
Abstract
Gastric cancer (GC) is one of the most common tumors; one of the reasons for its poor prognosis is that GC cells can resist normal cell death process and therefore develop distant metastasis. Cuproptosis is a novel type of cell death and a limited number of studies have been conducted on the relationship between cuproptosis-related genes (CRGs) in GC. The purpose of the present study was to establish a prognostic model of CRGs and provide directions for the diagnosis and treatment of GC. Transcriptome and clinical data of patients with GC were collected from The Cancer Genome Atlas and Gene Expression Omnibus datasets. Single sample gene set enrichment analysis (GSEA) and the randomized forest method were used to establish the prognostic model. Kaplan-Meier survival curve, receiver operating characteristics diagram and a nomogram were used to evaluate the reliability of the model. GSEA and gene set variation analysis (GSVA) were used to examine enrichment pathways between high and low risk groups. Finally, immunohistochemical analysis was used to examine ephrin 4 (EFNA4) expression in GC samples and determine the prognosis of patients with GC based on the expression pattern of EFNA4. A group of 7 predictive models (RTKN2, INO80B, EFNA4, ELF2, MUSTN, KRTAP4, and ARHGEF40) was established which were correlated with CRGs. This model can be used as an independent prognostic factor to predict the prognosis of patients with GC. GSEA and GSVA results indicated that high risk patients with GC were mainly associated with the enrichment of ANGIOGENESIS and TGF_BETA_SIGNALING pathways. Finally, EFNA4 expression in GC was significantly higher than that in normal tissues, and patients with GC and high EFNA4 expression exhibited improved prognosis. In conclusion, the prognosis model based on CRGs could be used as the basis for predicting the potential prognosis of patients with GC and provide new insights for the treatment of GC.
Similar content being viewed by others


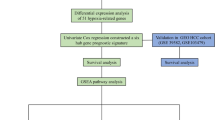
Introduction
Gastric cancer (GC) is the world’s fifth most common type of cancer. It is also the third most common cause of cancer-related death1. More than 1 million new cases of early GC occur each year. Early GC can be directly resected by endoscopy, while advanced GC can only be treated by surgery. The early GC postoperative 5-year survival rate can reach 90%, whereas the 5-year survival rate of stage IA and IB patients with GC can reach > 60%. Patients with stage III GC exhibit a 5-year survival rate of 18%2. Despite the high survival rate following early surgery, the overall survival rate for patients with GC worldwide is only 25%3. The reason for this is that most patients with GC are diagnosed at an advanced stage. Therefore, it is necessary to develop more effective prognostic models for this disease.
Copper is essential in the normal physiological activities of eukaryotes including energy metabolism, iron absorption, and signaling. An imbalance of copper in the body leads to the development of various diseases, such as anemia and centripenia, as well as the development and progression of tumors4,5. Cuprotosis is a newly discovered mode of cell death that differs from other known modes of death and is closely related to mitochondrial respiration. Specifically, copper causes toxic protein stress by binding to the fatty acylation component of the tricarboxylic acid cycle, which leads to cell death6. Considering the role of abnormal copper levels in tumors, several patients undergo treatment for their tumors via interference with the copper content in their body. The copper chelating agent Tetrathiomolybdate inhibits the transformation growth of melanoma cell lines, and the same effect also occurs in stem cell hepatoma7,8. The copper ion carrier disulfide binds to copper ions in breast cancer cells, thereby effectively inhibiting proteomase activity in these cells9. Therefore, the construction of cuproptosis-related genes (CRGs) can provide an improved basis for the prognosis of patients with tumors, as well as GC.
In the present study, the accuracy of the CRGS-based model was comprehensively investigated with regard to prognosis, clinical characteristics, receiver operating characteristics (ROC) and nomogram analysis of patients with GC in the training and validation sets. Moreover, the enrichment pathways of patients with GC were analyzed in the high and low risk groups. Finally, the expression levels of ephrin 4 (EFNA4) were investigated in each cell line by western blot analysis and in GC and normal tissues by immunohistochemical (IHC) analysis. Disease prognosis was determined according to the expression of EFNA4 in GC.
Materials and methods
Sample collection
The mRNA expression profile data of all samples were obtained from the The Cancer Genome Atlas (TCGA) and Gene Expression Omnibus (GEO) databases. GSE84437 served as the training set and TCGA-STAD and Gene Set Enrichment (GSE)26,253 served as the external verification set. To ensure the reliability of the analysis, paracancer tissues and samples with incomplete clinical information were deleted. Finally, 433 samples were included in GSE84437, 432 samples in GES26253, and 350 samples in TCGA-STAD (Table S2). Since the data for this study came from GEO and TCGA databases, we collected the maximum amount of data while deleting samples with paracarcinoma tissue and incomplete clinical information in order to ensure the reliability of the analysis. Peter et al.6 identified 10 copper death genes (FDX1, LIAS, LIPT1, DLD, DLAT, PDHA1, PDHB, MTF1, GLS, and CDKN2A) for subsequent analysis.
Model construction
In order to establish a reliable CRG signature, the CRGs were screened. Firstly, single sample gene set enrichment analysis (ssGSEA) was applied to calculate the cuproptosis score of each sample based on 10 CRGs10. Subsequently, based on the copper death fraction, the samples were divided into high and low copper death fraction groups and the differentially expressed genes in the two groups were screened. Subsequently, univariate Cox regression analysis was used to screen the genes associated with prognosis in patients with GC11. The following screening criteria were used: P < 0.05. Finally, the differentially expressed genes and the prognosis of the intersection of copper were considered to be death-related genes.
The random forest model was applied to establish the signature associated with cuproptosis in GC. The process was implemented based on the “randomForestSRC” package12. The random forest algorithm can greatly reduce the risk of overfitting by integrating the prediction results of multiple decision trees, thus improving the prediction accuracy, and can calculate a large number of data.Different signatures were established through arrangement and combination, and the signature with the lowest P value was regarded as the optimal signature. According to the Risk score =∑ Coefgene×Expgenes, Coefgene corresponds to the coefficient of each prognostic gene and Expgenes corresponds to the expression level of each gene. Based on median risk scores, the samples from the training and external verification sets were divided into high and low risk groups, respectively. In addition, the random survival forest algorithm was used to screen the importance of prognostic genes, and the data indicated the top 10 most important genes.
Verification of gene signature associated with cuproptosis
The Kaplan-Meier (KM) survival analysis was used to evaluate the survival of patients with high risk and low risk, a process completed based on “survival” and “survminer” packages13. Subsequently, the “survivalROC” package was used to plot a ROC curve to assess the specificity and sensitivity of the signature. In order to further evaluate a prognostic model for independent prognostic factors of GC, univariate and multivariate Cox regression analyses were used to assess the signature in the training and the external validation sets of the prognostic value. In addition, the prognostic value of this signature in patients with GC was analyzed with different clinical characteristics (age, sex, tumor size, and lymph node metastasis). Finally, nomograms were established based on signature and clinical features for clinical application. To ensure the accuracy of this nomogram, a calibration curve was drawn to assess the difference between the prediction and the real situation. The Nomogram itself is built on the basis of a prognostic model trained with big data, and does not involve migration problems. We only need to evaluate the actual situation of the patient to determine the prognosis of the patient.
Analysis pathways gene set variation analysis (GSVA) and gene set enrichment analysis (GSEA)
Both GSEA14 and GSVA15 assess the enrichment of gene sets based on gene expression. The difference between the two lies in that GSEA assesses the enrichment of gene sets based on the contribution of the genes in the gene sets, while GSVA uses ssGSEA to calculate the gene sets and “limma” to analyze the differences between groups. To assess the signaling pathways associated with the prognostic model, GSEA and GSVA were used to assess enrichment pathways in the high risk and low risk groups. GSEA (Version 4.3.2) was used for GSEA and “GSVA” package was used for GSVA analysis. P < 0.05 and FDR < 0.05 were considered to indicate statistically significant differences and were used to screen for signaling pathways. The Nomogram itself is built on the basis of a prognostic model trained with big data, and does not involve migration problems. We only need to evaluate the actual situation of the patient to determine the prognosis of the patient.
Cell culture, protein extraction, and western blot analysis
GES-1, HGC-27, AGS, and MKN-45 cells were purchased from the Cell Resource Center of Peking Union Medical College. All cells were cultured in a complete medium with RPMI-1640 (Gibco; Thermo Fisher Scientific, Inc.), 10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.), and 1% penicillin and streptomycin (Gibco; Thermo Fisher Scientific, Inc.) at a ratio of 100:10:1. All cells were cultured in the presence of 5% CO2at 37˚C17. The protein was extracted according to the instructions of the Abbkine ExKine™ total protein extraction kit and the target protein was detected by western blot analysis according to the procedure reported by Zhang et al.18. In the present study, the primary antibodies used were EFNA4 (1:1,000, R&D Systems, Inc.; cat. no. MAB369-SP) and GAPDH (rabbit, 1:10,000, Proteintech Group, Inc., cat. no. 60004-1-Ig).
Patients and specimens
All GC and normal specimens were collected from the Gansu Provincial Hospital and confirmed by pathological examination as patients with GC. A total of 88 patients from January 2016 to January 2023 were collected were enrolled.None of the patients received treatment prior to surgery. The Ethics Committee of Gansu Provincial Hospital approved the study (2023099). All patients in the cohort signed an informed consent form.
IHC analysis and outcome scoring
IHC operation was performed in accordance with the instructions provided by the manufacturer and Tan et al.19. The primary antibody used was anti-E (1:50, R&D Systems, Inc.; cat. no. MAB369-SP). IHC scores were evaluated independently by two pathologists. The staining area of EFNA4 was quantified according to the proportion of stained positive cells. The staining strength score consisted of 0 (no staining), 1 (weak), 2 (moderate), and 3 (strong), the expression intensity was weak at 0–25%, moderate at 25–50%, and strong at > 50%. The strength and percentage used were multiplied by 100 to calculate the final stain fraction. Finally, an organizational score of ≥ 150 was defined as high expression and < 150 as low expression.
Statistical analysis
SPSS software package (version 24.0, SPSS, Inc.) and GraphPad Prism (version 8.0, GraphPad Software, Inc.) were used for statistical analysis. All data are expressed as mean ± SEM. Statistical differences between experimental and control groups were analyzed using an unpaired two-tailed Student’s t-test (2 groups) or a one-way ANOVA (> 2 groups) followed by a multiple post hoc comparisons test (Dunnett’s test). The KM method was used to compare the GC survival time at different risk levels. Univariate Cox regression analysis was used to analyze the prognostic value of a single gene and Cox regression was used to analyze whether this factor could be used as an independent prognostic factor for GC. P < 0.05 was considered to indicate a statistically significant difference.
Results
Construction and genetic characteristics of a signature related to cuproptosis
Based on the copper death fraction, a total of 739 CRGs were screened by differential expression and univariate Cox regression analyses (Table S1). The random forest model was applied and a 7-gene signature was finally confirmed as the optimal signature (Fig. 1A). The following formula was used to estimate the risk score: Risk score = −0.0012×RTKN2 + −0.0027×INO80B + −0.0009×EFNA4 + 0.0029×ELF2 + 0.0050×MUSTN + −0.0050×KRTAP4-5 + −0.0054×ARHGEF40. By calculating the importance of the genes, it was found that it was ranked high for this signature (Fig. 1B). The position, similarity, forest map, and expression of the signature genes in the chromosomes were further explored. By analyzing the location of genes on chromosomes, it was found that these 7 genes were all located on autosomes (Fig. 1C). EFNA4, INO80B, and ELF2 were highly similar in terms of participating in the biological processes and molecular functions (Fig. 1D). Forest map results indicated that high expression levels of ELF2 and MUSTN1 suggested poor prognosis, while high expression levels of RTKN2, INO80B, ARHGEF40, KRTAP4-5, and EFNA4 were significantly correlated with improved prognosis (Fig. 1E). Differential expression analysis indicated that ELF2 and MUSTN1 demonstrated significantly high expression in the high risk group, while RTKN2, INO80B, ARHGEF40, KRTAP4-5, and EFNA4 exhibited significantly low expression in the high risk group (Fig. 1F).

Construction and genetic characteristics of gene signatures associated with copper-associated cellular death. (A) Expression of different gene signatures. (B) The importance of the signature genes was ranked through the random forest analysis. (C) The location of genes on chromosomes in the gene signatures. (D) Similarity of genes in molecular functions and biological processes in the gene signature. (E) The best gene signature selection using the forest analysis (F) Differences in gene signature expression between high and low risk groups.
Verification of gene signature associated with copper-associated cellular death
The parameters of gene expression were used to calculate the risk score in the patients, who were classified into high risk and low risk groups. It was found that both in the training set and in the external validation, the mortality rate from the high risk group was significantly higher than that of the low risk group (Fig. 2A and B). ROC curve analysis indicated that CRGs exhibited a high accuracy in the training set (GSE84437; Fig. 3A). Surprisingly, the gene signature was more accurate in the external validation sets TCGA-STAD (Fig. 3B) and GES26253 (Fig. 3C). KM survival analysis indicated that this prognostic signature differentiated prognosis in TCGA-STAD [overall survival (OS)], GSE84437 (OS), and GES26253 (relapse free survival; Fig. 3D).

The risk score distribution for each patient was showed in the training dataset and external validation dataset. (A) Distribution of risk score in the training set GSE84437. (B) Distribution of risk score in the external verification set TCGA-STAD. The top image represents the ranking of the risk score, the middle the distribution of the total survival time of patients with GC, and the bottom the expression of the gene signature. GSE, Gene Set Enrichment; GC, gastric cancer.

Prognostic value of the risk signature in GC. (A) 3-year and 5-year ROC analysis of TCGA-STAD. (B) 3-year and 5-year ROC analysis of GSE84437. (C) 3-year and 5-year ROC analysis of GES26253. (D) KM survival analysis based on OS in the TCGA-STAD queue. (E) KM survival analysis based on OS in the GSE84437 queue. (F) KM survival analysis based on RFS in GES26253. ROC, receiver operating characteristics; TCGA, The Cancer Genome Atlas; GSE, Gene Set Enrichment; KM, Kaplan-Meier; OS, overall survival; RFS, relapse free survival.
Relationship between the prognostic signature associated with cuproptosis and the clinical features
In order to identify genetic markers associated with cuproptosis, which can be used as independent prognostic factors for GC, univariate and multivariate Cox regression analyses were performed. The results of the univariate and multivariate Cox regression analyses indicated that this prognostic feature exhibited significant prognostic value in both the training set GSE84437 and the external validation set TCGA-STAD (Fig. 4A). Therefore, a signature associated with cuproptosis as an independent prognostic factor was established.

The independence identification of the risk model. (A) Univariate Cox regression analysis of the GSE84437 cohort. (B) Multivariate Cox regression analysis results of the GSE84437 cohort. (C) Univariate Cox regression analysis of the TCGA-STAD cohort. (D) The results of the multivariate Cox regression analysis of the TCGA-STAD cohort.
By using the analysis of high risk and low risk groups, the distribution of patients with different clinical features in the high risk and low risk groups was examined according to different age and sex distributions; the proportion of high risk patients significantly increased with the increase of pathological grading and according to the tumor node metastasis (TNM) stage. The proportion of high risk group patients in Stage IV was significantly increased (Fig. 5A). The results of KM survival analysis in the clinical subgroups indicated that prognosis was distinguishable in age, sex, and pathological_N subgroup except for the T1-T2 subgroup (Fig. 5B). This indicated that the prognostic signature associated with cuproptosis was closely related to the commonly used clinical pathological grade and TNM stage; in addition, its predictive ability was not affected by clinical characteristics.

Clinical characteristics of patients in high and low risk groups. (A) The correlations between the risk model and clinical factors. including Age, Gender, Histologic grade and TNM stage. (B) Kaplan-Meier survival curves for the high- and low-risk groups stratified by clinical factors. including Age. Gender and Stage.
Construction and evaluation of nomogram
A nomogram was established for clinicians to quantitatively determine patient outcomes based on clinical characteristics of prognostic variables associated with malformations and GC (Fig. 6A). Based on the age, gender, tumor size, lymph node metastasis, and the scores corresponding to the prognostic signature associated with copper-associated cellular death of specific patients with GC, their total scores were used to determine the 5-year and 8-year survival rates. By constructing a calibration curve, it was found that the predicted survival rate of the nomogram was very close to the actual survival rate (Fig. 6B).

Construction of the nomogram and calibration curve plot. (A) Establishment of a nomogram based on signature and clinical characteristics related to copper-associated cellular death. (B) Calibration curve plot for predicting 5-year and 8-year OS in TCGA dataset.
GSVA and GSEA. To further explore the potential molecular mechanism of the gene signature associated with cuproptosis, GSEA and GSVA analyses were performed. GSVA results indicated that, TGF_BETA_SIGNALING, UV_RESPONSE_DN, WNT_BETA_CATENIN_SIGNALING, APOPTOSIS, MITOTIC_SPINDLE, PI3K_AKT_MTOR_SIGNALING, NOTCH_SIG, NALING, and MYC_TARGETS_V2 signaling pathways were significantly enriched in the high risk group (Fig. 7A). GSEA enrichment results indicated that ANGIOGENESIS (ES = 0.32, P < 0.001), MYOGENESIS (ES = 0.29, P < 0.001), and PANCREAS_BETA_CELLS (ES = 0.26, P < 0.001) were significantly enriched in the high risk group (Fig. 7B). G2M_CHECKPOINT (ES=−0.37, P < 0.001), MITOTIC_SPINDLE (ES=−0.32, P = 0.003) and MYC_TARGETS_V2 (ES = −0.48, P < 0.001) were significantly enriched in the low risk group (Fig. 7C).

Results of GSVA and GSEA. (A) Top 16 GSVA enrichment results. (B) GSEA results were significantly enriched in the high risk group. (C) GSEA results were significantly enriched in the low risk group. GSVA, gene set variation analysis; GSEA, Gene Set Enrichment Analysis.
Expression of EFNA4 in tissues and cells
IHC analysis was used to detect the expression of EFNA4 in GC and normal tissues. EFNA4 expression was significantly lower in normal gastric tissues than in GC tissues (Fig. 8A); patients with GC and low expression exhibited improved prognosis (Fig. 8E). Western blot analysis indicated that the expression levels of EFNA4 in GC cells were the lowest in the normal gastric epithelial cell line GES-1, which were considerably lower than those of the other three GC cell lines. In tumor cells, the expression levels of MKN-45 were the highest (Fig. 8F).

Expression and prognostic value of EFNA4 in GC. (A-C) Negative staining of EFNA4 in normal tissues. (B-D) Positive staining of EFNA4 in GC tissues. (E) Survival analysis of EFNA4 low and high expression groups. (F) The expression levels of EFNA4 in GC cells. (G) relative expression of EFNA4 in GES-1, HGC-27, AGS, and MKN-45 cells. *P < 0.05, ***P < 0.001.
Discussion
Approximately 42% of GC cases worldwide occur in China and the majority of the patients are detected at a late stage, resulting in poor prognosis16. The existing prognostic evaluation criteria do not adequately meet clinical needs. Novel therapeutic strategies, such as immune checkpoint inhibitors have greatly improved the survival of patients with GC; however, the beneficiaries are limited17. Therefore, the development of a new prognostic criterion is urgently required.
Tumor cells are characteristic of the resistance to natural death, which comprises the basis of cancer occurrence and development; therefore, the cells cannot self destruct, which is considered to be related to the growth and metastasis of cancer18. Similar to other normal cell death patterns, ferroptosis aids gemcitabine to inhibit the resistance to pancreatic cancer19. Pyroptosis affects all stages of tumor carcinogenesis20. Similarly, the control of copper levels in the body can selectively kill tumor cells21. A new Schiff base copper (II) complex can inhibit the growth of multiple cancer cells and promote apoptosis22; this drug exhibits the same effect in GC cells23. Therefore, the application of the CRGs in assessing the occurrence and prognosis of GC has significant value.
A total of 10 CRGs were used as the main research database. The TCGA-STAD expression and survival data in the database were used to establish a prognostic model with cuproptosis abnormal gene expression differences related to the genetic variations and prognosis. Moreover, 7 CRG signatures were established using machine learning and the random forest model. EFNA4 is a component of the ephrin family; its members are widely expressed in a variety of cells by binding to cell membranes and are closely associated with the emergence of tumors24. EFNA4 has been extensively implicated in the progression of triple negative breast and ovarian cancer25, lung cancer26, and hepatocellular carcinoma24. Several articles analyzed the increased expression of EFNA4 in GC by means of literature searching; the results indicated that EFNA4 expression correlated with OS and disease free survival in patients with GC27,28, although the experimental verification was absent.The role of EFNA4 in the development of GC is currently unclear, and our study can only confirm that EFNA4 expression in GC is elevated and associated with poor prognosis in GC. INO80B, a subunit of INO80, is associated with multiple functions, such as DNA replication, transcriptional regulation, chromatin remodeling, and embryonic stem cell renewal29,30,31,32. By strongly mediating ATP-dependent chromatin remodeling, INO80 affects oncogenic transcription and tumor growth in melanoma33and non-small cell lung cancer34. The permanent division of embryonic stem cells resembles the characteristics of tumor cells. Therefore, the role of INO80B in tumor progression deserves further assessment. However, a limited number of studies have been conducted on the expression and prognosis of INO80B in GC. ELF2 also known as NERF (new Ets-related factor) is involved in the regulation of B and T cell development, cell cycle progression, and angiogenesis35. Overexpression of ELF2 can enhance various malignant phenotypes of nasopharyngeal carcinoma cells36and ELF2 can serve as an independent prognostic factor for non-small cell lung cancer37. Additional studies are required to clarify the relationship between ELF2 and GC. Rho guanine nucleotide exchange factor 40 (ARHGEF40) is a member of the Dbl-family of guanine nucleotide factor proteins, which participate in actin aggregation, cytoskeletal recombination, cell migration, and cell polarity38. ARHGEF40 regulates the progression of hereditary melanoma39and promotes proliferation and invasion of non-small cell lung cancer40. The role of ARHGEF40 in GC requires further verification. Rhotekin (RTKN) 2, a member of the RTKN protein family, is a Rho effector. The RTKN protein is involved in the regulation of key cellular functions, such as cell proliferation and cell cycle progression, cytoplasmic division, apoptosis, and transformation41. Reduction of RTKN2 expression in hepatocellular carcinoma42, colorectal cancer43, and non-small cell lung cancer44 leads to a corresponding decrease in cell proliferation and invasion. The expression of RTKN2 was increased in GC and its overexpression enhanced the proliferation of GC cells, promoted their migration and invasion and inhibited the induction of their apoptosis45. Keratin associated protein (KRTAP) 4–5, a member of the KRTAP gene family, is the main component of the hair proteome and plays an important role in hair formation. Therefore, the total KRTAP gene expression in hairy animals, such as sloths, is 10-fold higher than that noted in hairy animals, such as hedgehogs46. A limited number of reports have been noted on the association of KRTAP4-5 with tumors. Musculoskeletal nuclear protein (MUSTN) 1 is a key regulator of cell differentiation and tissue growth and is associated with normal embryonic development, bone, and skeletal muscle regeneration47. Previous studies on MUSTN1 have focused on cartilage and muscle tissue; studies on tumors are very limited and additional work is required.
GSVA converts the expression matrix between different samples into the expression matrix of the set of genes in the sample, thereby assessing whether different pathways are directly enriched in different samples. By using this analysis, the enrichment pathways of high risk patients with GC can be noted more directly and reference to the targeted clinical treatment can be provided. The TGF_BETA_SIGNALING pathway plays a key role in regulating the behavior of tumor cells48 and its role in GC has been well documented with multiple genes promoting the development of GC via TGF-β49,50,51. The WNT_BETA_CATENIN_SIGNALING pathway is widely implicated in human tumors and abnormal activation is strongly associated with increased prevalence, malignant progression of tumors, poor disease prognosis, and even increased cancer-related mortality52. The important role of this pathway in GC progression has also been demonstrated in multiple studies, and multiple genes have been identified to act via Wnt/β-catenin53,54. GSEA has been used as a pre-done variance analysis between the samples to further analyze and identify the gene set of interest. ANGIOGENESIS is the first gene set to be enriched in high risk patients with GC, and this process is one of the markers of cancer. Disorganized, immature, and permeable vascular networks often lead to poor perfusion creating a hypoxic environment more conducive to tumor invasion55. Angiogenic-related factors secreted by GC cells activate endothelial cells and the autocrine ring to stimulate GC development56, while angiogenic-related factors also play an integral role in enhancing GC immunotherapy57. Excessive muscle loss is often observed in patients with malignant tumors and has been shown to be associated with poor prognosis58. It has also been observed that high expression of multiple myogenic proteins in GC is associated with poor prognosis of this disease59,60. Therefore, it is beneficial to investigate the associated mechanisms of GC progression so as to develop effective treatment options.
The model of the present study has multiple advantages. Firstly, a gene signature was developed based on the copper death fraction in GC for the first time. Considering that cuproptosis is a novel cell death mode and depends on mitochondrial function, it may provide a new treatment scheme for GC. Secondly, the model of the present study provides improved results in the validation set, which is surprising. Thirdly, the predicted survival rate of the nomogram was basically consistent with the actual survival rate, indicating the accuracy and clinical application value of the model. In short, Using machine learning, we identified a new prognostic factor.A risk model created by combining ordinal prognostic factors with this new prognostic factor had better predictive power than conventional staging system. Then, it is necessary to speculate and discuss how it might be useful in actual clinical practice.
The present study exhibits several limitations. First of all, although the verification of our model was investigated by two external validation sets, the sample size of the patient specimens was insufficient, resulting in limited clinical information. This will be the focus of the subsequent studies. Secondly, the copper death fraction gene set was based on data analysis from public databases and was inevitably limited by selection bias, In order to reduce selection bias, we randomly selected one data set as the training set and the other two data sets as the verification set. This maximizes the reliability of our model construction and validation. Thirdly, the biological function of the related genes in GC has not been verified and will be verified sequentially in future studies.
Data availability
All dataset used and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Smyth, E. C., Nilsson, M., Grabsch, H. I., van Grieken, N. C. & Lordick, F. Gastric cancer. Lancet 396(10251), 635–648. https://doi.org/10.1016/S0140-6736(20)31288-5 (2020).
Sexton, R. E., Hallak, A., Diab, M. N., Azmi, A. S. & M., & Gastric cancer: A comprehensive review of current and future treatment strategies. Cancer Metastasis Rev. 39(4), 1179–1203. https://doi.org/10.1007/s10555-020-09925-3 (2020).
Rugge, M., Fassan, M. & Graham, D. Epidemiology of gastric Cancer. In Gastric Cancer (ed. Strong, V.) (Springer, 2015). https://doi.org/10.1007/978-3-319-15826-6_2.
Ruiz, L. M., Libedinsky, A. & Elorza, A. A. Role of copper on mitochondrial function and metabolism. Front. Mol. Biosci. 8, 711227. https://doi.org/10.3389/fmolb.2021.711227 (2021).
Babak, M. V. & Ahn, D. Modulation of intracellular copper levels as the mechanism of action of anticancer copper complexes: Clinical relevance. Biomedicines 9(8), 852. https://doi.org/10.3390/biomedicines9080852 (2021).
Tsvetkov, P. et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science 375(6586), 1254–1261. https://doi.org/10.1126/science.abf0529 (2022).
Brady, D. C., Crowe, M. S., Greenberg, D. N. & Counter, C. M. Copper chelation inhibits BRAFV600E-driven melanomagenesis and counters resistance to BRAFV600E and MEK1/2 inhibitors. Cancer Res. 77(22), 6240–6252. https://doi.org/10.1158/0008-5472.CAN-16-1190 (2017).
Davis, C. I. et al. Altered copper homeostasis underlies sensitivity of hepatocellular carcinoma to copper chelation. Metallomics Integr. Biometal Sci. 12(12), 1995–2008. https://doi.org/10.1039/d0mt00156b (2020).
Chen, D., Cui, Q. C., Yang, H. & Dou, Q. P. Disulfiram, a clinically used anti-alcoholism drug and copper-binding agent, induces apoptotic cell death in breast cancer cultures and xenografts via inhibition of the proteasome activity. Cancer Res. 66(21), 10425–10433. https://doi.org/10.1158/0008-5472.CAN-06-2126 (2006).
Yi, M., Nissley, D. V., McCormick, F. & Stephens, R. M. ssGSEA score-based Ras dependency indexes derived from gene expression data reveal potential Ras addiction mechanisms with possible clinical implications. Sci. Rep. 10(1), 10258. https://doi.org/10.1038/s41598-020-66986-8 (2020).
Emura, T., Matsui, S. & Chen, H. Y. compound.Cox: Univariate feature selection and compound covariate for predicting survival. Comput. Methods Programs Biomed. 168, 21–37. https://doi.org/10.1016/j.cmpb.2018.10.020 (2019).
Li, J. et al. A multicenter random forest model for effective prognosis prediction in collaborative clinical research network. Artif. Intell. Med. 103, 101814. https://doi.org/10.1016/j.artmed.2020.101814 (2020).
Liu, J. et al. Cancer genome atlas research network, an integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173(2), 400–416.e11. https://doi.org/10.1016/j.cell.2018.02.052 (2018).
Subramanian, A. et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102(43), 15545–15550. https://doi.org/10.1073/pnas.0506580102 (2005).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinform. 14, 7. https://doi.org/10.1186/1471-2105-14-7 (2013).
Gastric Cancer Association, China Anti-Cancer Association. Zhonghua wei chang wai ke za zhi = Chin. J. Gastrointest. Surg. 24(9), 741–748. https://doi.org/10.3760/cma.j.cn.441530-20210831-00351 (2021).
Kang, Y. K. et al. Nivolumab plus chemotherapy versus placebo plus chemotherapy in patients with HER2-negative, untreated, unresectable advanced or recurrent gastric or gastro-oesophageal junction cancer (ATTRACTION-4): A randomised, multicentre, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 23(2), 234–247. https://doi.org/10.1016/S1470-2045(21)00692-6 (2022).
Strasser, A. & Vaux, D. L. Cell death in the origin and treatment of cancer. Mol. Cell 78(6), 1045–1054. https://doi.org/10.1016/j.molcel.2020.05.014 (2020).
Yang, J. et al. Ferroptosis: At the crossroad of gemcitabine resistance and tumorigenesis in pancreatic cancer. Int. J. Mol. Sci. 22(20), 10944. https://doi.org/10.3390/ijms222010944 (2021).
Al Mamun, A. et al. Role of pyroptosis in cancer and its therapeutic regulation. Eur. J. Pharmacol. 910, 174444. https://doi.org/10.1016/j.ejphar.2021.174444 (2021).
Ge, E. J. et al. Connecting copper and cancer: From transition metal signalling to metalloplasia. Nat. Rev. Cancer 22(2), 102–113. https://doi.org/10.1038/s41568-021-00417-2 (2022).
Bao, R. D. et al. A new Schiff base copper(II) complex induces cancer cell growth inhibition and apoptosis by multiple mechanisms. J. Inorg. Biochem. 208, 111103. https://doi.org/10.1016/j.jinorgbio.2020.111103 (2020).
Xia, Y. et al. A new Schiff base coordinated copper(II) compound induces apoptosis and inhibits tumor growth in gastric cancer. Cancer Cell Int. 19, 81. https://doi.org/10.1186/s12935-019-0801-6 (2019).
Lin, J. et al. EFNA4 promotes cell proliferation and tumor metastasis in hepatocellular carcinoma through a PIK3R2/GSK3β/β-catenin positive feedback loop. Mol. Therapy Nucleic Acids. 25, 328–341. https://doi.org/10.1016/j.omtn.2021.06.002 (2021).
Damelin, M. et al. Anti-EFNA4 Calicheamicin conjugates effectively target triple-negative breast and ovarian tumor-initiating cells to result in sustained tumor regressions. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 21(18), 4165–4173. https://doi.org/10.1158/1078-0432.CCR-15-0695 (2015).
Zhao, X. et al. Oncogenic EFNA4 amplification promotes lung adenocarcinoma lymph node metastasis. Cancers 14(17), 4226. https://doi.org/10.3390/cancers14174226 (2022).
Ji, Y. et al. Identification of the hub genes and prognostic indicators of gastric cancer and correlation of indicators with tumor-infiltrating immune cell levels. J. Cancer 12(13), 4025–4038. https://doi.org/10.7150/jca.52105 (2021).
Xie, R., Yuan, M. & Jiang, Y. The Pan-cancer Crosstalk between the EFNA Family and Tumor Microenvironment for Prognosis and Immunotherapy of Gastric Cancer. Front. cell. Dev. Biol. 10, 790947. https://doi.org/10.3389/fcell.2022.790947 (2022).
Conaway, R. C. & Conaway, J. W. The INO80 chromatin remodeling complex in transcription, replication and repair. Trends Biochem. Sci. 34(2), 71–77. https://doi.org/10.1016/j.tibs.2008.10.010 (2009).
Wang, J., Gao, S., Peng, X., Wu, K. & Yang, S. Roles of the INO80 and SWR1 chromatin remodeling complexes in plants. Int. J. Mol. Sci. 20(18), 4591. https://doi.org/10.3390/ijms20184591 (2019).
Papamichos-Chronakis, M., Watanabe, S., Rando, O. J. & Peterson, C. L. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell 144(2), 200–213. https://doi.org/10.1016/j.cell.2010.12.021 (2011).
Wang, L. et al. INO80 facilitates pluripotency gene activation in embryonic stem cell self-renewal, reprogramming, and blastocyst development. Cell. Stem Cell. 14(5), 575–591. https://doi.org/10.1016/j.stem.2014.02.013 (2014).
Zhou, B. et al. INO80 governs superenhancer-mediated oncogenic transcription and tumor growth in melanoma. Genes Dev. 30(12), 1440–1453. https://doi.org/10.1101/gad.277178.115 (2016).
Zhang, S. et al. INO80 is required for oncogenic transcription and tumor growth in non-small cell lung cancer. Oncogene 36(10), 1430–1439. https://doi.org/10.1038/onc.2016.311 (2017).
Guan, F. H. et al. The antiproliferative ELF2 isoform, ELF2B, induces apoptosis in vitro and perturbs early lymphocytic development in vivo. J. Hematol. Oncol. 10(1), 75. https://doi.org/10.1186/s13045-017-0446-7 (2017).
Ma, X. & Li, Y. Circ_0028007 aggravates the malignancy of nasopharyngeal carcinoma by regulating miR-656-3p/ELF2 axis. Biochem. Genet. 60(6), 2069–2086. https://doi.org/10.1007/s10528-022-10205-8 (2022).
Jin, S. Q., Pu, Y. F., Qiu, Y., Aozasa, K. & Morii, E. Zhejiang da xue xue bao. Yi xue ban = Journal of Zhejiang University. Medical sciences 43(4), 413–419. https://doi.org/10.3785/j.issn.1008-9292.2014.06.001 (2014).
Reiner, D. J. & Lundquist, E. A. Small GTPases. WormBook: the online review of C. elegans biology 2018, 1–65. https://doi.org/10.1895/wormbook.1.67.2 (2018).
Campos, C. et al. High-throughput sequencing identifies 3 novel susceptibility genes for hereditary melanoma. Genes 11(4), 403. https://doi.org/10.3390/genes11040403 (2020).
Gu, J. et al. ARHGEF40 promotes non-small cell lung cancer proliferation and invasion via the AKT-Wnt axis by binding to RhoA. Mol. Carcinog. 61(11), 1016–1030. https://doi.org/10.1002/mc.23457 (2022).
Reid, T. et al. Rhotekin, a new putative target for rho bearing homology to a serine/threonine kinase, PKN, and rhophilin in the rho-binding domain. J. Biol. Chem. 271(23), 13556–13560. https://doi.org/10.1074/jbc.271.23.13556 (1996).
Wei, W., Chen, H. & Liu, S. Knockdown of Rhotekin 2 expression suppresses proliferation and invasion and induces apoptosis in hepatocellular carcinoma cells. Mol. Med. Rep. 13(6), 4865–4871. https://doi.org/10.3892/mmr.2016.5113 (2016).
Pang, X. et al. Knockdown of Rhotekin 2 expression suppresses proliferation and induces apoptosis in colon cancer cells. Oncol. Lett. 14(6), 8028–8034. https://doi.org/10.3892/ol.2017.7182 (2017).
Ji, L. et al. RTKN2 is Associated with unfavorable prognosis and promotes progression in Non-small-cell Lung Cancer. OncoTargets Ther. 13, 10729–10738. https://doi.org/10.2147/OTT.S260436 (2020).
Zhao, H. G. et al. RTKN2 enhances radioresistance in gastric cancer through regulating the Wnt/β-Catenin signalling pathway. Folia Biol. 68(1), 33–39 (2022).
Khan, I. et al. Mammalian keratin associated proteins (KRTAPs) subgenomes: Disentangling hair diversity and adaptation to terrestrial and aquatic environments. BMC Genom. 15(1), 779. https://doi.org/10.1186/1471-2164-15-779 (2014).
Hadjiargyrou, M. Mustn1: A developmentally regulated pan-musculoskeletal cell marker and regulatory gene. Int. J. Mol. Sci. 19(1), 206. https://doi.org/10.3390/ijms19010206 (2018).
Derynck, R., Akhurst, R. J. & Balmain, A. TGF-beta signaling in tumor suppression and cancer progression. Nat. Genet. 29(2), 117–129. https://doi.org/10.1038/ng1001-117 (2001).
Im, J. Y. et al. VGLL1 phosphorylation and activation promotes gastric cancer malignancy via TGF-β/ERK/RSK2 signaling. Biochim. Biophys. Acta Mol. Cell. Res. 1868(1), 118892. https://doi.org/10.1016/j.bbamcr.2020.118892 (2021).
Li, Y. et al. IGHG1 induces EMT in gastric cancer cells by regulating TGF-β/SMAD3 signaling pathway. J. Cancer. 12(12), 3458–3467. https://doi.org/10.7150/jca.56056 (2021).
Xiong, R., Yin, T., Gao, J. L. & Yuan, Y. F. HOXD9 activates the TGF-β/Smad signaling pathway to promote gastric cancer. OncoTargets Ther. 13, 2163–2172. https://doi.org/10.2147/OTT.S234829 (2020).
Yu, F. et al. Wnt/β-catenin signaling in cancers and targeted therapies. Signal. Transduct. Target. Therapy 6(1), 307. https://doi.org/10.1038/s41392-021-00701-5 (2021).
Tian, S. et al. SERPINH1 regulates EMT and gastric cancer metastasis via the Wnt/β-catenin signaling pathway. Aging 12(4), 3574–3593. https://doi.org/10.18632/aging.102831 (2020).
Guo, Q. et al. ADMA mediates gastric cancer cell migration and invasion via Wnt/β-catenin signaling pathway. Clin. Transl. Oncol. Off. Publ. Federation Span. Oncol. Soc. Natl. Cancer Inst. Mexico 23(2), 325–334. https://doi.org/10.1007/s12094-020-02422-7 (2021).
Viallard, C. & Larrivée, B. Tumor angiogenesis and vascular normalization: Alternative therapeutic targets. Angiogenesis 20(4), 409–426. https://doi.org/10.1007/s10456-017-9562-9 (2017).
Nienhüser, H. & Schmidt, T. Angiogenesis and anti-angiogenic therapy in gastric cancer. Int. J. Mol. Sci. 19(1), 43. https://doi.org/10.3390/ijms19010043 (2017).
Qing, X. et al. Molecular characteristics, clinical significance, and cancer immune interactions of angiogenesis-associated genes in gastric cancer. Front. Immunol. 13, 843077. https://doi.org/10.3389/fimmu.2022.843077 (2022).
Armstrong, V. S., Fitzgerald, L. W. & Bathe, O. F. Cancer-associated muscle wasting-candidate mechanisms and molecular pathways. Int. J. Mol. Sci. 21(23), 9268. https://doi.org/10.3390/ijms21239268 (2020).
Li, Y. F., Zhu, G. Y., Ma, Y. & Qu, H. Y. Expression and prognosis of MYOZ2 in gastric cancer. Eur. Rev. Med. Pharmacol. Sci. 22(18), 5920–5927. https://doi.org/10.26355/eurrev_201809_15921 (2018).
Shum, A. M. et al. Disruption of MEF2C signaling and loss of sarcomeric and mitochondrial integrity in cancer-induced skeletal muscle wasting. Aging 4(2), 133–143. https://doi.org/10.18632/aging.100436 (2012).
Acknowledgements
Not applicable.
Funding
This work was supported: The 2022 Natural Science Foundation of Gansu Province (2022-0405-JCC-0319, 20JR5RA145, 2022RCXM025); Key project of science and technology innovation platform fund of Gansu Provincial People’s Hospital (21gssya-4, 16gssy2-5); Postdoctoral Fund of Gansu Provincial People’s Hospital (ZX-62000001-2022-010, ZX-62000001-2022-055).
Author information
Authors and Affiliations
Contributions
Yulan Li and Pengxian Tao designed this study. Xiaolong Liu analyzed the data, wrote the manuscript and collected the data. Pengxian Tao analyzed the data. Yulan Li and He Su revised the manuscript. All authors approved the final version for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval and consent to participate
The study involving human subjects, including human tissues, was approved by the Human Research Ethics Committee of Gansu Provincial Hospital, Ref. 2023-099. All study participants confirmed and agreed in writing to the form of personally identifiable data. Our research is in line with the declaration of Helsinki (https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-in volving-human-subjects/).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, X., Tao, P., Su, H. et al. Machine learning-random forest model was used to construct gene signature associated with cuproptosis to predict the prognosis of gastric cancer. Sci Rep 15, 4170 (2025). https://doi.org/10.1038/s41598-025-88812-9
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-88812-9
