
Abstract
Aroideae is an important subfamily of the Araceae family and contains many plants with medicinal and edible value. It is difficult to identify and classify Aroideae species accurately on the basis of morphology alone because of their polymorphic phenotypic traits. The chloroplast genome (CPG) is useful for studying on plant taxonomy and phylogeny, and the analysis of codon usage bias (CUB) in CPGs provides further insights into the intricate phylogenetic relationships among Aroideae. The results showed that the codon third position of the chloroplast genome coding sequence in Aroideae was rich in A and T, with a GC content of 37.91%. The ENC-plot and PR2-plot revealed that the codon usage bias of Aroideae was influenced by multiple factors, with natural selection as the dominant factor. Thirteen to twenty optimal codons ending in A/T were identified in 61 Aroideae species. Additionally, the comparative analysis of CPGs revealed that two single copy regions and non-coding regions were variable in Aroideae. Eight highly divergent regions (Pi > 0.064) were identified (ndhF, rpl32, ccsA, ndhE, ndhG, ndhF-rpl32, ccsA-ndhD, and ndhE-ndhG) , in which ndhE have the potential to serve as a reliable DNA marker to discriminate chloroplasts in Aroideae subfamily. Furthermore, the maximum likelihood-based phylogenetic trees constructed from complete chloroplast genomes and protein-coding sequences presented similar topologies. Principal component clustering analysis based on relative synonymous codon usage values (RSCUs) revealed that Calla was clearly deviated from Montrichardia and Anubias, and that Alocasia was closer to Colocasieae than to Arisaemateae. These findings suggest that the use of RSCU for clustering analysis could offer new theoretical support for species classification and evolution. Our research could provide a theoretical foundation for the chloroplast genetic engineering, taxonomy, and phylogenetic relationships of Aroideae chloroplasts.
Similar content being viewed by others

Introduction
A Codon is a continuous three-base sequence on the messenger RNA chain that determines an amino acid. It plays a crucial role in the transmission of genetic information from messenger RNA to proteins in organisms1. In eukaryotes, there are 61 different codons encoding 20 amino acids2. All amino acids except tyrosine and methionine have multiple corresponding codons, known as synonymous codons. There is variation in the frequency of synonymous codon usage among species, which is recognised as codon usage bias (CUB)3. This bias significantly affects the efficiency of protein translation, with genes encoded by optimal codons tending to be highly expressed or having polymorphic sites, thereby maintaining genetic information stability and optimising function during evolution4,5. Codon usage bias is generally considered a comprehensive response to the drift of non-synonymous codon mutations and the selection pressure of optimal codons6. In previous studies, this bias was also influenced by multiple factors such as nucleotide composition, GC content7, gene expression levels8, and transfer RNA abundance9. Codon usage patterns vary among different species, but closely related species often share similar biases10. Therefore, studying codon usage patterns in plants helps reveal the adaptability of species to their environment and offers perspectives on gene expression regulation and species evolution2,11,12.
Chloroplasts are semi-autonomous organelles and an energy conversion systems unique to higher plants and algae, and play important roles in photosynthesis, biosynthesis, and carbon sequestration13. Since the chloroplast genome (CPG) sequences of tobacco14 and liverwort15 were first released in 1986, their structures and functions have received widespread attention. These sequences are of moderate length and are rich in genetic information16. In contrast to mitochondrial and nuclear genomes, CPGs are widely applied in molecular ecology and evolutionary studies because of their moderate nucleic acid substitution rates, conserved genome structures and gene composition, and the absence of paralogous homologous interference17,18,19. The study of codon usage patterns in CPGs serves as a fundamental step in understanding chloroplast function, while analysing base bias provides insights into genetic modification and transgenic systems of the chloroplast genome20.
Araceae, a large family with 144 accepted genera and 3645 species worldwide, is rich in resources and widely distributed21. Aroideae is the largest subfamily among Araceae, and is known for the significant medicinal and edible value of its plants. Pinellia ternata and Arisaema erubescens have been documented in the classic medical tome “Shennong Bencao Jing” of the Western Han Dynasty in China. Most of species in Aroideae have pharmacological actions including resolving phlegm, anti-inflammatory, and antitumour properties22,23. Furthermore, the bulb of Amorphophallus konjac has abundant glucomannan components, increasing its potential applications in the fields of medicine, food, and the chemical industry24. As a plant rich in starch and cellulose, Colocasia esculenta is considered an important food crop in some regions and plays a key role in local agricultural production and the food supply25.
In recent years, significant progress has been made in the research on the CPG of Aroideae. The CPGs of Arisaema erubescens and Pinellia ternata were published by Zhang et al.26 and Cai et al.27, respectively, and the phylogenetic analysis revealed that Arisaema and Pinellia were most closely related. The CPG of eight species in Dracunculus clade were published in 2021, further highlighting the molecular evolution of Aroideae28. The CPGs of several Amorphophallus species, including A. konjac29, A. yunnanensis30, and A. coaetaneus31, have been assembled and characterised. Our previous research published the CPG of three famous Aroideae species and conducted comparative analyses among 17 Aroideae species32. Currently, the NCBI database (https://www.ncbi.nlm.nih.gov/) contains CPG data for 61 Aroideae species. However, the codon usage bias and the phylogenetic relationships of these species remain unclear. In this study, the codon usage patterns of 61 Aroideae species were analysed by using their CPGs and the base composition, influencing factors, and optimal codons were identified. Furthermore, phylogenetic trees were constructed based on complete chloroplast genomes and protein-coding sequences, as well as a principal component clustering analysis (PCA) based on relative synonymous codon usage values, providing new insights into the evolution of Aroideae.
Results
Statistics of the chloroplast sequence data and codon composition analysis
The CPGs of 61 Aroideae species belonging to 15 tribes were downloaded from NCBI (https://www.ncbi.nlm.nih.gov/) (Supplementary Table 1). The CPG sizes of the 61 Aroideae species ranged from 158,177 bp (Anchomanes hookeri) to 177,076 bp (Amorphophallus muelleri). Several genes such as psbC, rpl16, cemA, and ycf68 are absent in some species, whereas others, such as rps16, ndhD, psbI, and atpH are found to be shorter than 300 bp. A total of 3,097 protein-coding genes were screened in the CPG of 61 Aroideae species, including 1,512 photosynthesis-related genes, 786 ribosomal protein-related genes, 244 self-replicating-related genes, 270 ycf genes and 291 other genes (Supplementary Table 2). According to functional classification, photosynthesis-related genes presented the highest values of total GC content (39.28%), and relatively high optimal codon frequency (FOP) and codon adaptation index (CAI) values, which suggested that these genes may be highly expressed. The effective number of codons (ENC) values of the chloroplast genes ranged from 37.17–59.64, with ENC values of rps18 and rps8 being less than 40, suggesting greater codon bias and greater stability for these two genes.
The codon usage patterns of the 61 Aroideae species were different (Fig. 1). Thomsonieae (tribe 2) and Arisaemateae (tribe 4) presented greater variation in codon numbers and GC values among the 15 tribes. Notably, Arisaema prazeri, Arisaema heterophyllum and Arisaema ringens had relatively low codon numbers, whereas Arisaema ringens, Arisaema prazeri, and Caladium bicolor had relatively high GC values. The GC contents at the first (GC1), second (GC2), and third (GC3) positions of the codon were 45.51% ~ 46.98%, 37.29% ~ 39.18% and 28.47% ~ 32.36% respectively, suggesting a preference for A/T bases over G/C bases in the codons of Aroideae. Moreover, the GC content at three codon positions followed the pattern GC1 > GC2 > GC3. The average GC content of the 61 Aroideae species was 37.91%. The AT content at the 3rd position of the codon was greater than the GC content, indicating a strong bias for codons ending with A/T bases (Supplementary Table 3).

Number of codons used, GC content, GC1, GC2, and GC3 analyses in Aroideae. X-axis: Different coloured blocks represent different tribes, and numbers 1–15 represent the corresponding 15 tribes. Y-axis: From top to bottom, it represents the number of codons used, the total GC content, and the GC content at the three positions of the codons.
Correlation analysis of chloroplast codon bias parameters in Aroideae
To explore the potential associations among codon composition, codon bias, and gene expression, a Pearson correlation analysis was performed using the values of codon-related parameters of the chloroplast genes in Aroideae (Fig. 2). The results revealed that the GC content was positively correlated with both GC12 (p < 0.0001, r = 0.92) and GC3 (p < 0.0001, r = 0.57), but the correlation between GC12 and GC3 was insignificant, suggesting that the GC contents at the three codon positions in the CPG of Aroideae were different. GC3 was positively correlated with ENC (p < 0.001, r = 0.41), suggesting that the 3rd base of the codon might influence the codon usage bias. As a measure of bias of preferred codons in highly expressed genes, the CAI was observed to be positively correlated with FOP (p < 0.0001, r = 0.7) and GC (p < 0.01, r = 0.3), suggesting that the frequency usage of optimal codon and GC contents was related to gene expression. Aromo was positively correlated with T3s (p < 0.01, r = 0.4), indicating that codons encoding aromatic amino acids prefer to end with T. Additionally, Gravy was positively correlated with T3s (p < 0.05, r = 0.37) and negatively correlated with A3s (p < 0.05, r = -0.4), suggesting that the hydrophobicity of proteins might be influenced by whether the 3rd position of the codon was A or T.

Correlation analysis of codon indices of chloroplast genes in Aroideae. A3s, T3s, G3s, C3s, composition at the third synonymous codon position; GC, overall GC content; GC3, GC content at the third position of the codons; GC12, average G/C contents of the 1st and 2nd positions of codons; CAI, codon adaptation index; CBI, codon bias index; FOP, optimal codon frequency; ENC, effective number of codons; Gravy, general average hydropathicity; Aromo, frequency of aromatic amino acids. *Significant at p < 0.05 (two-tailed); **Significant at p < 0.01 (two-tailed); ***Significant at p < 0.001 (two-tailed); ****Significant at p < 0.0001 (two-tailed) .
Analysis of factors influencing codon bias
The ENC-GC3 plot and the frequency distribution of ENC ratios are effective tools for analysing the factors affecting the codon usage bias of Aroideae. In the ENC-GC3 plots, the chloroplast genes of 61 Aroideae species deviated a deviation from the ENCexp standard curve (Fig. 3). Most genes, such as rps18, psbA, rpl16, and atpF were located in the lower region of the plot, indicating that the bias of these genes was influenced by natural selection. Only a few genes, such as psaA, ycf3, ycf68, and ndhE were located above the standard curve, indicating that mutations were the dominant factor leading to codon bias. In addition, the frequency distribution table of the ENC ratios revealed that 46.76% of the chloroplast genes fell within the range of -0.05–0.05, whereas 53.24% of the genes were outside this range (Supplementary Table 4). This further indicated that natural selection played a key role in the codon bias of most genes in Aroideae.

ENC-GC3 plots of chloroplast gene codons in 61 Aroideae species.
The PR2-plot analysis can reveal the usage frequency of A/T and C/G at the third codon position. If genes cluster in the centre of the PR2-plot plane, it indicates that the frequencies of A/T and G/C bases are similar, and the mutations are solely responsible for the codon bias. The distribution of chloroplast genes in Aroideae on the plane was uneven, with a majority of the genes distributed at the bottom of the plane (Fig. 4). These results suggested that the CPG of Aroideae preferred to use A/T bases, and the biased base usage implied that the codon usage patterns were more influenced by natural selection.

PR2 plots of chloroplast gene codons in 61 Aroideae species.
High frequency and optimal codons
A total of 30 codons with RSCU > 1 were identified in the CPG of 61 Aroideae species, and 29 codons (96.7%) ended in A/T with the exception of UUG. Conversely, the number of codons with RSCU < 1 was 32, and 90.6% of these ended in C/G. This result suggested that high-frequency synonymous codons (RSCU > 1) ended in A/T, whereas low-frequency synonymous codons (RSCU < 1) tended to end in C/G (Supplementary Table 5). A codon heatmap was generated using the RSCU values of 64 synonymous codons (Fig. 5). The codon colours in the map were similar across different species, suggesting that the RSCU values were stable in Aroideae, with a conservative evolutionary process. Based on the ENC value, codons that fulfilled the criteria of RSCU > 1 and △RSCU ≥ 0.08 were screened out (Fig. 6). The results revealed that the number of optimal codons for the CPG of 61 Aroideae species ranged from 13 (Alocasia navicularis) to 20 (Amorphophallus paeoniifolius). AGT(59),ATT(53),and TTA(55) were identified as optimal codons in more than 50 species, and CGT and TTT were found to be the optimal codons in 60 and 61 species, respectively.

Heatmap of the RSCU values of 61 Aroideae species.

Optimal codon analysis in 61 Aroideae species. Light blue background: RSCU > 1 and ΔRSCU ≥ 0.08; While background: RSCU > 1.
Genomic comparative and nucleotide diversity analyses
The divergence and conservation of the CPGs of Aroideae were studied using MultiPipMaker software, with reference to the annotated chloroplast genome sequences of Alocasia fornicata (Fig. 7). The alignments revealed that the large single-copy (LSC) and small single-copy (SSC) regions were more divergent than the inverted repeats (IRs) regions, and the non-coding regions were more divergent than the coding regions.

Structural comparison of the CPGs of 61 Aroideae species. Above the alignment, black arrows and bold black lines depict the orientation of genes, with each colour strip representing a different region: the blue strip for LSC, the orange strip for IRs, and the yellow strip for SSC. Peachblow strips indicate different chloroplast genomes, green bars indicate mismatches, and white bars represent insertions or deletions (indels).
To identify the sequence divergence hotspots in the CPG of Aroideae, DnaSP software was used to calculate nucleotide diversity (Pi) values within a 600 bp window (Fig. 8). The results revealed that the LSC and SSC regions (SCs) were more variable than the IRs regions. A total of eight regions with high variability (Pi > 0.064) were identified in SSC regions: five genes ndhF (0.072), rpl32 (0.068), ccsA (0.066), ndhE (0.064), and ndhG (0.07) and three intergenic regions: ndhF-rpl32 (0.069), ccsA-ndhD (0.065), and ndhE-ndhG (0.064). Four regions trnV-trnM (0.05), trnM-atpE (0.048), accD (0.05), and rpl36-rps8 (0.049), were identified as highly variable regions (Pi > 0.048) in the LSC region. These regions presented high variable sites (VSs), parsimony informative sites (Pins), discrimination success rate based on distance method (DSR), and average K-2P distances (Supplementary Table 6). Moreover, three variable regions (trnV-trnM, ndhE, and ndhF-rpl32) with DSR values higher than 85% were used to construct neighbor-joining phylogenetic trees. The results revealed that the ndhE region could discriminate different chloroplasts in Aroideae subfmily, indicating its potential to be developed as a valuable DNA barcode. Consistent with the results of the genomic alignments, the SC regions were more divergent than IRs regions. This could be attributed to a higher selective pressure during the evolution of SC regions, resulting in the accumulation of more .mutations.

Nucleotide diversity (Pi) analysis of the CPGs of 61 Aroideae species. Window length: 600 bp; step size: 200 bp.
Phylogenetic analysis
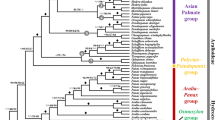
To investigate the phylogenetic relationships in Aroideae, we used the maximum likelihood method to construct chloroplast phylogenetic trees on the basis of CPGs and protein-coding sequences (CDSs) (Fig. 9). Lemna minor was used as an outgroup for both trees. The topologies of the two chloroplast phylogenies were similar, with most nodes showing high support values. In the Aroideae clade, three distinct clades (Zantedeschia, Amorphophallus and Ambrosina clade) could be identified and distinguished, and the positions of some species on the phylogenetic trees showed differed.

Phylogenetic analysis of 61 Aroideae species. Lemna minor was used as an outgroup. (a) phylogenetic tree based on filtered CDS; (b) phylogenetic tree based on CPG.
In the phylogenetic tree constructed from the CDS, Montrichardia arborescens was sister to Anubias heterophylla in Zantedeschia clade, and Calla palustris did not form a cluster with any other species (Fig. 9a). However, in the tree based on the CPG, Anubias heterophylla, Montrichardia arborescens, and C. palustris were grouped together (Fig. 9b). The branch of Alocasia, which includes with Alocasia fornicata , Alocasia navicularis , and Leucocasia gigantea, was clustered in the Arisaemateae clade in the tree based on CDS, but was clustered in Colocasieae clade in the tree based on CPG. The Caladieae species and Thomsonieae species were clustered in the Amorphophallus clade on both trees.
To further reveal the evolutionary relationships of Aroideae species, a PCA clustering analysis based on RSCU values was conducted in Zantedeschia, Calla and Ambrosina clades (Fig. 10). The results revealed that Calla palustris was separated from Anubias heterophylla and Montrichardia arborescens (Fig. 10a). In the Ambrosina clade, Alocasia was more closely related to Colocasieae than to the Arisaemateae (Fig. 10b).

PCA clustering analysis based on RSCU values. (a) PCA clustering analysis of species in the Zantedeschia and Calla clades; (b) PCA clustering analysis of species in the Ambrosina clade.
Discussion
In this study, the the codon usage patterns of the chloroplast genome of 61 Aroideae species were analysed according to the above relevant experimental methods. The protein-coding sequences of Aroideae were rich in A or T, with an average GC content of 37.91%. Arisaema ringens, Arisaema prazeri, and Caladium bicolor presented relatively high GC contents. A trend towards increasing GC values from GC3 to GC2 to GC1 was identified in 61 Aroideae species, and similar results were also reported in Aconitum and Juglandaceae species33,34. Correlation analysis revealed that CAI was significantly positively correlated with GC and FOP, suggesting that genes with high GC contents presented greater codon usage bias and higher expression levels. This was consistent with previous research findings35. In the CPG of Aroideae, the highest CAI (0.18) and GC values (39.28%) were found for photosynthesis-related genes, implying that these genes presented strong codon usage bias and were highly expressed. It could be inferred that photosynthesis-related genes may have played crucial roles in the evolutionary process of Aroideae adapting to natural environments.
Codon usage bias results from a combination of multiple factors36. Explaining the causes of this phenomenon in the chloroplast genomes of different species can help to further understand the evolutionary mechanisms of plant37. The analyses of the ENC plot, ENC ratios, and PR2 plot revealed that natural selection contributed the most to the codon usage bias in the CPG of Aroideae. Similar results have been reported in Gynostemma38, Miscanthus39 and Euphorbiaceae40. However, in Medicago truncatula41, mutation is the main reason for bias. In Mesona chinensis42, mutation pressure is relatively balanced with the influence of natural selection.
The RSCU value is a key index for evaluating the degree of codon usage bias. An RSCU that exceeds 1 indicates a high frequency of codon usage and a strong bias 43. Heatmap plotting and cluster analysis of the RSCU revealed that 64 synonymous codons could be divided into two groups: codons ending with G or C and codons ending with A or T. Twenty-nine high-frequency codons (96.67%) were clustered in the group in which codons ending with A or T, suggesting that the chloroplast genes of Aroideae tend to use codons ending with A or T. Furthermore, AGT, ATT, TTA, TTT, and CGT were identified as optimal codons in more than 50 Aroideae species, which follow the “NNA” and “NNT” patterns. Studies have shown that codons with this pattern can effectively improve the efficiency of transcription and translation during gene expression44,45. Therefore, selecting these optimal codons is expected to improve gene expression efficiency in chloroplast genetic engineering46.
Mutation hotspots are regions of the genome that are prone to mutation and play crucial roles in understanding evolutionary mechanisms47. Using MultiPipMaker and nucleotide diversity analysis, eight highly variable regions were identified including five gene regions (ndhF, rpl32, ccsA, ndhE, and ndhG) and three intergenic regions (ndhF-rpl32, ccsA-ndhD, and ndhE-ndhG). The ndhE-ndhG region, for example, has been shown to be a particularly useful marker for predicting phylogeny among related species of Spathiphyllum in the Araceae family48. Moreover, the ndhF-rpl32, ccsA-ndhD, and ndhE-ndhG intergenic regions have been identified as suitable DNA barcodes for species identification and phylogenetic analysis in a range of plant species, including Dracocephalum49, Siraitia Merrill50, Magnolia polytepala51, and Gynopodium52. The ndhF gene region is another noteworthy hypervariable fragment, particularly in Lagerstroemia53, Lirianthe54, and Dalbergia species55. Collectively, these highly variable regions serve as tools for the identification and evaluation of germplasm resources, genetic diversity analysis, and population evolution in Aroideae.
In the analysis of codon usage patterns, an exploration of RSCU values in different species can aid in understanding their evolutionary relationships56,57. The phylogenetic positions of Calla palustris and Alocasia were ambiguous in the chloroplast phylogenies (Fig. 9). In nuclear analysis, Calla palustris, Anubias heterophylla and Montrichardia arborescens were found to form a sister group58, but the support of this relationship in the mitochondrial phylogeny and molecular and morphological data was weak59,60. According to our PCA results based on the RSCU value, Calla clearly deviated from Montrichardia and Anubias, providing an evidence that supports the possibility of independent evolution of the Calla clade and the Zantedeschia clade. Alocasia was more closely related to Colocasieae than to Arisaemateae according to the PCA, and the same results were also found in the previously reported RAxML tree based on mitochondrial sequences and IQ-tree based on CPG61,62. In general, our ML analysis based on the CDS yielded outcomes that were generally consistent with those obtained from CPG. For example, Pinellia species are located most closely to Arisaema, Sauromatum and Typhonium in the Arisaemateae clade63. Thomsonieae were clustered together with Caladieae in Amorphophallus clade64,65. Therefore, the RSCU values of chloroplast protein-coding sequences could play an important role in the study of the phylogenetic relationships and taxonomy of Aroideae species.
Conclusion
In this study, the codon usage patterns of chloroplast genome was analysed in 61 Aroideae species. Our results revealed that Aroideae chloroplast genes preferred codons ending in A or T, and natural selection was the primary force driving codon usage bias. AGT (Ser), ATT (Ile), TTA (Leu), TTT (Phe), and CGT (Arg) were the five optimal codons shared by more than 50 samples. We also identified a highly variable gene region (ndhF, rpl32, ccsA, ndhE and ndhE) that could serve as a reliable DNA barcode for species identification and genetic diversity studies of Aroideae. Furthermore, principal component clustering analysis from RSCU values can help to better understand the phylogenetic relationships among Aroideae species and may serve as a tool in species identification and classification.
Materials and methods
Sequence data
The original protein-coding sequences (CDSs) of chloroplast genomes (CPGs) of 61 Aroideae species were obtained from the National Center for Biotechnology Information (NCBI) (https://www.ncbi.nlm.nih.gov/) on April 15, 2024. To reduce sampling bias and improve the accuracy of the analysis, repeated sequences and sequences less than 300 bp in length were excluded66. The CDS must have both a start codon and a termination codon. Finally, a total of 3,103 eligible coding sequences were screened for subsequent analysis (Supplementary Table 1).
Analysis of the codon usage index
CodonW 1.4.2 software (http://codonw.sourceforge.net/) was used to obtain the codon usage indices, including (1) relative synonymous codon usage (RSCU); (2) effective number of codons (ENC); (3) the codon adaptation index (CAI); (4) the codon bias index (CBI); (5) the total number of amino acids (L_aa); (6) the optimal codon frequency (FOP); (7) the number of synonymous codons (L_sym); (8) the general average hydropathicity (Gravy); (9) the frequency of aromatic amino acids (Aromo); (9) the GC content at the third position of the synonymous codons (GC3s); and (10) the composition at the third synonymous codon position (A3s, T3s, G3s, and C3s). The GC content including the overall GC content (GC), the GC content at the first (GC1), second (GC2) and third codon positions (GC3) was calculated using MEGA-X software38, and the average G/C contents of the 1st and 2nd positions of codons (GC12) were calculated by Excel 2019. SPSS 26.0 software was used for correlation analysis of the above parameters, and the graphs were created using Origin 2022.
ENC plot analysis
The ENC is an effective index for quantifying the degree of synonymous codon bias67. By comparing the expected ENC value with the GC3 value, the ENC-plot can be used to investigate the influence of base composition on the codon usage bias. The ENC-plot was constructed with the GC3 value as the abscissa and the ENC value as the ordinate. The standard curve indicated that the determinant of codon usage bias was mutation pressure, and the formula of the standard curve was as followed: ENC = 2 + GC3s + 29/[GC3s 2 + (1-GC3s)2]66. To accurately evaluate the difference between the observed value (ENCobs) and the expected value (ENCexp), the ENC ratio was calculated using the formula “ENC ratio = (ENCexp—ENCobs)/ENCexp”, and the difference between ENCobs and ENCexp was quantified according to the distribution of ENC ratios68.
Parity rule 2 (PR2) plot analysis
PR2 plot analysis is widely used to explain the influence of mutational pressure and natural selection on the nucleotide composition of double-stranded DNA. With G3s/(G3s + C3s) as the abscissa and A3s/(A3s + T3s) as the ordinate, the centre of the plane is the position where A3s equals T3s and where G3s equals C3s, indicating that there is no mutational pressure or natural selection bias69. If G3s and C3s or A3s and T3s are close, the codon usage bias in the CPG is affected only by mutation pressure; if there is a large difference between G3s and C3s or A3s and T3s, the bias is attributed primarily to natural selection70.
RSCU and optimal codon analysis
Relative synonymous codon usage values (RSCUs) refer to the ratio of the observed frequency of codon usage to the expected frequency under an unbiased usage. A codon with RSCU > 1 is considered a high-frequency codon, RSCU > 2 indicates that the codon is used with extremely high frequency, and RSCU < 1 indicates that the codon is a low-frequency codon43. A heatmap of the average RSCU values for all synonymous codons in the CPGs of 61 Aroideae species was constructed via TBtools v1.10871. Excel 2019 software was used to sort ENC values according to their size, and the genes in the top 10% and bottom 10% of ENC values were chosen to create high-expression gene datasets and low-expression gene datasets. The RSCU values of the two datasets were calculated according to previous research, and the ΔRSCU values were obtained by subtraction43. Codons with a ΔRSCU ≥ 0.08 and RSCU > 1 were defined as optimal codons1.
Genomic variation analysis
The CPGs of 61 Aroideae species were aligned via the Multiple Sequence Alignment Program (MAFFT v. 7 427)72. The online MultiPipMaker software (http://pipmaker.bx.psu.edu/pipmaker/)with default parameters was subsequently used to make alignments among the 61 CPGs of Aroideae, with the annotated chloroplast genome of Alocasia fornicata used as a reference73. To determine the level of nucleotide variability (Pi) within the Aroideae, a sliding window analysis was conducted via the DnaSP v5.10 program with a step size of 200 bp and a window length of 600 bp74. To develop molecular makers for the identification of different chloroplasts in Aroideae, the variable sites (VSs), parsimony informative sites (Pins), discrimination success rate based on distance method (DSR), and average K-2P distance of polymorphism sites were analysed, and the neighbor joining phylogenetic trees were constructed via the divergent regions with DSR values higher than 85%. Statistical analysis was conducted using Excel 2019.
Phylogenetic analysis
To clarify the phylogenetic relationships among the 61 Aroideae species, the maximum likelihood (ML) phylogenetic trees were constructed using the RaxML version which is based on CPGs and filtered CDSs75, and the sequences were aligned with MAFFT v. 7 427. Bootstrap replication was conducted with 1,000 replicates, and the other parameters were set to their defaluts. The phylogenetic trees were visualised using ChiPlot76. Moreover, we also performed principal component analysis (PCA) via Origin 2022,whcih is based on RSCU value, to further investigate the genetic relationships within Aroideae.
Data availability
The datasets of the 61 Aroideae plants analysis are available in the NCBI (https://www.ncbi.nlm.nih.gov/).
References
Liu, H. B., Lu, Y. Z., Lan, B. L. & Xu, J. C. Codon usage by chloroplast gene is bias in Hemiptelea davidii. J. Genet. 99, 8. https://doi.org/10.1007/s12041-019-1167-1 (2020).
Quax, T. E., Claassens, N. J., Söll, D. & van der Oost, J. Codon bias as a means to fine-tune gene expression. Mol. Cell. 59, 149–161. https://doi.org/10.1016/j.molcel.2015.05.035 (2015).
Andargie, M. & Zhu, C. Y. Genome-wide analysis of codon usage in sesame (Sesamum indicum L.). Heliyon. 8, e08687. https://doi.org/10.1016/j.heliyon.2021.e08687 (2021).
Frumkin, I. et al. Codon usage of highly expressed genes affects proteome-wide translation efficiency. Proc. Natl. Acad. Sci. USA 115, E4940–E4949. https://doi.org/10.1073/pnas.1719375115 (2018).
Duan, X. J. et al. High-level expression of codon-optimized Thielavia terrestris cutinase suitable for ester biosynthesis and biodegradation. Int. J. Biol. Macromol. 135, 768–775. https://doi.org/10.1016/j.ijbiomac.2019.05.173 (2019).
Behura, S. K. & Severson, D. W. Codon usage bias: Causative factors, quantification methods and genome-wide patterns: With emphasis on insect genomes. Biol. Rev. Camb. Philos. Soc. 88, 49–61. https://doi.org/10.1111/j.1469-185X.2012.00242.x (2013).
Niu, Y., Luo, Y. Y., Wang, C. L. & Liao, W. B. Deciphering codon usage patterns in genome of cucumis sativus in comparison with nine species of Cucurbitaceae. Agronomy 11, 2289. https://doi.org/10.3390/agronomy11112289 (2021).
Blake, W. J., Kærn, M., Cantor, C. R. & Collins, J. J. Noise in eukaryotic gene expression. Nature 422, 633–637. https://doi.org/10.1038/nature01546 (2003).
Schwark, D. G., Schmitt, M. A., Biddle, W. & Fisk, J. D. The Influence of competing tRNA abundance on translation: Quantifying the efficiency of sense codon reassignment at rarely used codons. Chembiochem 21, 2274–2286. https://doi.org/10.1002/cbic.202000052 (2020).
Christianson, M. L. Codon usage patterns distort phylogenies from or of DNA sequences. Am. J. Bot. 92(8), 1221–1233. https://doi.org/10.3732/ajb.92.8.1221 (2005).
Sharp, P. M., Emery, L. R. & Zeng, K. Forces that influence the evolution of codon bias. Philos. Trans. R. Soc. Lond. B Biol. Sci. 365, 1203–1212. https://doi.org/10.1098/rstb.2009.0305 (2010).
Zhou, J. H. et al. The effect of multiple evolutionary selections on synonymous codon usage of genes in the Mycoplasma bovis genome. PloS one. 9, e108949. https://doi.org/10.1371/journal.pone.0108949 (2014).
Daniell, H., Lin, C. S., Yu, M. & Chang, W. J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 17, 134. https://doi.org/10.1186/s13059-016-1004-2 (2016).
Shinozaki, K. et al. The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J. 5, 2043–2049. https://doi.org/10.1002/j.1460-2075.1986.tb04464.x (1986).
Ohyama, K. et al. Chloroplast gene organization deduced from complete sequence of liverwort Marchantia polymorpha chloroplast DNA. Nature 322, 572–574 (1986).
Raubeson, L. A. et al. Comparative chloroplast genomics: Analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genom. 8, 174. https://doi.org/10.1186/1471-2164-8-174 (2007).
Du, F. K. et al. An improved method for chloroplast genome sequencing in non-model forest tree species. Tree Genet Genom. 11, 114. https://doi.org/10.1007/s11295-015-0942-2 (2015).
Nakandala, U., Furtado, A., Smith, M. W., Williams, D. C. & Henry, R. J. Phylogenetic relationships among Australian native citrus species based upon complete chloroplast genomes and single copy nuclear genes. Trop. Plants. 2, 21. https://doi.org/10.48130/TP-2023-0021 (2023).
Liang, H. et al. Comparative chloroplast genome analysis of Camellia oleifera and C. meiocarpa: Phylogenetic relationships, sequence variation and polymorphic markers. Trop. Plants. 3, e023 (2024).
Scotti, N. et al. High-level expression of the HIV-1 Pr55gag polyprotein in transgenic tobacco chloroplasts. Planta. 229, 1109–1122. https://doi.org/10.1007/s00425-009-0898-2 (2009).
Boyce, P. C. & Croat, T. B. The Überlist of Araceae, Totals for published and estimated number of species in Aroid Genera. http://www.aroid.org/genera/180211uberlist.pdf (2011).
Gao, S. M. et al. Traditional uses, phytochemistry, pharmacology and toxicology of Codonopsis: A review. J. Ethnopharmacol. 219, 50–70. https://doi.org/10.1016/j.jep.2018.02.039 (2018).
Ren, W. et al. Research advance on Qingfei Paidu decoction in prescription principle, mechanism analysis and clinical application. Front. Pharmacol. 11, 589714. https://doi.org/10.3389/fphar.2020.589714 (2021).
Cescutti, P., Campa, C., Delben, F. & Rizzo, R. Structure of the oligomers obtained by enzymatic hydrolysis of the glucomannan produced by the plant Amorphophallus konjac. Carbohydr. Res. 337, 2505–2511. https://doi.org/10.1016/s0008-6215(02)00332-4 (2002).
Huang, A. S., Titchenal, C. A. & Meilleur, B. A. Nutrient composition of taro corms and breadfruit. J. Food Compos. Anal. 13, 859–864. https://doi.org/10.1006/jfca.2000.0936 (2000).
Zhang, Y., Guo, X. & Yan, B. Characterization of the complete chloroplast genome of Arisaema erubescens (Wall.) Schott, a traditional Chinese medicinal herb. Mitochond. DNA B Resour. 5, 3149–3150. https://doi.org/10.1080/23802359.2020.1797577 (2020).
Cai, Z., Wang, H. & Wang, G. Complete chloroplast genome sequence of Pinellia ternata (Thunb.) Breit, a medicinal plants to China. Mitochond. DNA B Resour. 5, 2107–2108. https://doi.org/10.1080/23802359.2020.1765207 (2020).
Abdullah, et al. Chloroplast genome evolution in the Dracunculus clade (Aroideae, Araceae). Genomics 113, 183–192. https://doi.org/10.1016/j.ygeno.2020.12.016 (2021).
Hu, H., Liu, J. G., Wang, B. L., An, J. X. & Wang, Q. Characterization of the complete chloroplast genome of Amorphophallus konjac (Araceae) and its phylogenetic analysis. Mitochond. DNA Part B. 4, 1658–1659. https://doi.org/10.1080/23802359.2019.1606683 (2019).
Yin, S. & Gao, Y. Characterization of the complete chloroplast genome assembly of Amorphophallus yunnanensis Engler, Pflanzenr (Araceae) from southwestern China. Mitochond. DNA B Resour. 8, 1445–1449. https://doi.org/10.1080/23802359.2023.2294896 (2023).
Gao, Y., Dong, K., Xiao, P., Wu, W. & Yin, S. Complete assembly of the chloroplast genome of Amorphophallus coaetaneus S. Y. Liu & S. J. Wei 1986 (Araceae) from southwestern China. Mitochond. DNA B Resour. 8, 766–770. https://doi.org/10.1080/23802359.2023.2238939 (2023).
Li, B. et al. Complete chloroplast genome sequences of three aroideae species (Araceae): Lights into selective pressure, marker development and phylogenetic relationships. BMC Genom. 23, 218. https://doi.org/10.1186/s12864-022-08400-3 (2022).
Xia, C., Wang, M., Guan, Y. & Li, J. Comparative analysis of the chloroplast genome for Aconitum Species: Genome structure and phylogenetic relationships. Front. Genet. 13, 878182. https://doi.org/10.3389/fgene.2022.878182 (2022).
Zeng, Y. J., Shen, L. W., Chen, S. Q., Qu, S. & Hou, N. Codon usage profiling of chloroplast genome in Juglandaceae. Forests 14, 378. https://doi.org/10.3390/f14020378 (2023).
Guo, X., Bao, J. & Fan, L. Evidence of selectively driven codon usage in rice: Implications for GC content evolution of Gramineae genes. FEBS Lett. 581, 1015–1021. https://doi.org/10.1016/j.febslet.2007.01.088 (2007).
Huang, X. et al. Analysis of transcriptome data reveals multifactor constraint on codon usage in Taenia multiceps. BMC Genom.. 18, 308. https://doi.org/10.1186/s12864-017-3704-8 (2017).
Li, G., Zhang, L. & Xue, P. Codon usage pattern and genetic diversity in chloroplast genomes of Panicum species. Gene 802, 145866. https://doi.org/10.1016/j.gene.2021.145866 (2021).
Zhang, P., Xu, W., Lu, X. & Wang, L. Analysis of codon usage bias of chloroplast genomes in Gynostemma species. Physiol. Mol. Biol. Plants. 27, 2727–2737. https://doi.org/10.1007/s12298-021-01105-z (2021).
Sheng, J., She, X., Liu, X., Wang, J. & Hu, Z. Comparative analysis of codon usage patterns in chloroplast genomes of five Miscanthus species and related species. PeerJ. 9, e12173. https://doi.org/10.7717/peerj.12173 (2021).
Wang, Z. et al. Comparative analysis of codon usage patterns in chloroplast genomes of six Euphorbiaceae species. PeerJ. 8, e8251. https://doi.org/10.7717/peerj.8251 (2020).
Yang, G. F., Su, K. L., Zhao, Y. R., Song, Z. B. & Sun, J. Analysis of codon usage in the chloroplast genome of Medicago truncatula. Acta Prataculturae Sinica. 24, 171–179 (2015).
Yuan, X. L., Li, Y. Q., Wang, Y. & Zhang, J. F. Analysis of codon usage in the chloroplast genome of Betula alnoides. Genom. Appl. Biol. 39(12), 5758–5764 (2020).
Sharp, P. M. & Li, W. H. An evolutionary perspective on synonymous codon usage in unicellular organisms. J. Mol. Evol. 24, 28–38. https://doi.org/10.1007/BF02099948 (1986).
Tang, D. et al. Analysis of codon usage bias and evolution in the chloroplast genome of Mesona chinensis Benth. Dev. Genes Evol. 231, 1–9. https://doi.org/10.1007/s00427-020-00670-9 (2021).
Klump, H. H. Correlation between genome size, observed codon preference and Gibbs energy of codon–anticodon interaction. Pure Appl. Chem. 65, 1947–1950. https://doi.org/10.1351/PAC199365091947 (1993).
Pluhar, W. AT2-AT3-profiling: A new look at synonymous codon usage. J. TheorBiol. 243, 308–321. https://doi.org/10.1016/j.jtbi.2006.07.004 (2006).
Novoa, E. M. & Ribas de Pouplana, L. Speeding with control: Codon usage, tRNAs, and ribosomes. Trends Genet. 28, 574–581. https://doi.org/10.1016/j.tig.2012.07.006 (2012).
Martin, A. & Orgogozo, V. The Loci of repeated evolution: A catalog of genetic hotspots of phenotypic variation. Evolution. 67, 1235–1250. https://doi.org/10.1111/evo.12081 (2013).
Henriquez, C. L. et al. Molecular evolution of chloroplast genomes in Monsteroideae (Araceae). Planta 251, 72. https://doi.org/10.1007/s00425-020-03365-7 (2020).
Fu, G. et al. Characterization of the complete chloroplast genome of the dragonhead herb, Dracocephalumheterophyllum (Lamiaceae), and comparative analyses with related species. Diversity 14, 110. https://doi.org/10.3390/d14020110 (2022).
Shi, H. et al. Complete chloroplast genomes of two Siraitia Merrill species: Comparative analysis, positive selection and novel molecular marker development. PloS one 14, e0226865. https://doi.org/10.1371/journal.pone.0226865 (2019).
Sun, L. et al. The complete chloroplast genome of Magnolia polytepala: Comparative analyses offer implication for genetics and phylogeny of Yulania. Gene 736, 144410. https://doi.org/10.1016/j.gene.2020.144410 (2020).
Xie, H. et al. Comparative analysis of the complete chloroplast genomes of six threatened subgenus Gynopodium (Magnolia) species. BMC Genom. 23, 716. https://doi.org/10.1186/s12864-022-08934-6 (2022).
Zheng, G. et al. Comparative analyses of chloroplast genomes from 13 Lagerstroemia (Lythraceae) species: Identification of highly divergent regions and inference of phylogenetic relationships. Plant Mol. Biol. 102, 659–676. https://doi.org/10.1007/s11103-020-00972-6 (2020).
Wu, T. et al. Comparative analysis of the chloroplast genomes of eight species of the genus Lirianthe Spach with its generic delimitation implications. Int. J. Mol. Sci. 25, 3506. https://doi.org/10.3390/ijms25063506 (2024).
Li, C., Liu, Y., Lin, F., Zheng, Y. & Huang, P. Characterization of the complete chloroplast genome sequences of six Dalbergia species and its comparative analysis in the subfamily of Papilionoideae (Fabaceae). PeerJ. 10, e13570. https://doi.org/10.7717/peerj.13570 (2022).
Li, G. et al. Analysis of synonymous codon usage of chloroplast genome in Porphyra umbilicalis. Genes Genom. 41, 1173–1181 (2019).
Li, C. et al. Codon usage bias and genetic diversity in chloroplast genomes of Elaeagnus species (Myrtiflorae: Elaeagnaceae). Physiol. Mol. Biol. Plants. 29, 239–251. https://doi.org/10.1007/s12298-023-01289-6 (2023).
Chartier, M., Gibernau, M. & Renner, S. S. The evolution of pollinator-plant interaction types in the Araceae. Evolution 68, 1533–1543. https://doi.org/10.1111/evo.12318 (2014).
Henriquez, C. L., Arias, T., Pires, J. C., Croat, T. B. & Schaal, B. A. Phylogenomics of the plant family Araceae. Mol. Phylogenet. Evol. 75, 91–102. https://doi.org/10.1016/j.ympev.2014.02.017 (2014).
Cusimano, N. et al. Relationships within the Araceae: Comparison of morphological patterns with molecular phylogenies. Am. J. Bot. 98, 654–668. https://doi.org/10.3732/ajb.1000158 (2011).
Abdullah, et al. Comparison of chloroplast genomes among species of unisexual and bisexual clades of the monocot family Araceae. Plants 9, 737. https://doi.org/10.3390/plants9060737 (2020).
Cui, N., Chen, W., Li, X. & Wang, P. Comparative chloroplast genomes and phylogenetic analyses of Pinellia. Mol. Biol. Rep. 49, 7873–7885. https://doi.org/10.1007/s11033-022-07617-5 (2022).
Nauheimer, L., Metzler, D. & Renner, S. S. Global history of the ancient monocot family Araceae inferred with models accounting for past continental positions and previous ranges based on fossils. New Phytol. 195, 938–950. https://doi.org/10.1111/j.1469-8137.2012.04220.x (2012).
Gao, Y. & Yin, S. The complete chloroplast genome assembly of Amorphophallus kiusianus makino 1913 (araceae) from Southern China. Mitochon. DNA B Resour. 9, 522–526. https://doi.org/10.1080/23802359.2024.2342934 (2024).
Wright, F. The “effective number of codons” used in a gene. Gene 87, 23–29. https://doi.org/10.1016/0378-1119(90)90491-9 (1990).
Jiang, Y., Deng, F., Wang, H. & Hu, Z. An extensive analysis on the global codon usage pattern of baculoviruses. Arch. Virol. 153, 2273–2282. https://doi.org/10.1007/s00705-008-0260-1 (2008).
Kawabe, A. & Miyashita, N. T. Patterns of codon usage bias in three dicot and four monocot plant species. Genes Genet. Syst. 78, 343–352. https://doi.org/10.1266/ggs.78.343 (2003).
Sueoka, N. Directional mutation pressure, mutator mutations, and dynamics of molecular evolution. J. Mol. Evol. 37, 137–153. https://doi.org/10.1007/BF02407349 (1993).
Sueoka, N. Translation-coupled violation of Parity Rule 2 in human genes is not the cause of heterogeneity of the DNA G+C content of third codon position. Gene 238, 53–58. https://doi.org/10.1016/s0378-1119(99)00320-0 (1999).
Chen, C. et al. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant. 13, 1194–1202. https://doi.org/10.1016/j.molp.2020.06.009 (2020).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. https://doi.org/10.1093/molbev/mst010 (2013).
Schwartz, S. et al. PipMaker–a web server for aligning two genomic DNA sequences. Genome Res. 10, 577–586. https://doi.org/10.1101/gr.10.4.577 (2000).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. https://doi.org/10.1093/bioinformatics/btp187 (2009).
Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. https://doi.org/10.1093/bioinformatics/btu033 (2014).
Xie, J. et al. Tree Visualization By One Table (tvBOT): A web application for visualizing, modifying and annotating phylogenetic trees. Nucleic Acids Res. 51, W587–W592. https://doi.org/10.1093/nar/gkad359 (2023).
Acknowledgements
The authors expresses gratitude to all the reviewers who provided suggestions for this manuscript.
Funding
This work supported by National Natural Science Foundation of China (32060683), Jiangxi Key Research and Development Program Project (20212BBF61001), Jiangxi Provincial Natural Science Foundation (20242BAB25381), and The Postdoctoral Research Funding program of Jiangxi Province (2021KY38).
Author information
Authors and Affiliations
Contributions
X.B.J. and J.Q.W. analyzed the data and finished the first draft. Y.W. and C.H.produced charts and figures. P.C., D.C., and Y.F. collected the literature. Q.H. checked the data analysis results and revised the manuscript. Y.J. and Q.L. guided the writing of the manuscript and approved the final manuscript. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
Not applicable.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Jia, X., Wei, J., Chen, Y. et al. Codon usage patterns and genomic variation analysis of chloroplast genomes provides new insights into the evolution of Aroideae. Sci Rep 15, 4333 (2025). https://doi.org/10.1038/s41598-025-88244-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-88244-5
Keywords
This article is cited by
-
Comprehensive analysis of chloroplast genome evolution in Poaceae: codon usage patterns, selection pressures, and phylogenomic relationships
BMC Genomics (2026)
-
The complete chloroplast genome of Curcuma cochinchinensis Gagnep, 1907 (Zingiberaceae): insights into phylogeny and comparative genomics
Genetic Resources and Crop Evolution (2026)
-
Plastome diversity and phylogenetic insights among modern Egyptian wheat cultivars: Genome-Wide and Gene-Level perspectives
BMC Plant Biology (2025)
-
Plastome phylogenomics of the Korean endemic plant Arisaema thunbergii subsp. geomundoense (Araceae)
Scientific Reports (2025)
-
Comparative plastome analysis reveals evolutionary dynamics and codon usage patterns in Bidens (Asteraceae)
Functional & Integrative Genomics (2025)
