
Abstract
Emerging evidence has indicated that m5C modification plays a vital role in cancer development. However, the function of m5C-lncRNAs in PTC has never been reported. This study aims to explore the regulation mechanism of m5C RNA methylation-related long noncoding RNAs (m5C-lncRNAs) in papillary thyroid cancer (PTC). Bioinformatics analysis was used to investigate the role of m5C-lncRNAs in the prognosis and tumor immune microenvironment of PTC. Subsequently, we preliminarily verified the regulation mechanisms of m5C-lncRNAs in vivo and in vitro experiments. A total of six m5C-lncRNAs and five immune cell types were selected to construct the risk score and immune risk score (IRS) model, respectively. Patients with a high-risk score had a worse prognosis and the ROC indicated a reliable prediction performance (AUC = 0.796). As expected, the ESTIMATE and immune scores were higher (P < 0.001) and the tumor purity (P < 0.05) was significantly lower in the low-risk subgroup. CIBERSORT analysis showed Tregs, M0 macrophages, dendritic cells resting, and eosinophils were positively correlated to the risk score. Moreover, the expression levels of PD-1, PD-L1, CTLA-4, TIM-3, LAG-3, and KLRB1 were lower in the high-risk subgroup. Importantly, patients in high-risk subgroup tended to have a better response to immunotherapy than those in low-risk subgroup (P = 0.022). Similar to the above risk score, the IRS model also showed favorable prognosis predictive performance (AUC = 0.764). An integrated nomogram combining risk score, IRS, and age exhibited good prognostic predictive performance. Additionally, we validate the downregulation of PPP1R12A-AS1 promotes proliferation and metastasis by activating the MAPK signaling pathway. Our research confirms that m5C-lncRNAs not only contribute to evaluating the prognosis of patients with PTC but also help predict immune cell infiltration and immunotherapy response.
Similar content being viewed by others


Introduction
Papillary thyroid cancer (PTC) is a common malignant endocrine tumor, and its incidence has increased over the past decades1. In China, thyroid cancer has become the fastest-growing malignant tumor among women under 30 years old2,3. Most patients with PTC (PTCs) have a favorable prognosis by surgical resection, followed by either radioactive iodine or observation. However, a small number of PTCs still suffer from radioactive iodine refractory or progress to distant metastasis, resulting in high mortality rates and poor life quality4,5. Hence, elucidating the underlying molecular mechanisms of PTC progression, and developing novel therapeutic targets for PTC treatment is crucial.
5-methylcytosine (m5C) is a common RNA modification of mRNA or non-coding RNAs (ncRNAs), which plays an essential role in tRNA stability, ribosome assembly, RNA export, and translation6,7,8. At the post-transcriptional level, m5C RNA modifications can be reversibly and dynamically controlled by methyltransferases (writers), demethylases (erasers), and binding proteins (readers)9,10. The methyltransferase complex includes NOL1/NOP2/sun family (NSUN1-7), DNA methyltransferase (DNMT) member (DNMT1, DNMT2, DNMT3A, and DNMT3B). TET2 is an important “eraser” protein which acts as a demethylase. The main “reader” is ALYREF, which can read the m5C motif. Existing evidence has indicated that dysregulation of m5C modification plays an important role in tumor initiation, progression, and drug responses11,12. For instance, aberrant NSUN2 promotes pathogenesis by targeting the m5C methylation site in the untranslated region of HDGF3 in human bladder urothelial carcinoma13. Moreover, NSUN2 was also demonstrated to promote tumor progression and increase drug resistance by methylation of NMR in esophageal squamous cell carcinoma14. Additionally, studies showed that the downregulation of TET3 promotes glioblastoma tumorigenesis15. These studies reveal that m5C regulators are highly involved in tumor progression and metastasis, thus serving as useful therapeutic targets with promising prognostic values. However, the role of m5C in PTC is still unclear.
In recent years, increasing evidence has revealed that tumor microenvironment (TME) plays a critical role in PTC aggressiveness and progression, especially in radioiodine-refractory and advanced PTC16,17. Novel therapies are necessary for these PTCs. Antitumor efficacy of immune checkpoint blockade (ICB) therapy, targeting cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) and/or programmed cell death protein 1 (PD-1)/PD-ligand 1 (PD-L1), has been observed promising clinical results in non-small-cell lung carcinoma, metastatic melanoma, and renal cancer18,19. In the light of these successes, there has been strong interest in radioiodine-refractory and advanced PTCs. To optimize this therapeutic approach, there is a critical need to identify high-risk PTCs that are likely to achieve benefits in the early stage but currently perform poorly in PTCs.
In this study, we performed in-depth sequence analyses to identify the prognostic significance of m5C-related lncRNA (m5C-lncRNAs) in PTCs based on The Cancer Genome Atlas (TCGA) dataset. Corresponding risk models and immune risk score (IRS) of m5C-lncRNAs prognostic signature were established to improve risk stratification and the possible response to ICB therapy of patients. Importantly, the vitro and vivo functional assay to confirm the biological behavior of m5C-lncRNAs. This study sought to provide new insight into the regulatory mechanisms associated with TME and the strategies for PTC immunotherapy.
Materials and methods
Gene datasets and clinical data collection
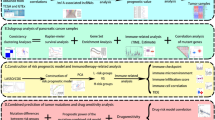
We downloaded the RNA-seq data from the UCSC Xena browser (http://xena.ucsc.edu/), which contained 58 normal and 472 thyroid cancer samples with complete clinical information. The expression data was normalized by transcript per million (TPM). The corresponding patients’ clinical information included sex, age, laterality, multifocality, TNM stage, lymph node metastasis, and survival time (Table S1). According to published literature6,13, there were fourteen m5C related regulators, including NSUN1-7, DNMT1, DNMT2 (TRDMT1), DNMT3A, DNMT3B, ALYREF, TRDMT1, TET2, and YBX1. Next, differential expression analysis of these m5C related regulators between PTC and normal samples by the limma package. Subsequently, Spearman correlation coefficient analysis was used to determine the relationship of m5C related regulators. The work flow was shown in (Fig. 1).

Study flow chart.
Protein-protein interaction (PPI) network
To further analyze the potential PPI relationship, we use the STRING database (https://string-db.org/) to analyze the m5C related regulators and construct the PPI network, The threshold of genes at the center of the PPI network was the minimum gene interaction score > 0.4.
Construction of risk assessment model construction
Pearson’s correlation method was performed to filter the m5C-lncRNAs based on the threshold criteria of Pearson’s coefficient |R| >0.5 and P < 0.001. Then, univariable and multivariable Cox analyses were used to identify the prognostic significance of m5C-lncRNAs. A total of nine prognostic m5C-lncRNAs were extracted for further analysis. The least absolute shrinkage and selection operator (LASSO) Cox regression was employed to select candidate prognostic m5C-lncRNAs by the glmnet package. The optimal penalty parameter λ was set as the minimum 10-fold cross-validation. The risk score for each patient was calculated according to the following algorithm: Risk score =\(\:{\sum\:}_{i=1}^{n}coef(i)*a\left(i\right)\). The αi represented the enrichment scores of m5C-lncRNAs in the risk score model and the enrichment scores of immune cells in the IRS model, respectively. Whereas \(\:coef(i)\) represents the coefficient of each m5C-lncRNA and immune cells, respectively. Thereafter, the patients were divided into high- and low-risk groups based on the median risk score. Kaplan–Meier curves with the log-rank test were used to draw the survival curves.
Evaluation of immune infiltration and enrichment functional analysis
Single-sample gene set enrichment analysis (ssGSEA) was used to calculate the enrichment scores of types of infiltrating immune cells with the GSVA and GSEABase packages.
The ESTIMATE algorithm (https://bioinformatics.mdanderson.org/estimate/) was employed to calculate the immune and stroma scores for each patient, and tumor purity was calculated by genomic methods. Furthermore, CIBERSOFT and MCP counter algorithms were performed to assess the immune response of immune cells and components. The response of PTCs to ICB therapy was predicted by ImmuCellAI (http://bioinfo.life.hust.edu.cn/ImmuCellAI/)20. Differentially expressed genes (DEGs) between high- and low-immune risk score (IRS) subgroups were screened by the limma package based on P values < 0.05 and |log2FC| ≥1. GO enrichment and KEGG21,22 pathway analysis were realized through Metascape (http://metascape.org).
Tissues, cell lines, and cell transfection
In this study, four human PTC cell lines (BCPAP, K1, TPC-1, IHH4) and a human normal thyroid epithelial cell line (Nthy-ori3-1) were used. All of the cell lines were purchased from the Chinese Academy of Science Cell Bank and cultured in a complete medium composed of 10% fetal bovine serum and 90% RPMI1640or DMEM (Gibco, Carlsbad, USA). These cells were cultured at 37 °C with 5% CO2.
We collected 20 PTC tissue samples and paired adjacent tissues from patients who underwent thyroidectomy in the Thyroid Surgery Department of Xiangya hospital from March 2020 to June 2020. The fresh tissues were stored at -80 °C. Informed consent was obtained from all the participants and approved by the Ethics Committee of Xiangya Hospital of Central South University. All research was performed in accordance with relevant guidelines and regulations.
Small interfering RNA (siRNA) targeting PPP1R12A-AS1 was synthesized and designed by RiboBio company (Guangzhou, China). The TPC-1 cells were infected with a concentrated vector according to our previous description23. The sequences of siRNAs are listed in Table S2.
Quantitative real-time polymerase chain reaction validation
Total RNA was extracted using the AG RNAex Pro RNA Extraction Reagent (AG21102, Accurate Biology, China) and reverse-transcribed into cDNA with an Evo M-MLV RT Mix Kit with gDNA Clean for qPCR Ver.2 (AG11728, Accurate Biology, China) that included random primers suitable for lncRNA reverse transcription. qPCR experiments were performed using the SYBR Green Premix Pro Taq HS qPCR Kit (AG11701, Accurate Biology, China). The lncRNA primers were designed by Accurate Biology and validated for specificity using Primer-BLAST (Table S2).
Western blotting
Cells were lysed in RIPA lysis buffer (Solarbio, China) with a protease inhibitor cocktail. Total proteins were separated by 10% SDS-PAGE and transferred onto nitrocellulose membranes (Millipore, USA), the membranes were blocked with 5% skimmed milk and then incubated with antibodies at 4 °C overnight. The following antibodies were used: ERK (66192-1-Ig, Proteintech, China), p-ERK (28733-1-AP, Proteintech, China), p-P38 (AP1508, ABclonal, China), P38 (A4771, ABclonal, China), p-AKT (#9271, CST, USA), AKT (#9272, CST, USA), and GAPDH (GB15002, Servicebio, China).
Functional assays in vitro
The procedures for colony formation, EDU assay, cell counting kit (CCK-8), transwell migration and invasion assay, and flow cytometry analysis were conducted as previously described24.
Animal experiments
Animal experiments were performed in accordance with ARRIVE guidelines and approved by the Animal Welfare Ethical Review Committee of Central South University. All experiments were performed in accordance with relevant guidelines and regulations. For xenograft models, 4–6 weeks-old male BALB/c nude mice were purchased from Slack King of Laboratory Animal Co., Ltd. (Hunan, China). Approximately 5 × 106 TPC-1 cells from the si-NC or si-PPP1R12A-AS1 group were injected into the flank of mice. During the 30-day observation, the tumor volume was measured (width2 ×length×0.5). After 30 days of subcutaneous inoculation, the nude mice were euthanized using CO2 inhalation followed by cervical dislocation, and the tumor tissues were separated for weighing and photographing.
Statistical analysis
Statistical tests were performed using R software (version 4.3.1). Chi-square and T-test were performed to compare the differences between the two subgroups. Kruskal–Wallis test was used to compare immune scores, stroma scores, tumor purity, and ESTIMATE scores among different risk score subgroups. The statistical significance was shown as followed: P < 0.05(*), P < 0.01(**), P < 0.001(***), P < 0.0001(****).
Results
The profile of m5C-lncRNA regulators
To clarify the interaction and related functions of m5C regulators in PTC, we analyzed 472 PTC tissues and 58 normal tissues from the TCGA dataset. The results showed that the expression of NSUN3, NSUN4, NSUN6, NSUN7, DNMT3B, ALYREF, TRDMT1, TET2, and YBX1 were significantly down-regulated in PTC samples compared with normal tissues (all P < 0.05). While NSUN1 (NOP2), NSUN5, and DNMT1 were significantly up-regulated in PTCs (both P < 0.001). However, the expression of NSUN2 and DNMT3A showed no significant differences (Fig. 2A). Next, to identify the relationship among fourteen m5C regulators, we used the STRING database to build a PPI network (Fig. 2B), and revealed the TRDMT1 as a hub gene among m5C regulators that interacted with the other 12 m5C regulators (Fig. 2C). However, TRDMT1 did not show a strong correlation with other m5C regulators in the subsequent correlation analysis. Interestingly, NSUN3 and TET2 had the strongest correlation, and their expression was significantly associated with other eight m5C regulators (Fig. 2D). These results indicated that NSUN3 may be a key gene that is involved in the development of PTC.

The profile of m5C-lncRNAs regulators. (A) Differential expression of 14 m5C regulators; (B,C) PPI network shows the interaction between the m5C regulators. (D) Pearson correlation analysis of the m5C regulators. (E) The correlations between m5C-lncRNAs and m5C regulators. (F) Differential expression of 9 m5C-lncRNAs regulators. *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
Then, Pearson correlation analysis was employed to search m5C-lncRNAs based on the criterion of Pearson R > 0.5 and p < 0.001, and 556 m5C-lncRNAs were uncovered. Integrated with the prognostic information, univariable and multivariable Cox regression to screen m5C-lncRNAs prognostic signatures. Finally, 9 m5C-lncRNAs were significantly related with the overall survival. Figure 2E showed that most of the m5C regulators positively correlated with the expression level of lncRNAs. Among 9 m5C-lncRNAs prognostic signatures, the expression of ZNF451-AS1, AC018647.2, POLR2J4, PVT1, AL359532.1, A2M.AS1, PPP1R12A-AS1, and AC090515.3 showed significantly lower in PTC samples than in normal tissues (p < 0.05). Whereas only AC024257.3 was up-regulated in PTCs (Fig. 2F).
Risk signature was associated with PTC prognosis and tumor immune microenvironment
To evaluate the prognostic value of m5C-lncRNAs in PTC, the LASSO Cox analysis showed that 6 m5C-lncRNAs exhibited strong prognostic value (Figure S1). The risk score for each patient was calculated based on the expression profile of 6 m5C-lncRNAs weighted by their regression coefficients. Risk score = (4.0 × ZNF451-AS1) + (0.68 × AC018647.2) + (2.02 ×POLR2J4) + (0.53 × PVT1) - (4.64 × AC024257.3) + (0.58 × PPP1R12A-AS1). The patients were divided into low- and high-risk subgroups based on the median risk scores, and further survival analysis showed that patients in the low-risk subgroups had better prognosis than high-risk subgroups (P = 0.016) (Fig. 3A), indicating that the risk model had a prognostic value. Also, the number of death cases in the low-risk subgroup was less than that in the high-risk subgroup (Fig. 3B). To assess the prognostic accuracy of the risk score, the AUC of ROC analysis was 0.796 (Fig. 3C). At the same time, principal component analysis could distinguish the distribution of the two risk groups (Figure S2). As for the TME of PTC, compared with the high-risk subgroup, the ESTIMATE and immune scores were higher (P < 0.001) and the tumor purity (P < 0.05) was significantly lower in the low-risk subgroup (Fig. 3D). CIBERSORT analysis showed Tregs, M0 macrophages, dendritic cells resting, and eosinophils were positively correlated to the risk score (Fig. 3E). Further, elevated levels of T cells, cytotoxicity score, myeloid dendritic cells, and decreased levels of neutrophil and endothelial cells were found in the low-risk subgroup when compared with the high-risk subgroup (Fig. 3F). These results indicate that the risk score model has a promising prognostic predictive value and is closely associated with TME in PTCs.

The relationship between risk score and PTC prognosis and tumor immune microenvironment. (A) Kaplan-Meier curves showed that patients in the low-risk subgroup had a better prognosis than those in the high-risk subgroup (P = 0.016). (B) Distributions of risk scores and survival status of PTCs. (C) The area under the ROC curve (AUC) of the risk score. (D–F) The immune cell infiltration landscape in the risk score subgroups was analyzed using ESTIMATE, CIBERSOFT, and MCP counter, respectively. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, and ns: no significance.
In addition, the results of the ssGSEA analysis demonstrated that 26 cell types showed significant differences in low-risk and high-risk groups (Fig. 4A). With the aid of the LASSO Cox regression analysis, we generated five cell types to calculate the immune risk score for each patient. Immune risk score (IRS) = (44.37 × central memory CD4 T cell) + (19.83 × eosinophil) + (60.06 ×monocyte) - (49.01 × T follicular helper cell) - (29.86 × type17 T helper cell). Similar to the above risk score, patients were also classified into low- and high-IRS subgroups based on the median IRS. The heatmap results indicated that central memory CD4 T cell, eosinophil, and monocyte were highly enrichment in the high-IRS group, whereas the low-IRS group had higher infiltration levels of T follicular helper cell and type17 T helper cell. Moreover, IRS was positively correlated with risk score (Fig. 4B). Kaplan–Meier analysis showed that the overall survival of high-IRS was poorer than the low-IRS subgroup (P = 0.0049) (Fig. 4C). The AUC value for the IRS model was 0.764 (Fig. 4D), which showed favorable prognostic predictive performance. Combining clinical features, we found that age, TNM stage, T stage, N stage, M stage, laterality, and multifocality had no significant differences between the two IRS groups, and only sex was associated with IRS (P < 0.05) (Figure S3). Besides, the ESTIMATE and stroma scores were notably higher (P < 0.001), whereas the tumor purity was significantly lower in the high-IRS than those in the low-IRS subgroup (P < 0.05) (Fig. 4E). Utilizing the CIBERSORT algorithm, we found the level of B cell naive, plasma cell, M1 macrophages, and mast cells activated were positively correlated to the IRS, while Tregs, T cells gamma delta, M1 macrophages, mast cells resting, monocytes, and neutrophils exhibited negative correlations (Fig. 4F). Further, MCP-counter analysis showed that B cells, monocytes, myeloid dendritic cells, and cancer associated fibroblast were enrichment in the high-IRS subgroup (Fig. 4G).

Construction of the immune risk model and evaluation of the immune infiltration. (A) Comparison of 28 immune cell types between high- and low-risk subgroups. (B) The heatmap of five cell types. (C) Kaplan-Meier analysis showed that the overall survival of high-IRS was poorer than that of the low-IRS subgroup (p = 0.0049). (D) The area under the ROC curve (AUC) of the immune risk score. (E–G) The immune cell infiltration landscape in the immune risk score subgroups was analyzed using ESTIMATE, CIBERSOFT, and MCP counter, respectively. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, and ns: no significance.
Molecular basis of the IRS signature
To better comprehend the potential biological mechanisms between the high- and low-IRS subgroups, we performed gene enrichment analysis by Metascape to explore their mechanism pathways. The top 6 GO terms used were TNFα signaling via NF-kB, epithelial mesenchymal transition, response to growth factor, blood vessel development, leukocyte differentiation, and KRAS signaling up (Fig. 5).

The functional and pathway enrichment analysis of m5C-lncRNAs by Metascape. (A) Bar graph of enriched terms, colored by p-values. (B) Network of enriched terms: colored by term ID, where nodes that share the same term ID are typically close to each other. (C) Network of enriched terms: colored by p-value, where terms containing more genes tend to have a more significant p-value.
Association between risk score and response to immunotherapy
To explore the role of the risk score in the immunotherapy response of PTCs. We compared the expression levels of PD-1, PD-L1, CTLA-4, TIM-3, LAG-3, and KLRB1 between high- and low-risk subgroups. The expression of the above immune checkpoints in the low-risk subgroup was higher than in the high-risk subgroup (Fig. 6A). Meanwhile, we found that patients in high-risk subgroup have better to immunotherapy compared with those in low-risk subgroup (P = 0.022) (Fig. 6B). Besides, we separated patients into response and no response groups according to the predicted response to ICB. Interestingly, the ssGSEA scores including central memory CD4 T cell, eosinophil, monocyte, T follicular helper cell, and type17 T helper cell in the no response groups were significantly higher than the response groups (P < 0.001) (Fig. 6C).

Prediction of the response to immunotherapy. (A) The expression levels of six immune checkpoints between high- and low-risk subgroups. (B) The rate of response to immune checkpoints blockade therapy. (C) The ssGSEA scores of five immune cells were lower in the response group. **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Construction of the m5C-lncRNAs prognosis nomogram
To further improve the accuracy of our survival prognostic model, we developed an integrated nomogram by combining the immune risk model with several predictable clinical features. Univariable and multivariable Cox regression analysis showed that age (HR = 1.15, 95% CI = 1.08–1.23, P < 0.001), risk score (HR = 2.72, 95% CI = 1.54–4.79, P < 0.001), and IRS (HR = 1.69, 95% CI = 1.13–2.54, P = 0.011) were the independent prognostic factors for PTCs (Table S3). Subsequently, an integrated nomogram was established (Fig. 7A). The C-index and AUC of the nomogram were 0.968 and 0.969 (Fig. 7B), respectively, indicating good discrimination. Moreover, the calibration plots showed excellence in predicting the 3- and 5-year overall survival (Fig. 7C). Besides, decision curve analysis demonstrated the integrated nomogram could achieve a greater net benefit than any others (Fig. 7D). These data suggested the advantageous usability of our prognostic prediction model in clinical practice.

Construction of the m5C-lncRNAs prognosis nomogram. (A) A nomogram was developed to predict the survival probability. (B) The AUC value of the nomogram. (C) The calibration plot of the nomogram model. (D) Decision curve analysis and clinical impact curve of the nomogram.
Downregulation of PPP1R12A-AS1 promotes proliferation and metastasis by activating the MAPK signaling pathway
To verify the expression level of m5C-lncRNAs in the PTC tissues and cells for subsequent functional and molecular experiments, we performed RT-qPCR analysis on m5C-lncRNAs involved in the construction of nomogram. Unfortunately, the primer sequence of AC018647.2 and AC024257.3 are difficult to design and could not be found on the Lncipedia website (https://lncipedia.org/), and the expression level of POLR2J4 and PVT1 have no significant difference. While the ZNF451-AS1 and PPP1R12A-AS1 were down-regulated in PTC tissues compared with normal samples (Fig. 8A). Furthermore, we further utilized 4 PTC cell lines to verify ZNF451-AS1 and PPP1R12A-AS1 expression at the RNA level (Fig. 8B and C). The expression of PPP1R12A-AS1 was significantly lower in PTC cells, especially in the TPC-1 cells. Therefore, PPP1R12A-AS1 was chosen for further experiments.

Downregulation of PPP1R12A-AS1 promotes proliferation and metastasis by activating the MAPK signaling pathway. (A–C) The expression level of m5C-lncRNAs in 20 pairs of PTC tissues and 5 cell lines. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, and ns: no significance. (D) The efficiency of knocking down PPP1R12A-AS1 was verified by qRT-PCR. (E,F) Colony formation and EdU assay showed that silenced PPP1R12A-AS1 dramatically enhanced cell proliferation. (G) PPP1R12A-AS1 knockdown promotes cell viability by CCK-8 assay. (H) Cell cycle phase distribution in TPC-1 cells transfected with si-NC and si-PPP1R12A-AS1. (I) Invasive and migratory capacity of TPC-1 cells after transfection with si-NC and si-PPP1R12A-AS1. (J–L) Tumor weight and volume of nude mice in si-NC and si-PPP1R12A-AS1 groups. (M) The expression levels of related signaling pathway proteins were detected by WB assay. *P < 0.05, **P < 0.01, ***P < 0.001, and ns: no significance.
To further elucidate the biological functions of PPP1R12A-AS1 in PTC. To this end, we knocked down endogenous PPP1R12A-AS1 by using three siRNAs in TPC-1 cells, the reduction effect was confirmed by qRT-PCR (Fig. 8D). Colony formation and EdU assay showed that silenced PPP1R12A-AS1 dramatically enhanced cell proliferation (Fig. 8E, F). The CCK-8 assay revealed that PPP1R12A-AS1 knockdown significantly increased cell viability (Fig. 8G). Likewise, cell cycle analysis showed that PPP1R12A-AS1 silenced significantly increased the ratio of S phase compared with the control group (P < 0.05) (Fig. 8H). For the transwell assays, as shown in Fig. 8I, cell migration and invasion were significantly enhanced when PPP1R12A-AS1 knockdown (P < 0.05). To further explore the effect of PPP1R12A-AS1 in vivo, a subcutaneous tumor xenograft model was established. Knocking down PPP1R12A-AS1 significantly promoted tumor growth (Fig. 8J–L). Taken together, these data indicate that PPP1R12A-AS1 could suppress thyroid cancer growth in vivo and in vitro.
The MAPK and PI3K/AKT pathways are important for the proliferation and invasion of thyroid cancer cells25. Western blot analysis demonstrated that downregulation of PPP1R12A-AS1 markedly increased p38, p-p38, and ERK expression levels. However, no difference was observed in AKT and p-AKT (Fig. 8M). These results indicated that ablation of PPP1R12A-AS1 led to the activation of the MAPK signaling pathway in thyroid cancer.
Discussion
Despite with relatively favorable prognosis, PTCs still have a risk of disease recurrence and persistence5. Identifying the high-risk patients in the early stage is vital for clinicians to adopt more aggressive treatment. Currently, the risk stratification and prognostic prediction for PTCs are mainly dependent on the AJCC staging system26,27. Considering the heterogeneity of PTCs, it is limited to the recognition of patients with progression in the early phase. Hence, developing novel molecular indicators that effectively predict the prognosis of PTCs is necessary and urgent. In this study, we found that m5C regulators were significantly lower expressed in PTC, and then used six m5C-lncRNAs signatures to construct a prognostic risk model and IRS model. Meantime, we analyzed the role of the risk score in tumor immune infiltration and immunotherapy response of PTCs. Finally, an integrated nomogram was established, based on risk score, IRS, and patients’ age, which yielded a superior performance. To our knowledge, this is the first study to report the role of m5C-lncRNAs in PTC.
m5C is one of the common post-transcriptional modifications, which is closely related to cancer tumorigenesis and disease pathogenesis6,8. Previous studies have demonstrated that m5C modification was implicated in the initiation and progression of liver carcinoma28,29. Also, there was existing evidence showing that m5C related regulators could be recognized as diagnostic and prognostic biomarkers of carcinoma30,31,32. As mentioned above, m5C is found in a wide range of RNAs in all life domains but has also been detected in lncRNA and small ncRNAs33. m5C deposition in ncRNA and affects its processing into regulatory small RNA34,35. In one report, conventional bisulfite sequencing revealed a marked disappearance of known target m5C methylation in the non-coding RPPH1 when NSUN2 was knocked down36. More recently, another study reported that aberrant NSUN2 mediated methylation of H19 lncRNA by recruiting the G3BP1 oncoprotein and thus affected tumor proliferation and invasion28. Besides, Pan et al.37. implemented bioinformatic analysis to mining m5C related prognostic lncRNAs in TCGA datasets and successfully established a risk score model to forecast the overall survival of lung adenocarcinoma. Taken together, these researches confirmed that m5C methylation modification can target or modulate lncRNAs to influence carcinoma development. In this study, we provide a new insight into the interplay between lncRNAs and m5C modification in PTCs.
In addition, the tumor immune microenvironment has received extensive attention. The TME consists of stromal cells, immune cells, extracellular matrix, lymphatic and vascular system, and bioactive molecules, which play a key role in cancer occurrence, immune escape, and therapeutic efficacy38,39. Several studies have shown a complexity and strong interrelationship between TME infiltrating immune cells and m5C modification. Schoeler et al.40. demonstrated that TET enzymes promoted a permissive chromatin state in antibody-mediated immunity. TET3 and TET2 guided the transition of germinal center B cells to antibody-secreting plasma cells. In breast cancer, the aberrant NSUN2 was associated with the up-regulation of macrophages/monocytes and Tregs in the TME41. To investigate the role of the immune microenvironment on prognosis. We constructed a prognostic IRS model, and the AUC value was 0.764, indicating favorable discrimination performance and IRS was positively correlated with the risk score, which is consistent with the above results. Besides, the ESTIMATE and stroma scores were notably higher, whereas the tumor purity was significantly lower in the high-IRS than those in the low-IRS subgroup. The abundance levels and proportions of B cell naive, plasma cell, M1 macrophages, and mast cells activated were higher in the high-IRS subgroup. Similarly, MCP-counter algorithms analysis showed that the level of B cells, monocytes, and myeloid dendritic cells was higher in the high-IRS subgroup. The above results demonstrated that high-IRS patients have a lower proportion of antitumor immune cells and a higher proportion of tumor-promoting immune cells.
Currently, immune checkpoint inhibitors (ICB), such as anti-CTLA-4 and anti-PD-L1/PD-1 antibodies, elicit durable and effective responses in some solid tumors42,43. However, the efficacy and side effects of each patient in the treatment show individual differences44. Therefore, it is necessary to identify patients who might benefit from ICB therapy. In our study, the expression levels of PD-1, PD-L1, CTLA-4, TIM-3, LAG-3, and KLRB1 were significantly lower in high-risk subgroups, and patients with high-risk scores tended to have better responses to ICB. This finding indicates that ICB therapy might be more effective for this specific population. Therefore, the m5C-lncRNAs risk score may be a useful immunotherapy predictor of PTC outcome in clinical practice. The underlying mechanism of the relationship between m5C modification and ICB requires further exploration.
Furthermore, to improve the accuracy of our survival prognostic model, we developed an integrated nomogram based on risk score, IRS, and age. The revised 8th TNM stage updated the age cutoff from 45 to 55 years, which reclassified a large number of DTC patients into lower stages45,46. Although the TNM 8th edition has made great improvements in predicting prognosis on the basis of the TNM 7th edition47, there is still no concept of molecular profile for prognostic risk stratification and insufficient to distinguish the risk of mortality in PTCs. In this study, the C-index and AUC of the nomogram indicated good discrimination of the model. Moreover, the calibration plots showed excellence in predicting the 3- and 5-year OS, and decision curve analysis demonstrated the integrated nomogram could achieve a greater net benefit than any others.
Finally, we validated the role of PPP1R12A-AS1 in TC cell proliferation and invasion. To our knowledge, this is the first study that confirmed the tumor suppressor role of PPP1R12A-AS1 in thyroid cancer. Tumor progression is a series of molecular events that lead to abnormal phenotypes and genetic instability, and the change of the tumor immune microenvironment is also an important factor for metastasis. As mentioned earlier, m5C-lncRNAs were significantly associated with immune cells and immune checkpoint inhibitors. PPP1R12A known as the Myosin Phosphatase Targeting Subunit 1 (MYPT1), also belongs to the Myosin Phosphatase Targeting Protein (MYPT) family. In cancer cells, PPP1R12A considers as a key regulator of the Wnt/β-catenin pathway48,49. PPP1R12A also plays a critical role in the Hippo pathway which is triggered by external stimuli like TGβ1 or EGF, it acts on the stability of YAP/TAZ, thereby controlling cell proliferation and growth50. In this study, we identified that PPP1R12A-AS1 suppressed proliferation and metastasis by activating the MAPK signaling pathway. MAPK activation plays a crucial role in regulating cell proliferation, dedifferentiation, and thyroid tumorigenesis. The mutated genes that affect this pathway encode intracellular signal transducers RAS and BRAF and cell-membrane receptor tyrosine kinases NTRK1 and RET, which leads to progression of PTC51. Treatment with MAPK pathway inhibitors can increase the sensitivity of RAI52. This finding might provide a potential therapeutic target for advanced thyroid cancer.
However, there are also several limitations in the present study. Firstly, our main findings were based on accessed databases, resulting in an inevitable selection bias in clinical and genetic data. Secondly, the cutoff value of the risk score model was defined as the median risk score. Actually, this definition lacks rigor and requires larger clinical patient cohorts to verify. Lastly, the research on the mechanism of immune difference is based on the inference of a variety of algorithms and has not been verified by experiments. Hence, future clinical and experimental studies are necessary to validate the application of our survival prediction model in clinical practice.
Conclusion
With the aid of bioinformatics analysis methods, this study proved that m5C-lncRNAs could predict the clinical prognostic risk and regulate the TME in PTCs. Besides, a risk score model based on the expression level of m5C-lncRNAs could be used to screen suitable patients who can benefit from for ICB therapy. Furthermore, an integrated nomogram combining infiltrating immune cells with m5C-lncRNAs exhibited good predictive performance. More importantly, the downregulation of PPP1R12A-AS1 promotes proliferation and metastasis by activating the MAPK signaling pathway. These findings indicated that m5C-lncRNAs have a promising value in tumor prognosis and individual immunotherapy.
Data availability
The datasets used and analyzed during the current study are available from the corresponding author upon reasonable request.
References
Wang, W., Yang, Z. & Ouyang, Q. A nomogram to predict skip metastasis in papillary thyroid cancer. World J. Surg. Oncol. 18 (1), 167 (2020).
Zhang, S. et al. Cancer incidence and mortality in China, 2015. J. Natl. Cancer Cent. 1 (1), 2–11 (2021).
Wang, W., Meng, C., Ouyang, Q., Xie, J. & Li, X. Magnesemia: an independent risk factor of hypocalcemia after thyroidectomy. Cancer Manag. Res. 11, 8135–8144 (2019).
Wang, W. et al. Identification and validation of potential novel biomarkers to predict distant metastasis in differentiated thyroid cancer. Annals Transl. Med. 9 (13), 1053–1053 (2021).
Ding, Y. & Zhou, R. Distant lymph node metastasis in differentiated thyroid cancer: a population-based cohort study. Surgeon (2024).
Nombela, P., Miguel-Lopez, B. & Blanco, S. The role of m(6)A, m(5)C and psi RNA modifications in cancer: novel therapeutic opportunities. Mol. Cancer 20 (1), 18 (2021).
Dominissini, D. & Rechavi, G. 5-methylcytosine mediates nuclear export of mRNA. Cell. Res. 27 (6), 717–719 (2017).
Trixl, L. & Lusser, A. The dynamic RNA modification 5-methylcytosine and its emerging role as an epitranscriptomic mark. Wiley Interdiscip. Rev. RNA 10 (1), e1510 (2019).
Pan, J., Huang, Z. & Xu, Y. m5C RNA methylation regulators predict prognosis and regulate the immune microenvironment in lung squamous cell carcinoma. Front. Oncol. 11, 657466 (2021).
Chen, H. et al. M(5)C regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in lung adenocarcinoma. Transl. Lung Cancer Res. 10 (5), 2172–2192 (2021).
Li, Q. et al. NSUN2-mediated m5C methylation and METTL3/METTL14-mediated m6A methylation cooperatively enhance p21 translation. J. Cell. Biochem. 118 (9), 2587–2598 (2017).
Chen, H. et al. M(5)C modification of mRNA serves a DNA damage code to promote homologous recombination. Nat. Commun. 11 (1), 2834 (2020).
Chen, X. et al. 5-methylcytosine promotes pathogenesis of bladder cancer through stabilizing mRNAs. Nat. Cell. Biol. 21 (8), 978–990 (2019).
Li, Y. et al. Novel long noncoding RNA NMR promotes tumor progression via NSUN2 and BPTF in esophageal squamous cell carcinoma. Cancer Lett. 430, 57–66 (2018).
Carella, A. et al. Epigenetic downregulation of TET3 reduces genome-wide 5hmC levels and promotes glioblastoma tumorigenesis. Int. J. Cancer 146 (2), 373–387 (2020).
Ferrari, S. M. et al. Immune and inflammatory cells in thyroid cancer microenvironment. Int. J. Mol. Sci. 20 (18). (2019).
Hurst, Z. et al. Risk haplotypes uniquely associated with radioiodine-refractory thyroid cancer patients of high African ancestry. Thyroid 29 (4), 530–539 (2019).
Motzer, R. J. et al. Molecular subsets in renal cancer determine outcome to checkpoint and angiogenesis blockade. Cancer Cell. 38 (6), 803–817e804 (2020).
Prat, A. et al. Immune-related gene expression profiling after PD-1 blockade in non-small cell lung carcinoma, head and neck squamous cell carcinoma, and melanoma. Cancer Res. 77 (13), 3540–3550 (2017).
Miao, Y. R. et al. ImmuCellAI: a unique method for comprehensive T-cell subsets abundance prediction and its application in cancer immunotherapy. Adv. Sci. 7 (7), 1902880 (2020).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44 (D1), D457–462 (2016).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28 (1), 27–30 (2000).
Wang, W., Bai, N. & Li, X. Comprehensive analysis of the prognosis and drug sensitivity of differentiation-related lncRNAs in papillary thyroid cancer. Cancers (Basel) 14 (5). (2022).
Ding, Y., Liu, M., Wang, W. & Li, X. YTHDF3-induced degradation of P4HA2 mRNA inhibits glycolysis in papillary thyroid cancer through hippo signaling pathway. Int. J. Biol. Macromol. 291, 139150 (2024).
Su, X. et al. Vitamin C kills thyroid cancer cells through ROS-dependent inhibition of MAPK/ERK and PI3K/AKT pathways via distinct mechanisms. Theranostics 9 (15), 4461–4473 (2019).
Ghaznavi, S. A. et al. Using the American thyroid association risk-stratification system to refine and individualize the American joint committee on cancer eighth edition disease-specific survival estimates in differentiated thyroid cancer. Thyroid 28 (10), 1293–1300 (2018).
Wang, W. et al. The role of m6A RNA methylation-related lncRNAs in the prognosis and tumor immune microenvironment of papillary thyroid carcinoma. Front. Cell. Dev. Biol. 9, 719820 (2021).
Sun, Z. et al. Aberrant NSUN2-mediated m(5)C modification of H19 lncRNA is associated with poor differentiation of hepatocellular carcinoma. Oncogene 39 (45), 6906–6919 (2020).
Xue, C., Zhao, Y., Li, G. & Li, L. Multi-omic analyses of the m(5)C Regulator ALYREF reveal its essential roles in hepatocellular carcinoma. Front. Oncol. 11, 633415 (2021).
Xiang, S. et al. M(5)C RNA methylation primarily affects the ErbB and PI3K-Akt signaling pathways in gastrointestinal cancer. Front. Mol. Biosci. 7, 599340 (2020).
Sun, L. et al. Large-scale transcriptome analysis identified RNA methylation regulators as novel prognostic signatures for lung adenocarcinoma. Ann. Transl Med. 8 (12), 751 (2020).
Wang, J., Yu, F., Shang, Y., Ping, Z. & Liu, L. Thyroid cancer: incidence and mortality trends in China, 2005–2015. Endocrine 68 (1), 163–173 (2020).
Amort, T. et al. Long non-coding RNAs as targets for cytosine methylation. RNA Biol. 10 (6), 1002–1008 (2014).
Hussain, S. et al. NSun2-mediated cytosine-5 methylation of vault noncoding RNA determines its processing into regulatory small RNAs. Cell. Rep. 4 (2), 255–261 (2013).
Sajini, A. A. et al. Loss of 5-methylcytosine alters the biogenesis of vault-derived small RNAs to coordinate epidermal differentiation. Nat. Commun. 10 (1), 2550 (2019).
Squires, J. E. et al. Widespread occurrence of 5-methylcytosine in human coding and non-coding RNA. Nucleic Acids Res. 40 (11), 5023–5033 (2012).
Pan, J., Huang, Z. & Xu, Y. m5C-related lncRNAs predict overall survival of patients and regulate the tumor immune microenvironment in lung adenocarcinoma. Front. Cell. Dev. Biol. 9, 671821 (2021).
Binnewies, M. et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 24 (5), 541–550 (2018).
Xu, X. et al. Association of germline variants in natural killer cells with tumor immune microenvironment subtypes, tumor-infiltrating lymphocytes, immunotherapy response, clinical outcomes, and cancer risk. JAMA Netw. Open 2 (9), e199292 (2019).
Schoeler, K. et al. TET enzymes control antibody production and shape the mutational landscape in germinal centre B cells. FEBS J. 286 (18), 3566–3581 (2019).
Mou, W. et al. Expression of Sox2 in breast cancer cells promotes the recruitment of M2 macrophages to tumor microenvironment. Cancer Lett. 358 (2), 115–123 (2015).
Lei, Q., Wang, D., Sun, K., Wang, L. & Zhang, Y. Resistance mechanisms of anti-PD1/PDL1 therapy in solid tumors. Front. Cell. Dev. Biol. 8, 672 (2020).
Kumagai, S. et al. The PD-1 expression balance between effector and regulatory T cells predicts the clinical efficacy of PD-1 blockade therapies. Nat. Immunol. 21 (11), 1346–1358 (2020).
Akin Telli, T. et al. PD-1 and PD-L1 inhibitors in oesophago-gastric cancers. Cancer Lett. 469, 142–150 (2020).
Nam, S. H. et al. A comparison of the 7th and 8th editions of the AJCC staging system in terms of predicting recurrence and survival in patients with papillary thyroid carcinoma. Oral Oncol. 87, 158–164 (2018).
Shteinshnaider, M. et al. Reassessment of differentiated thyroid cancer patients using the eighth TNM/AJCC classification system: a comparative study. Thyroid 28 (2), 201–209 (2018).
Kim, T. H. et al. Prognostic value of the eighth edition AJCC TNM classification for differentiated thyroid carcinoma. Oral Oncol. 71, 81–86 (2017).
Zhou, L. et al. Merlin-deficient human tumors show loss of contact inhibition and activation of Wnt/beta-catenin signaling linked to the PDGFR/Src and Rac/PAK pathways. Neoplasia 13 (12), 1101–1112 (2011).
Benvenuto, G. et al. Expression profiles of PRKG1, SDF2L1 and PPP1R12A are predictive and prognostic factors for therapy response and survival in high-grade serous ovarian cancer. Int. J. Cancer 147 (2), 565–574 (2020).
Serrano, I., McDonald, P. C., Lock, F., Muller, W. J. & Dedhar, S. Inactivation of the Hippo tumour suppressor pathway by integrin-linked kinase. Nat. Commun. 4, 2976 (2013).
Nikiforov, Y. E. & Nikiforova, M. N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 7 (10), 569–580 (2011).
Chakravarty, D. et al. Small-molecule MAPK inhibitors restore radioiodine incorporation in mouse thyroid cancers with conditional BRAF activation. J. Clin. Investig. 121 (12), 4700–4711 (2011).
Funding
This work was supported by the Postdoctoral Fellowship Program of CPSF (GZC20233167), China Postdoctoral Science Foundation (2024M763707).
Author information
Authors and Affiliations
Contributions
YD, CL and WW designed the study. Entire experiments and data analysis were mainly performed by YD and partially by WW, XL and CL. The initial draft of the manuscript was written by YD and XL and revised by WW and CL. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
Informed consent was obtained from all the participants and approved by the Ethics Committee of Xiangya Hospital of Central South University (No. 202004192).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Ding, Y., Li, X., Wang, W. et al. Integrative analysis of 5-methylcytosine associated signature in papillary thyroid cancer. Sci Rep 15, 4405 (2025). https://doi.org/10.1038/s41598-025-88657-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-88657-2
Keywords
This article is cited by
-
Identification of prognostic genes related to T cell proliferation in papillary thyroid cancer based on single-cell RNA sequencing and bulk RNA sequencing data
Clinical and Experimental Medicine (2025)
