other signals on the variation region and explore more effective features to enable single nucleotide polymorphism variation detection in CNVs (e.g., GC content). Furthermore, an intelligent optimization algorithm can be adopted to improve the precision and recall of the algorithm. Intelligent optimization algorithms, such as genetic algorithm or particle swarm optimization, have shown great potential in optimizing complex processes and decision-making. Fine-tuning the parameters of intelligent optimization algorithms can improve the accuracy of variant calling. However, analyzing multiple signals and incorporating them into CNV detection will increase the computational complexity of the algorithm. This may require significant computational resources and efficient algorithms to handle the increased complexity.
Although extending CNV detection types and incorporating intelligent optimization algorithms present challenges, the potential benefits of more accurate and comprehensive CNV detection are significant. This will lead to advancements in our understanding of genetic variations and their impact on various diseases and traits.
Data availability
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.
References
Li, J. et al. Genomic Copy Number Variation Study of Nine Macaca Species provides New insights into their genetic divergence, adaptation, and Biomedical Application. Genome Biol. Evol. 12, 2211–2230. https://doi.org/10.1093/gbe/evaa200 (2020).
Xie, K. et al. IhybCNV: an intra-hybrid approach for CNV detection from next-generation sequencing data. Digit. Signal. Process. 121, 103304 (2021).
Dharanipragada, P., Vogeti, S., Parekh, N. & iCopyDAV Integrated platform for copy number variations-Detection, annotation and visualization. PLoS One. 13, e0195334. https://doi.org/10.1371/journal.pone.0195334 (2018).
Neveling, K. et al. Next-generation cytogenetics: Comprehensive assessment of 52 hematological malignancy genomes by optical genome mapping. Am. J. Hum. Genet. 108, 1423–1435. https://doi.org/10.1016/j.ajhg.2021.06.001 (2021).
Pinto, D. et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466, 368–372. https://doi.org/10.1038/nature09146 (2010).
Soemedi, R. et al. Contribution of global rare copy-number variants to the risk of sporadic congenital heart disease. Am. J. Hum. Genet. 91, 489–501. https://doi.org/10.1016/j.ajhg.2012.08.003 (2012).
Zare, F., Dow, M., Monteleone, N., Hosny, A. & Nabavi, S. An evaluation of copy number variation detection tools for cancer using whole exome sequencing data. BMC Bioinform. 18, 286. https://doi.org/10.1186/s12859-017-1705-x (2017).
Zhang, T. et al. CNV-PCC: an efficient method for detecting copy number variations from next-generation sequencing data. Front. Bioeng. Biotechnol. 10, 1000638. https://doi.org/10.3389/fbioe.2022.1000638 (2022).
Miller, C. A., Hampton, O., Coarfa, C. & Milosavljevic, A. ReadDepth: a parallel R package for detecting copy number alterations from short sequencing reads. PLoS One. 6, e16327. https://doi.org/10.1371/journal.pone.0016327 (2011).
Smith, S. D., Kawash, J. K. & Grigoriev, A. GROM-RD: resolving genomic biases to improve read depth detection of copy number variants. PeerJ 3, e836. https://doi.org/10.7717/peerj.836 (2015).
Boeva, V. et al. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 28, 423–425. https://doi.org/10.1093/bioinformatics/btr670 (2012).
Liu, G., Zhang, J., Yuan, X. & Wei, C. R. K. D. O. S. C. N. V. A local Kernel density-based Approach to the detection of Copy Number variations by using next-generation sequencing data. Front. Genet. 11, 569227. https://doi.org/10.3389/fgene.2020.569227 (2020).
Abyzov, A., Urban, A. E., Snyder, M. & Gerstein, M. CNVnator: an approach to discover, genotype, and characterize typical and atypical CNVs from family and population genome sequencing. Genome Res. 21, 974–984. https://doi.org/10.1101/gr.114876.110 (2011).
Vardhanabhuti, S., Jeng, X. J., Wu, Y. & Li, H. Parametric modeling of whole-genome sequencing data for CNV identification. Biostatistics (Oxford England). 15, 427–441. https://doi.org/10.1093/biostatistics/kxt060 (2014).
Mohiyuddin, M. et al. MetaSV: an accurate and integrative structural-variant caller for next generation sequencing. Bioinformatics 31, 2741–2744. https://doi.org/10.1093/bioinformatics/btv204 (2015).
Banuelos, M., Almanza, R., Adhikari, L., Mareia, R. F. & Sindi, S. in 2016 IEEE Statistical Signal Processing Workshop (SSP). 1–5.
Guan, P. & Sung, W. K. Structural variation detection using next-generation sequencing data: a comparative technical review. Methods 102, 36–49. https://doi.org/10.1016/j.ymeth.2016.01.020 (2016).
Handsaker, R. E., Korn, J. M., Nemesh, J. & McCarroll, S. A. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat. Genet. 43, 269–276. https://doi.org/10.1038/ng.768 (2011).
Rausch, T. et al. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28, i333–i339. https://doi.org/10.1093/bioinformatics/bts378 (2012).
Layer, R. M., Chiang, C., Quinlan, A. R. & Hall, I. M. LUMPY: a probabilistic framework for structural variant discovery. Genome Biol 15, R84, (2014). https://doi.org/10.1186/gb-2014-15-6-r84
Chen, X. et al. Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. https://doi.org/10.1093/bioinformatics/btv710 (2016).
Liu, G., Yang, H. & Yuan, X. A shortest path-based approach for copy number variation detection from next-generation sequencing data. Front. Genet. 13, 1084974. https://doi.org/10.3389/fgene.2022.1084974 (2022).
Wang, X. et al. PEcnv: accurate and efficient detection of copy number variations of various lengths. Brief. Bioinform. 23 https://doi.org/10.1093/bib/bbac375 (2022).
Ma, J. & Perkins, S. in Proceedings of the International Joint Conference on Neural Networks, 1741–1745 vol.1743. (2003).
Li, Y. et al. SM-RCNV: a statistical method to detect recurrent copy number variations in sequenced samples. Genes Genomics. 41, 529–536. https://doi.org/10.1007/s13258-019-00788-9 (2019).
Walter, V., Nobel, A. B. & Wright, F. A. DiNAMIC: a method to identify recurrent DNA copy number aberrations in tumors. Bioinformatics 27, 678–685. https://doi.org/10.1093/bioinformatics/btq717 (2011).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. https://doi.org/10.1093/bioinformatics/btp324 (2009).
Li, H. et al. The sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. https://doi.org/10.1093/bioinformatics/btp352 (2009).
Dong, J., Qi, M., Wang, S. & Yuan, X. D. I. N. T. D. Detection and inference of Tandem duplications from short sequencing reads. Front. Genet. 11, 924. https://doi.org/10.3389/fgene.2020.00924 (2020).
Yuan, X. et al. Detecting Copy Number Variation and genotyping deletion zygosity from single tumor samples using sequence data. IEEE/ACM Trans. Comput. Biol. Bioinform. 17, 1141–1153. https://doi.org/10.1109/tcbb.2018.2883333 (2020).
Yuan, T. et al. DTDHM: detection of tandem duplications based on hybrid methods using next-generation sequencing data. PeerJ 12, e17748. https://doi.org/10.7717/peerj.17748 (2024).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255. https://doi.org/10.1093/bioinformatics/btz891 (2020).
Yuan, X. et al. CNV_IFTV: an isolation forest and total variation-based detection of CNVs from short-read sequencing data. IEEE/ACM Trans. Comput. Biol. Bioinform. 18, 539–549. https://doi.org/10.1109/tcbb.2019.2920889 (2021).
Dohm, J. C., Lottaz, C., Borodina, T. & Himmelbauer, H. Substantial biases in ultra-short read data sets from high-throughput DNA sequencing. Nucleic Acids Res. 36, e105. https://doi.org/10.1093/nar/gkn425 (2008).
Yoon, S., Xuan, Z., Makarov, V., Ye, K. & Sebat, J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 19, 1586–1592. https://doi.org/10.1101/gr.092981.109 (2009).
Rudin, L. I., Osher, S. & Fatemi, E. Nonlinear total variation based noise removal algorithms. Phys. D: Nonlinear Phenom. 60, 259–268 (1992).
Duan, J., Zhang, J. G., Deng, H. W. & Wang, Y. P. CNV-TV: a robust method to discover copy number variation from short sequencing reads. BMC Bioinform. 14, 150. https://doi.org/10.1186/1471-2105-14-150 (2013).
Condat, L. A direct algorithm for 1-D total variation denoising. IEEE. Signal. Process. Lett. 1054–1057. https://doi.org/10.1109/lsp.2013.2278339 (2013).
Pu, G., Wang, L., Shen, J. & Dong, F. A hybrid unsupervised clustering-based anomaly detection method. Tsinghua Sci. Technol. 26, 146–153. https://doi.org/10.26599/TST.2019.9010051 (2021).
Brereton, R. G. & Lloyd, G. R. Support vector machines for classification and regression. Analyst 135, 230–267. https://doi.org/10.1039/b918972f (2010).
Pattnaik, S., Gupta, S., Rao, A. A. & Panda, B. SInC: an accurate and fast error-model based simulator for SNPs, indels and CNVs coupled with a read generator for short-read sequence data. BMC Bioinform. 15, 40. https://doi.org/10.1186/1471-2105-15-40 (2014).
Jia, D., Dong, J., Jiang, H., Zhao, Z. & Jiang, X. TD-COF: a new method for detecting tandem duplications in next generation sequencing data. SoftwareX 27, 101881 (2024).
Hyman, S. E. Use of mouse models to investigate the contributions of CNVs associated with schizophrenia and autism to disease mechanisms. Curr. Opin. Genet. Dev. 68, 99–105. https://doi.org/10.1016/j.gde.2021.03.004 (2021).
Author information
Authors and Affiliations
Contributions
Mengjiao zhou and Jinxin Dong wrote the main manuscript text and Zuyao Zhao invetigated the study. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhou, M., Dong, J., Jiang, H. et al. A copy number variation detection method based on OCSVM algorithm using multi strategies integration. Sci Rep 15, 3526 (2025). https://doi.org/10.1038/s41598-025-88143-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-88143-9
