
Abstract
Observational studies have reported an association between lipoprotein(a) (Lp(a)) and immune-mediated inflammatory diseases (IMIDs). This study used Mendelian Randomization (MR) and multivariable MR (MVMR) to explore the causal relationship between lipoprotein(a) [Lp(a)] and immune-mediated inflammatory diseases (IMIDs). We performed a bidirectional two-sample mendelian randomization analyses based on genome-wide association study (GWAS) summary statistics of Lp(a) and nine IMIDs, specifically celiac disease (CeD), Crohn’s disease (CD), ulcerative colitis (UC), inflammatory bowel disease (IBD), multiple sclerosis (MS), psoriasis (Pso), rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), type 1 diabetes (T1D), and summary-level data for lipid traits. Furthermore, we performed MVMR to examine the independence of relationship between Lp(a) and IMIDs after controlling other lipid traits, namely high-density lipoprotein cholesterol (HDL-C), low-density lipoprotein cholesterol (LDL-C) and triglycerides (TG). We didn’t observe a causal association between Lp(a) and the risk of IMIDs in univariable and multivariable MR analysis, challenging previous observational studies. However, genetically predicted lipid traits HDL-C was associated with increased risk of Type 1 diabetes (T1D). The identification of potential mechanisms underlying the observed associations in observational studies necessitates further investigation.
Similar content being viewed by others

Introduction
Lipoprotein(a) is synthesized by the covalent linkage of apolipoprotein A to apolipoprotein B via disulfide bonds, and its concentration demonstrates an inverse association with the size of apolipoprotein A. Moreover, this concentration is predominantly influenced by genetic factors at a level of 90%1. The significance of Lp(a) as a risk factor for cardiovascular diseases2,3, degenerative aortic stenosis4, atrial fibrillation5 and heart failure6 has gained recognition. Lp(a) has a tendency to undergo oxidative alterations and produce oxidized phospholipids (OxPLs). Lp(a) carries more than 80% OxPLs in its particles, and OxPLs induce inflammatory responses by increasing secretion of inflammatory cytokines by macrophages, such as IL-1β, TNF-α and IL-6, multiplying the inflammatory effect7,8. Lipoprotein(a) also carries monocyte chemoattractant protein-1 (MCP-1), a key chemokine in the initiation and progression of vascular inflammationm, and Lp(a)-associated MCP-1 enhances recruitment of monocytes to the vascular wall9. On the other hand, despite the genetic determination of Lp(a) levels, several studies have demonstrated that chronic inflammation disrupts Lp(a) expression and elevates plasma levels of Lp(a)10,11. Collectively, the available evidence suggests a reciprocal association between Lp(a) and inflammation.
Immune-mediated inflammatory diseases encompass a diverse array of pathological conditions, such as celiac disease, Crohn’s disease and inflammatory bowel disease. Multiple observational studies have consistently indicated increased concentrations of Lp(a) in individuals diagnosed with different inflammatory disorders, such as rheumatoid arthritis, systemic lupus erythematosus, and acquired immunodeficiency syndrome12,13,14,15,16,17,18,19,20. However, the majority of clinical investigations are limited in scope and rely on an observational design. While epidemiological studies indicate potential links between Lp(a) and autoimmune disorders, the underlying causality of these connections remains uncertain. The presence of unmeasured covariates and reverse causation in these studies introduces bias, making it challenging to establish a definitive causal relationship. Nonetheless, exploring the potential causal linkage between Lp(a) and autoimmune disorders could offer valuable insights into specific biological pathways and contribute to the development of preventive strategies.
Mendelian randomization is a methodologically robust approach to establish causal associations between exposures and outcomes in epidemiological studies. In MR analyses, genetic variants, predominantly single nucleotide polymorphisms (SNPs), serve as instrumental variables (IVs) for putative risk factors. The principle of MR is grounded in Mendel’s second law, which posits that during gametic formation, gene alleles segregate independently when DNA is transmitted from parent to offspring. As these variants are randomly assigned during conception, MR has the potential to mitigate bias arising from environmental confounders when conducted appropriately21,22,23. In this study, we employed bidirectional and multivariable MR approaches to investigate the causal association between immune-mediated inflammatory diseases and lipoprotein(a), employing summary statistics obtained from GWAS conducted on European populations for both characteristics. Our study seeks to offer fresh perspectives and empirical data regarding the correlation between lipoprotein(a) and IMIDs.
Methods
Study design
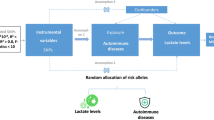
To ascertain the causal direction between lipoprotein(a) and immune-mediated inflammatory diseases, we conducted univariable and multivariable MR analyses using GWAS datasets24. Firstly, we conducted univariable Mendelian randomization (UVMR) analyses to establish a bidirectional causal link between Lp(a) and IMIDs. Additionally, multivariable Mendelian randomization analyses25 were performed on lipid traits (Lp(a), HDL-C, LDL-C, TG) for autoimmune diseases to assess the independent association of Lp(a) with autoimmune diseases. The overall design of this study is depicted in Fig. 1 through a comprehensive flow chart. The assumptions outlined below were applied to all MR analyses and are collectively described for both the UVMR and MVMR approaches. We adopted three fundamental hypotheses of classical MR analysis, as follows: (1) IVs exhibit a direct relationship with exposure. (2) Confounding variables do not affect the independence of IVs. (3) IVs solely influence the outcomes through exposure22,26. We didn’t pre-register any study protocol or details.

Diagram of the univariable and multivariable Mendelian randomization study for the association between lipoprotein(a) and risk of IMIDs.
Data sources
To perform our MR analyses, we used summary-level data from the publicly available GWAS for each trait27,28,29,30,31,32,33,34. Genetic IVs for HDL-C, LDL-C and TG were obtained through genome-wide association studies conducted in the UK Biobank35. GWAS data for exposure and outcomes were obtained from different databases to ensure minimal overlap. All study participants were of European descent, avoiding racial differences. The original publications provide comprehensive information regarding recruitment procedures and diagnostic criteria. The detail information of used GWAS datasets was listed in Table 1.
Instrumental variable selection
Initially, we identified single nucleotide polymorphism associated with each trait using a threshold of p = 5 × 10 − 8 based on the comprehensive summary-level GWAS statistics. Subsequently, to ensure the independence among the SNPs, a strict linkage disequilibrium (LD) threshold of r2 = 0.001 was applied when clustering IVs within 10 Mb. We ensured the effect estimates were standardized for exposure and outcome, while excluding any alleles that could potentially cause incompatibility or palindromic SNPs. For consistency, we used only SNPs that were tested for the trait as IVs and did not use proxies to replace SNPs that were missing in the outcome data. Additionally, we eliminated any SNPs linked to the confounding variable affecting the result using the PhenoScanner. For Lp(a) as the exposure and IMIDs as the outcomes, we regard the C-reactive protein levels (CRP) as the confounding variable. For IMIDs as the exposures and Lp(a) as the outcome, we regard the fat content, blood lipid and the coronary disease as the confounding variables. We employed F statistics (beta2/se2)36 to evaluate the robustness of genetically determined instrumental variables, with a threshold of F > 10 in accordance with the first assumption of Mendelian randomization and to avoid bias towards weak IVs37,38.
Mendelian randomization analysis
We utilized the statistical software R (V4.4.1, http://www.r-project.org) and employed the TwoSampleMR (http://gittub.com/MRCIEU/TwoSampleMR) and MR-PRESSO (http://gittub.com/rondolab/MR-PRESSO) packages for conducting all analyses39. Multiple MR methods, including inverse variance weighting (IVW)40, weighted median (WM)41, MR-Egger42, and MR-pleiotropy residual sum and outlier (MR-PRESSO)42 were utilized for deducing causal connections between lipoprotein(a) and IMIDs. The IVW method was primarily employed for fundamental causal estimates, which would provide the most precise results when all selected SNPs were valid IVs. The IVW method calculates a weighted average of Wald ratio estimates. Under the assumption of Instrument Strength Independent of Direct Effect (InSIDE), the MR-Egger regression executes a weighted linear regression and yields a consistent causal estimate, even though the genetic IVs are all invalid42. However, it exhibits low precision and is susceptible to outlying genetic variants. Additionally, we utilized the weighted median technique, which computes the midpoint of the weighted approximations and ensures consistent effects even in scenarios where 50% of instrumental variables exhibit pleiotropy. The Weighted Median regression method, which does not demand the InSIDE hypothesis, calculates a weighted median of the Wald ratio estimates and is robust to horizontal pleiotropic bias41. It is confirmed that the Weighted Median method has some advantages over the MR-Egger regression, as it provides lower type I error and higher causal estimate power. MR-PRESSO identifies and eliminates outliers in IVW linear regression to offer refined MR estimations. To evaluate the potential for horizontal pleiotropy of the SNPs, we used MR Egger regression. In addition, we performed a sensitivity analysis using the “leave-one-out” method to detect any SNPs that may have a significant impact. In this method, every SNP was methodically eliminated and its impact on the correlation was evaluated43. The heterogeneity of selected SNPs was assessed using the Cochrane’s Q test (P < 0.05). In instances where significant heterogeneity was observed, we employed the random effects IVW test to obtain more cautious and reliable estimates42. The mr_funnel_plot function was utilized to generate funnel plots for visualizing the heterogeneity of IVs. We additionally estimated FDR corrected P values for the multivariable analyses to adjust for the multiple tests performed on each exposure.
Results
Instrumental variables
For lipoprotein(a) as the exposure, IVs were chosen as SNPs linked to lipoprotein(a) (4 SNPs for CeD, 8 SNPs for CD, 7 SNPs for IBD, 52 SNPs for MS, 65 SNPs for Pso, 46 SNPs for RA, 57 SNPs for SLE, 77 SNPs for T1D, 8 SNPs for UC). For lipoprotein(a) as the outcome, IVs were chosen as SNPs linked to IMIDs (10 SNPs for CeD, 44 SNPs for CD, 40 SNPs for IBD,12 SNPs for MS, 6 SNPs for Pso, 15 SNPs for RA, 9 SNPs for SLE, 21 SNPs for T1D, 29 SNPs for UC). The absence of weak instrument bias was indicated by all F-statistics > 10. The comprehensive details regarding the instrumental variables can be found in Table S1–S3.
Causal estimates of genetic susceptibility to lipoprotein(a) and IMIDs risk
The findings from the MR analysis exploring the causal association between lipoprotein(a) and nine IMIDs traits are depicted in Fig. 2. Lipoprotein(a) exhibited no causal association with CeD (OR = 0.797, 95% CI 0.498–1.274), CD (OR = 1.231, 95% CI 0.519–2.920), IBD (OR = 1.026, 95% CI 0.804–1.309), MS (OR = 1.007, 95% CI 0.822–1.232), Pso (OR = 1.059, 95% CI 0.910–1.231), RA (OR = 0.936, 95% CI 0.782–1.120), SLE (OR = 1.039, 95% CI 0.801–1.349), T1D (OR = 1.065, 95% CI 0.931–1.218), and UC (OR = 1.016, 95% CI 0.707–1.461). This discovery aligns with the outcomes derived from alternative MR techniques, including MR Egger and weighted median.

MR Estimates from Mendelian randomization analysis of lipoprotein(a) and risk of IMIDs.
There was noticeable heterogeneity observed in our instrumental variables for Lp(a) in relation to CD (Q P.val = 7.65E−11), SLE (Q P.val = 3.66E−5), MS (Q P.val = 0.0001), Pso (Q P.val = 0.0043) and T1D (Q P.val = 0.0051) as outcome (Table S4). We observed that all MR-Egger regression intercepts were not significantly different from zero, indicating no indication of horizontal pleiotropy between the Lp(a) instrumental variables and IMIDs (intercept p > 0.05), except for T1D where a marginal deviation was found (intercept p = 0.021) (Table S4). The MR-PRESSO analysis revealed significant horizontal pleiotropy in certain analysis. Nevertheless, the causal estimates of Lp(a) with MS, Pso, SLE, and T1D remained consistent even after conducting outlier-corrected analyses (Table S5).
In addition, the sensitivity analysis plots indicated that no individual SNP was expected to have a substantial impact on the causal relationship, thus affirming the reliability of our findings (Fig. S1–S4). Taken collectively, these findings provide compelling evidence supporting the absence of a causal association between Lp(a) and IMIDs.

MR Estimates from Mendelian randomization analysis of IMIDs and risk of lipoprotein(a).
Causal estimates of genetic susceptibility to IMIDs and lipoprotein(a) levels
Furthermore, conducting reverse studies investigating the association between exposure to the risk of 9 IBIDs and the outcome of Lp(a) levels, we found no significant association between CeD (OR = 0.998, 95% CI 0.995–1.002), CD (OR = 1.000, 95% CI 0.991–1.008), IBD (OR = 0.995, 95% CI 0.987–1.003), MS (OR = 0.992, 95% CI 0.983–1.001), Pso (OR = 1.003, 95% CI 0.989–1.018), RA (OR = 1.003, 95% CI 0.998–1.009), SLE (OR = 0.998, 95% CI 0.993–1.004), T1D (OR = 0.998, 95% CI 0.994–1.002), UC (OR = 1.004, 95% CI 0.994–1.013) and Lp(a) in the IVW analysis results (Fig. 3). The outcomes obtained from each of the three MR techniques exhibited concurrence. There was noticeable diversity observed in our instrumental variables for CD (Q P.val = 1.60E−6), and UC (Q P.val = 0.0024 (Table S6). The presence of imbalanced horizontal pleiotropy was not indicated by the MR-Egger intercept, as it exhibited a central tendency around zero in all MR analyses (Table S6). Although in certain analyses, MR-PRESSO revealed the presence of substantial horizontal pleiotropy, the causal estimates of Lp(a) with UC and CD remained robust after outlier-corrected analyses (Table S7). Additionally, the sensitivity analysis plots, which employed a leave-one-out approach, indicated that the individual impact of each SNP on the causal association was not significant. This finding further strengthens our conclusions (Figs. S5–S8).
Multivariable MR
To account for potential pleiotropic pathways arising from the relationship between different lipid traits, we employed a multivariable Mendelian randomization model incorporating Lp(a), HDL-C, LDL-C, and TG as joint exposures for each IMIDs outcome. Following adjustment for HDL-C, LDL-C, and TG, genetically elevated Lp(a) showed no causal association with the onset of IMIDs, consistent with the findings of univariable MR analysis. Additionally, The association between genetically predicted HDL-C and type 1 diabetes remained marginally significant even after adjusting for multiple lipid traits (ORMVMR = 0.80, 95% CI 0.68–0.95; adjust P = 0.043). Furthermore, no significant associations were observed between other lipid traits and the IMID diseases of concern (Table S8).
Discussion
The correlation between levels of lipoprotein(a) and inflammatory conditions has garnered increasing attention. However, to our knowledge, this study represents the first systematic exploration of potential causal relationships between lipoprotein(a) levels and IMIDs using MR methods. Our results didn’t observe a causal relationship between genetic susceptibility to lipoprotein(a) and the risk of IMIDs. To differentiate between a genuine adverse outcome and the lack of validity in the MR studies, we conducted various sensitivity analyses to ensure adherence to the three MR assumptions. Taking into account the consistency of our MR findings across these diverse methodologies, we possess confidence regarding the veracity of our MR analyses.
Although most studies have suggested that Lp(a) is associated with inflammatory levels, whether a causal relationship exists between lipoprotein(a) and autoimmune diseases remains undetermined. Numerous observational studies have consistently reported elevated levels of Lp(a) in active autoimmune diseases, including rheumatoid arthritis14,44 and systemic lupus erythematosus45. However, Holm et al. performed a cross-sectional observational investigation and found no statistically significant disparities in serum Lp(a) levels among individuals with coronary artery disease, regardless of the presence or absence of inflammatory rheumatic disease46. Regarding the underlying mechanisms behind most of these observations, several investigations have indicated that Lp(a) itself may promote inflammation in diverse cellular populations, encompassing endothelial cells, monocytes and macrophages, through its association with oxidized phospholipids, mediating the increased cardiovascular disease risk17,47,48. In addition to the oxidation of lipoprotein (a), its glycosylated form, such as beta2-GPI-Lp(a), was detected in patients with rheumatoid arthritis49 and nephrotic syndrome50. Furthermore, elevated levels of Lp(a) are correlated with increased concentrations of acute phase proteins. In patients with rheumatoid arthritis, Lp(a) levels are positively associated with elevated C-reactive protein and erythrocyte sedimentation rate, indicating its significant role in the inflammatory cascade during the acute phase14. The formation of antibodies against Lp(a), which be related to its oxidation and glycosylation, appears to be triggered by autoimmune disease. Anti-malondialdehyde (MDA)-Lp(a) was detected in patients with antiphospholipid syndrome51. Some studies suggested that autoimmune processes may occur particularly in individuals with inherited high Lp(a) levels and certain HLA Class II genotypes, triggered by concurrent infections52.
Our findings suggest that there is no evidence from Mendelian randomization studies supporting a causal relationship between lipoprotein(a) levels and the risk of immune-mediated inflammatory diseases. The inconsistencies between our findings and previous observational studies that have reported a causal relationship can be attributed to confounding factors or the presence of reverse causality. Traditional observational research is susceptible to clinical confounding factors. The unmeasured variables associated with both the exposure and outcome lead to biased estimates of the effect of the exposure on the outcome. which can impact both exposure and outcome variables, thereby weakening the ability of observational studies to accurately establish causality. Second, these studies may be affected by reverse causality, where the outcome influences the exposure (rather than vice versa)53. Consequently, even if an observational study reports a strong correlation, it cannot definitively prove the existence of a direct causal association and the direction of relationship.
A considerable body of research indicates that conditions associated with abnormal blood lipid composition may influence immune-related diseases, and vice versa. In our analysis, after adjusting for HDL-C, LDL-C, and TG as covariates, only marginally statistical significance was observed between HDL-C and T1D. Here’s the possible explanation that the matter of lipids is complex as the composition and distribution of different kinds of lipids described in distinct organism species, organs, tissues, cells, and even cellular organelles is highly variable. In the future, by including more lipid characteristics and analyzing their relationship with inflammation-related diseases through lipidomics and other methods, it will provide more understanding of the relationship between lipid disorders and inflammation-related diseases.
Mendelian randomization overcomes this limitation by utilizing genetic instrumental variables to mitigate the influence of confounding factors and provide a relatively accurate assessment of causality. With the growing availability of large genetic data sets, MR has become a powerful and accessible tool for studying the risk factors for diseases. However, similar to other observational study designs, Mendelian Randomization possesses inherent limitations. Besides potential violations of the core MR assumptions, additional sources of bias may have influenced the study outcomes. Weak instruments, which are not strongly associated with the exposure of interest, can lead to biased MR estimates. Genetic instruments may be correlated with variants that are associated with the outcome of interest due to linkage disequilibrium, violating the MR assumptions54. In addition, diverse populations have been underrepresented in genomics research. Overall, while MR alone can never prove a causal relationship beyond reasonable doubt, MR offers a rigorous approach for investigating possible causal relationships in observational data and has the potential to transform our understanding of the etiology and treatment of diseases.
We would like to emphasize several strengths of our study and acknowledge certain limitations. Firstly, this study represents the inaugural investigation aimed at evaluating the bidirectional causal association between lipoprotein(a) and immune-mediated inflammatory diseases using a two sample Mendelian randomization approach. This methodology offers enhanced resilience against confounding factors, reverse causation, and non-differential exposures compared to observational studies. Secondly, we meticulously selected a diverse range of relatively prevalent autoimmune diseases along with their associated genome-wide association study data comprising large sample sizes. Lastly, we performed a sensitivity analysis to maintain the coherence of causal estimation and verify the reliability of our results. Our study also has several limitations. First and foremost, it is crucial to acknowledge that this research was carried out specifically within a European demographic. Consequently, prudence must be exercised when extrapolating our discoveries to alternative populations. Additionally, it is worth considering the possibility of other autoimmune diseases associated with lipoprotein(a) that were not encompassed within the scope of our analysis. Additionally, some of our Mendelian randomization analyses had inadequate statistical power for detecting minor impacts owing to the restricted variability explained by the single nucleotide polymorphism instruments or the relatively small sample sizes in the GWAS for outcome traits. In this regard, the exclusion of ambiguous or palindromic SNPs may have potentially compromised the statistical power of our MR study. The implementation of larger genome-wide association studies on autoimmune traits will markedly augment the statistical power of subsequent MR studies intended to detect and establish associations between these traits and potential risk factors or comorbidities.
Conclusion
Our investigate doesn’t observe the presence of a bilateral causal link between lipoprotein(a) levels and the susceptibility to immune-mediated inflammatory diseases in Europeans. This suggests that the observed associations could be attributed to shared genetic factors or confounding environmental influences. Future studies, especially those using other MR techniques or experimental models, could further explore the relationship and potential mechanism.
Data availability
All data generated used in the current study are available in this published article and supplementary file associated with it.
References
Marcovina, S. M. & Koschinsky, M. L. Lipoprotein(a) as a risk factor for coronary artery disease. Am. J. Cardiol. 82, 57U–66U. https://doi.org/10.1016/s0002-9149(98)00954-0 (1998).
Kotani, K., Serban, M. C., Penson, P., Lippi, G. & Banach, M. Evidence-based assessment of lipoprotein(a) as a risk biomarker for cardiovascular diseases - some answers and still many questions. Crit. Rev. Clin. Lab. Sci. 53, 370–378. https://doi.org/10.1080/10408363.2016.1188055 (2016).
Erqou, S. et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. Jama 302, 412–423. https://doi.org/10.1001/jama.2009.1063 (2009).
Guddeti, R. R. et al. Lipoprotein(a) and calcific aortic valve stenosis: a systematic review. Prog Cardiovasc. Dis. 63, 496–502. https://doi.org/10.1016/j.pcad.2020.06.002 (2020).
Mohammadi-Shemirani, P. et al. Elevated lipoprotein(a) and risk of Atrial Fibrillation: an observational and mendelian randomization study. J. Am. Coll. Cardiol. 79, 1579–1590. https://doi.org/10.1016/j.jacc.2022.02.018 (2022).
Kamstrup, P. R. & Nordestgaard, B. G. Elevated lipoprotein(a) levels, LPA risk genotypes, and increased risk of Heart failure in the General Population. JACC Heart Fail. 4, 78–87. https://doi.org/10.1016/j.jchf.2015.08.006 (2016).
van der Valk, F. M. et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation 134, 611–624. https://doi.org/10.1161/circulationaha.116.020838 (2016).
Buechler, C. et al. Lipoprotein (a) downregulates lysosomal acid lipase and induces interleukin-6 in human blood monocytes. Biochim. Biophys. Acta. 1642, 25–31. https://doi.org/10.1016/s0167-4889(03)00083-1 (2003).
Wiesner, P. et al. MCP-1 binds to oxidized LDL and is carried by lipoprotein(a) in human plasma. J. Lipid Res. 54, 1877–1883. https://doi.org/10.1194/jlr.M036343 (2013).
Nordestgaard, B. G. & Langsted, A. Lipoprotein (a) as a cause of cardiovascular disease: insights from epidemiology, genetics, and biology. J. Lipid Res. 57, 1953–1975. https://doi.org/10.1194/jlr.R071233 (2016).
Tomic Naglic, D., Manojlovic, M., Pejakovic, S., Stepanovic, K. & Prodanovic Simeunovic, J. Lipoprotein(a): role in atherosclerosis and new treatment options. Biomol. Biomed. 23, 575–583. https://doi.org/10.17305/bb.2023.8992 (2023).
Govindan, K. P., Basha, S., Ramesh, V., Kumar, C. N. & Swathi, S. A comparative study on serum lipoprotein (a) and lipid profile between rheumatoid arthritis patients and normal subjects. J. Pharm. Bioallied Sci. 7, S22–25. https://doi.org/10.4103/0975-7406.155767 (2015).
Borba, E. F. et al. Lipoprotein(a) levels in systemic lupus erythematosus. J. Rheumatol. 21, 220–223 (1994).
Dursunoğlu, D. et al. Lp(a) lipoprotein and lipids in patients with rheumatoid arthritis: serum levels and relationship to inflammation. Rheumatol. Int. 25, 241–245. https://doi.org/10.1007/s00296-004-0438-0 (2005).
García-Gómez, C. et al. Lipoprotein(a) concentrations in rheumatoid arthritis on biologic therapy: results from the CARdiovascular in rheuMAtology study project. J. Clin. Lipidol. 11, 749–756e743. https://doi.org/10.1016/j.jacl.2017.02.018 (2017).
Koutroubakis, I. E. et al. Increased levels of lipoprotein (a) in Crohn’s disease: a relation to thrombosis? Eur. J. Gastroenterol. Hepatol. 13, 1415–1419. https://doi.org/10.1097/00042737-200112000-00004 (2001).
Missala, I., Kassner, U. & Steinhagen-Thiessen, E. A Systematic Literature Review of the Association of Lipoprotein(a) and Autoimmune diseases and atherosclerosis. Int. J. Rheumatol. 2012 (480784). https://doi.org/10.1155/2012/480784 (2012).
Kronenberg, F. et al. Multicenter study of lipoprotein(a) and apolipoprotein(a) phenotypes in patients with end-stage renal disease treated by hemodialysis or continuous ambulatory peritoneal dialysis. J. Am. Soc. Nephrol. 6, 110–120. https://doi.org/10.1681/asn.v61110 (1995).
Thillet, J. et al. Am. J. Kidney Dis. 23 620–621 (1994).
Van den Hof, M. et al. Elevated lipoprotein(a) in perinatally HIV-Infected Children compared with healthy ethnicity-matched controls. Open. Forum Infect. Dis. 6, ofz301. https://doi.org/10.1093/ofid/ofz301 (2019).
Emdin, C. A., Khera, A. V. & Kathiresan, S. Mendelian Randomization Jama 318, 1925–1926 https://doi.org/10.1001/jama.2017.17219 (2017).
Davies, N. M., Holmes, M. V. & Davey Smith, G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. Bmj 362, k601. https://doi.org/10.1136/bmj.k601 (2018).
Larsson, S. C., Butterworth, A. S. & Burgess, S. Mendelian randomization for cardiovascular diseases: principles and applications. Eur. Heart J. 44, 4913–4924. https://doi.org/10.1093/eurheartj/ehad736 (2023).
Hemani, G., Tilling, K. & Davey Smith, G. Orienting the causal relationship between imprecisely measured traits using GWAS summary data. PLoS Genet. 13, e1007081. https://doi.org/10.1371/journal.pgen.1007081 (2017).
Burgess, S. & Thompson, S. G. Multivariable mendelian randomization: the use of pleiotropic genetic variants to estimate causal effects. Am. J. Epidemiol. 181, 251–260. https://doi.org/10.1093/aje/kwu283 (2015).
Sanderson, E. et al. Mendelian randomization. Nat. Rev. Methods Primers. 2 https://doi.org/10.1038/s43586-021-00092-5 (2022).
Trynka, G. et al. Dense genotyping identifies and localizes multiple common and rare variant association signals in celiac disease. Nat. Genet. 43, 1193–1201. https://doi.org/10.1038/ng.998 (2011).
Liu, J. Z. et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 47, 979–986. https://doi.org/10.1038/ng.3359 (2015).
Multiple sclerosis genomic. Map implicates peripheral immune cells and microglia in susceptibility. Science 365 https://doi.org/10.1126/science.aav7188 (2019).
Stuart, P. E. et al. Transethnic analysis of psoriasis susceptibility in South asians and europeans enhances fine-mapping in the MHC and genomewide. HGG Adv. 3 https://doi.org/10.1016/j.xhgg.2021.100069 (2022).
Okada, Y. et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 506, 376–381. https://doi.org/10.1038/nature12873 (2014).
Bentham, J. et al. Genetic association analyses implicate aberrant regulation of innate and adaptive immunity genes in the pathogenesis of systemic lupus erythematosus. Nat. Genet. 47, 1457–1464. https://doi.org/10.1038/ng.3434 (2015).
Chiou, J. et al. Interpreting type 1 diabetes risk with genetics and single-cell epigenomics. Nature 594, 398–402. https://doi.org/10.1038/s41586-021-03552-w (2021).
Barton, A. R., Sherman, M. A., Mukamel, R. E. & Loh, P. R. Whole-exome imputation within UK Biobank powers rare coding variant association and fine-mapping analyses. Nat. Genet. 53, 1260–1269. https://doi.org/10.1038/s41588-021-00892-1 (2021).
Richardson, T. G. et al. Evaluating the relationship between circulating lipoprotein lipids and apolipoproteins with risk of coronary heart disease: a multivariable mendelian randomisation analysis. PLoS Med. 17, e1003062. https://doi.org/10.1371/journal.pmed.1003062 (2020).
Bowden, J. et al. Improving the accuracy of two-sample summary-data mendelian randomization: moving beyond the NOME assumption. Int. J. Epidemiol. 48, 728–742. https://doi.org/10.1093/ije/dyy258 (2019).
Davey Smith, G. & Hemani, G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum. Mol. Genet. 23, R89–98. https://doi.org/10.1093/hmg/ddu328 (2014).
Pierce, B. L. & Burgess, S. Efficient design for mendelian randomization studies: subsample and 2-sample instrumental variable estimators. Am. J. Epidemiol. 178, 1177–1184. https://doi.org/10.1093/aje/kwt084 (2013).
Hemani, G. et al. The MR-Base platform supports systematic causal inference across the human phenome. Elife 7 https://doi.org/10.7554/eLife.34408 (2018).
Slob, E. A. W. & Burgess, S. A comparison of robust mendelian randomization methods using summary data. Genet. Epidemiol. 44, 313–329. https://doi.org/10.1002/gepi.22295 (2020).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent estimation in mendelian randomization with some Invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314. https://doi.org/10.1002/gepi.21965 (2016).
Bowden, J., Davey Smith, G. & Burgess, S. Mendelian randomization with invalid instruments: effect estimation and bias detection through Egger regression. Int. J. Epidemiol. 44, 512–525. https://doi.org/10.1093/ije/dyv080 (2015).
Burgess, S., Bowden, J., Fall, T., Ingelsson, E. & Thompson, S. G. Sensitivity analyses for robust causal inference from mendelian randomization analyses with multiple genetic variants. Epidemiology 28, 30–42. https://doi.org/10.1097/ede.0000000000000559 (2017).
García-Gómez, C. et al. Conventional lipid profile and lipoprotein(a) concentrations in treated patients with rheumatoid arthritis. J. Rheumatol. 36, 1365–1370. https://doi.org/10.3899/jrheum.080928 (2009).
Sari, R. A., Polat, M. F., Taysi, S., Bakan, E. & Capoğlu, I. Serum lipoprotein(a) level and its clinical significance in patients with systemic lupus erythematosus. Clin. Rheumatol. 21, 520–524. https://doi.org/10.1007/s100670200127 (2002).
Holm, S. et al. Levels of lipoprotein (a) in patients with coronary artery disease with and without inflammatory rheumatic disease: a cross-sectional study. BMJ Open. 9, e030651. https://doi.org/10.1136/bmjopen-2019-030651 (2019).
Hoogeveen, R. C. & Ballantyne, C. M. Residual Cardiovascular risk at low LDL: remnants, Lipoprotein(a), and inflammation. Clin. Chem. 67, 143–153. https://doi.org/10.1093/clinchem/hvaa252 (2021).
Swerdlow, D. I. et al. Treatment and prevention of lipoprotein(a)-mediated cardiovascular disease: the emerging potential of RNA interference therapeutics. Cardiovasc. Res. 118, 1218–1231. https://doi.org/10.1093/cvr/cvab100 (2022).
Zhang, C. et al. Increased serum levels of β₂-GPI-Lp(a) complexes and their association with premature atherosclerosis in patients with rheumatoid arthritis. Clin. Chim. Acta. 412, 1332–1336. https://doi.org/10.1016/j.cca.2011.03.029 (2011).
Zhang, C. et al. Elevated serum β₂-GPI-Lp(a) complexes levels in children with nephrotic syndrome. Clin. Chim. Acta. 413, 1657–1660. https://doi.org/10.1016/j.cca.2012.05.006 (2012).
Romero, F. I. et al. Autoantibodies against malondialdehyde-modified lipoprotein(a) in antiphospholipid syndrome. Arthritis Rheum. 42, 2606–2611 (1999).
Dahlén, G. H. Lp(a) lipoprotein in cardiovascular disease. Atherosclerosis 108, 111–126. https://doi.org/10.1016/0021-9150(94)90106-6 (1994).
Levin, M. G. & Burgess, S. Mendelian randomization as a Tool for Cardiovascular Research: a review. JAMA Cardiol. 9, 79–89. https://doi.org/10.1001/jamacardio.2023.4115 (2024).
Lawlor, D. A., Harbord, R. M., Sterne, J. A. & Timpson, N. Davey Smith, G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat. Med. 27, 1133–1163. https://doi.org/10.1002/sim.3034 (2008).
Funding
This work was supported by the Taishan Scholars Program of Shandong Province (tsqn202312327), the National Natural Science Foundation of China (82100279), and the Chinese Cardiovascular Association-Access fund (2021-CCA-ACCESS-142). The study funders/sponsors had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Author information
Authors and Affiliations
Contributions
LJ, TY and BP conceived the study, participated in its design and coordination. HY searched the databases. QX and XY reviewed the GWAS datasets and finished the data collection. ZQ finished the data analysis. LJ drafted the manuscript and XD critically revised the manuscript. LJ, TY and XD had full access to all the data collection, analysis, and interpretation.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ti, Y., Xu, D., Qin, X. et al. Mendelian randomization analysis does not support a causal influence between lipoprotein(A) and immune-mediated inflammatory diseases. Sci Rep 15, 3834 (2025). https://doi.org/10.1038/s41598-025-88375-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-88375-9
