
Abstract
Clinical samples are vital for understanding diseases, but their scarcity requires refined research methods. Emerging single-cell technologies offer detailed views of tissue heterogeneity but need sufficient fully characterized tissues. We developed an optimized single-nuclei RNA sequencing (snRNA-seq) protocol to extract nuclei from just 15 mg of cryopreserved human tissue. Applied to four cancer tissues (brain, bladder, lung, prostate), it profiled 1550–7468 nuclei per tissue, revealing heterogeneity comparable to public single-cell atlases. This method enhances the use and sharing of rare, cryopreserved biospecimens, supporting research where sample quantity is limited and full tissue characterization is needed.
Similar content being viewed by others

Introduction
Recent advances in single-cell technologies have unveiled an unprecedented level of detail and insight into the heterogeneity and complexity of biological systems. The creation of comprehensive atlases for various organs and tissues has provided a vast repository of information, enabling the intricate deconvolution of cell populations and a deeper understanding of their diversity1,2,3. The application of single-cell technologies has facilitated the prediction of cellular trajectories, the identification of rare cell populations, a more profound comprehension of malignant cell transformation and numerous other discoveries4,5,6,7. Despite the significant progress achieved, the clinical translation of these technologies still encounters several challenges that must be addressed to fully realize their potential and expand their applications.
Current approaches to investigate the heterogeneity in human tissues are costly or demand access to large amounts of material while optimized for a small number of tissue types8,9,10,11. Moreover, most of these methodologies have been developed with fresh tissues as the starting material, inherently assuming that samples can be immediately processed. This poses significant logistical challenges, particularly because samples are often collected from multiple locations distant from research laboratories thereby increasing costs and restricting availability. Consequently, maintaining the integrity of fresh samples during transport becomes a critical issue. Additionally, the process of dissociating tissues into a single-cell suspension typically involves extensive mechanical and enzymatic steps over several hours to isolate live cells12. This procedure induces alterations in transcriptional profiles and introduces sequencing noise as well as cell-type dissociation biases, thereby compromising the quality of downstream analyses12,13,14,15. Moreover, for many research projects, the use of fresh samples is not feasible because a comprehensive pathological characterization is required to select appropriate samples for analysis. The detailed characterization of the tissue often precludes its immediate processing, making it difficult to adhere to protocols designed for fresh specimens. Therefore, expanding the applicability of single-cell technologies to low-input cryopreserved material in repositories would be beneficial for researchers.
Numerous protocols and methods have been developed to efficiently extract nuclei from diverse tissues prior to single-nuclei RNA sequencing (snRNA-seq)16,17. Transcriptomic analyses from isolated nuclei have demonstrated equivalency to those obtained from live cells isolated from fresh tissues, suggesting that nuclei are a reliable proxy18,19,20,21,22,23. Additionally, snRNA-seq has been shown to provide datasets that more accurately reflect the composition of cell populations within the tissue8,13,24,25,26. These methodologies have proven successful in profiling samples that have been cryopreserved for extended periods of time, although they typically require a minimum starting material of around 30 to over 100 mg to provide reliable results9,18,27,28. While some approaches have been adapted for low-input material, most protocols are optimized for specific tissue types8,29,30 complicating their application across different tissues. Optimizing conditions for nuclei extraction is crucial for a successful snRNA-seq experiment. The process requires careful mechanical homogenization and an optimal balance between high-quality nuclei and minimal cellular debris31,32. Standardizing these approaches to efficiently use low-input cryopreserved material for snRNA-seq is crucial to fully leverage the potential of clinical samples and expand the range of tissues studied with these techniques.
Developing an adaptable and versatile method compatible with multiple tissues for generating snRNA-seq data is a significant challenge due to the diverse biophysical characteristics of different tissues. In this study, we propose a method to extract high-quality nuclei from low-input cryopreserved human tissues. Our approach optimizes the solubilization process for cancer tissues with distinct biophysical properties (brain, bladder, lung, and prostate). By incorporating nuclei sorting to purify samples before tagging and sequencing, we ensure that only the most intact nuclei are used for analysis. Our results were comparable to those from existing cellular atlases, suggesting that our approach is both robust and suitable for a wide range of tissues with varying biophysical characteristics.
Methods
The detailed protocol is available as supplementary information.
Tissue specimen
The research conducted within this study involved the use of human cancer samples. Prior to the collection of all human samples, the research protocols were reviewed and approved by the Institutional Review Boards (Comité d’éthique de la recherche du CHU de Québec-Université Laval and Comité d'éthique de la recherche de l’Institut universitaire de cardiologie et de pneumologie de Québec – Université Laval). Informed consent was obtained from all participants. Bladder and prostate samples were obtained from the URO-1 biobank of the CHU de Québec-Université Laval (Projects No. 2012-1002 and 2024-6885). Brain samples were obtained from the CHU de Québec – Université Laval (Project No. 2023-6392 and 2024-6885). Lung samples were obtained from the Biobanque du Réseau de recherche en santé respiratoire du Québec (biobanque.ca) – site Institut universitaire de cardiologie et de pneumologie de Québec – Université Laval (Project No. MP-10-2024-4071). All experiments were performed in accordance with relevant guidelines and regulations.
Tissue homogenization
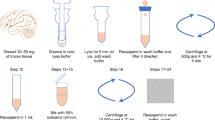
Cryopreserved samples were minced in a pre-cooled mortar on dry ice using a scalpel, transferred into 15 mL tubes prior to adding 3 mL of ice-cold lysis buffer (10 mM Tris–HCl pH 7.4, 10 mM NaCl 10, 3 mM MgCl2.6H2O, 0.05% NP-40). A Dounce homogenizer was used with different pestles: pestle A (loose, clearance: 0.0025–0.0055 inches, Fisher Scientific K885301-0002) or pestle B (tight, clearance: 0.0005–0.0025 inches, Fisher Scientific K885303-0007). The number of strokes and the selection of the pestle were optimized for each tissue (Table 1). Following homogenization, samples were transferred, and 2 mL of ice-cold lysis buffer were added on ice and incubated for 5 min. The lysis reaction was stopped with the addition of 5 mL of ice-cold nuclei washing buffer (0.5X PBS, 5% BSA, 0.25% Glycerol, 40 units/mL Protector RNAse inhibitor). Following filtration on 30 µm MACS strainers (Miltenyi Biotech, Bergisch Gladbach,Germany), samples were centrifuged for 10 min at 1000 g (4 °C). Pellets were resuspended in 1 mL of nuclei washing buffer and then 1 mL of a 50% (wt/vol) solution of iodixanol (Optiprep Millipore Sigma, Burlington, USA) was added. The nuclei suspension was gently transferred on top of a 2 mL cushion of 29% (wt/vol) iodixanol. The pellet was resuspended in 300 µL of nuclei washing buffer. Microscopy images were obtained using the ZEIZZ Axio Observer 7 (Carl-Zeiss-Stiftung, Oberkochen, Germany) and the Fiji image processing package (v 1.54j).
Nuclei sorting
Following homogenization, nuclei were stained with 7-AAD (Thermo Fisher Scientific Waltham, USA, Cat. No. A1310) for 10 min before sorting using a BD FACSAria Fusion (Becton Dickinson, New Jersey, USA). A 70 µm nozzle with a 4-way purity mask was used to increase the quality of the sorted nuclei and reduce the final volume. To estimate the relative size of the nuclei, non-fluorescent flow cytometry size calibration beads were used with sizes of 7.88,10.1, and 16.4 uM (Spherotech Cat. No. PPS-6 k). Fluorescent-positive events within size limits were collected and centrifuged at 1000 g for 10 min at 4 °C. Approximately 70 µL of the supernatant was kept to resuspend the purified nuclei before quantification and quality control by microscopy. Sorting plots were generated by FCSExpress (v7 DeNovo Software, Pasadena, USA).
Single-nuclei sequencing
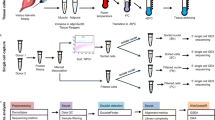
Tagging of single-nuclei and library preparation was performed through Gel Beads-in-emulsion following the 10X Genomics (10 × Genomics, Pleasanton, USA) guidelines (CG000204 Rev. D) by the Genomics platform from the Centre de Recherche du CHUQ de Québec – Université Laval. An estimated 15,300 to 30,300 nuclei were loaded per run. The quality of cDNA was assessed with High Sensitivity D5000 ScreenTape Assay on 4200 TapeStation (Agilent, Santa Clara, USA). Samples that passed the quality control were sequenced on a NovaSeq 6000 (Illumina, San Diego, USA) to a minimal depth of 20,000 paired-end reads (100 bp) per expected nuclei.
Data analysis
Following sequencing, FastQ files were used as input for Cell Ranger (v7.1.0)33. Single-nuclei matrices were examined and filtered to remove ambient RNA using SoupX (v1.6.2)34. Central processing of snRNA-seq was made using Seurat (v5.1.0)35. Values for total number of transcripts (UMI) detected in a single nucleus (nCount_RNA), the number of unique features detected in a single nucleus (nFeature_RNA), and the mitochondrial percentage precent in each dataset was computed. The following filters were applied to ensure quality data: nCount_RNA ≥ 300, nFeatures between 250 and 2500 and mitochondrial percentage < 10%. Additionally, we examined the percentage of ribosomal and hemoglobin genes and the ratio of genes per UMIs (Figure S1). Following filtrations, the dataset was processed to detect and eliminate doublets with DoubletFinder (v2.0.4)36. Processed and filtered datasets were integrated with single-cell atlases downloaded from CZ CELLxGENE (Table S1)37. Prior to integration the SCTransform (SCT) method was applied to each dataset for normalization35. Then integration anchors were identified between the datasets to find common cell types or states. The identified anchors were used to integrate the datasets into a single Seurat object. Batch effects were corrected using the Harmony (v1.2.0) algorithm38. Principal component analysis (PCA) was performed on the integrated data to reduce dimensionality and identify key components. Clustering and dimensionality reduction through Uniform Manifold Approximation and Projection (UMAP) were performed for downstream analyses and visualizations.
For cell type annotations, the nomenclature of cellular populations found within single-cell atlases was used as a reference for manual annotation. Next, we performed curation to refine and validate the annotation by cross-referencing the preliminary labels with single-cell atlases to ensure proper cell type assignment. Discrepancies between the gene expression patterns and marker genes were resolved to ensure robust and biologically meaningful classification of the cell populations. Plots were generated by Seurat (v5.1.0)35, and ggplot2 (v3.5.1)39.
Results
Isolation of single-nuclei starting from low-input cryopreserved tissues
To determine the feasibility of generating quality single-nuclei data from low-input cryopreserved material, we initially tested traditional nuclei isolation involving solubilization and centrifugation. However, significant material loss during the process made this approach unsuitable for low-input material (Figure S2). To develop a more efficient and versatile approach, we investigated alternative methods aiming to work with less than 15 mg of cryopreserved tissue, an amount typically found in a standard biopsy for solid tumors. Through iterative testing and refinement, we developed a protocol optimized for low-input cryopreserved human tissues for single-nuclei transcriptomics (Fig. 1). This protocol is divided into four modules: (1) Homogenization; (2) Nuclei Sorting; (3) Quality Control, and (4) Tagging and Sequencing. We validated the protocol using four tissues of significant interest to cancer research: brain, bladder, lung, and prostate. These tissues exhibit a range of biophysical properties, from low stiffness (brain) to high stiffness (prostate)40,41,42. In the following sections, we will detail the necessary steps to achieve high-quality results.

Workflow for single-nuclei isolation starting with low-input cryopreserved tissues. Outline of the main steps of the process for isolating and preparing single-nuclei for transcriptomics analyses starting with low-input cryopreserved human tissues. Module 1—Homogenization: Tissue samples are mechanically homogenized to release single nuclei while preserving nuclear integrity using a Dounce homogenizer. Module 2—Nuclei Sorting: The homogenized mixture is subjected to fluorescence-activated cell sorting (FACS) to isolate and purify the nuclei based on integrity and size. Module 3—Quality Control: Isolated nuclei undergo quality control assessment with microscopic examination to confirm their integrity and purity. Module 4—Tagging and Sequencing: Nuclei are tagged with unique molecular identifiers (UMIs) and barcoded using the Chromium platform. The tagged nuclei are then subjected to library preparation and high-throughput sequencing followed by data analysis.
Homogenization of cryopreserved tissues requires optimization
Tissue homogenization is the first step of our method to isolate nuclei from cryopreserved tissues. A Dounce homogenizer was used to solubilize the tissue, using specific conditions. The type of pestle and the number of strokes were optimized for each tissue. Two metrics were used to assess nuclei recovery: the number of nuclei per mg of tissue (Fig. 2A–D) and the percentage of good-quality nuclei (Fig. 2E–H). The number of nuclei per mg of tissue ranged between ~ 10,000 (lung) and ~ 75,000 (brain). For single-nuclei applications, we aimed for minimally ~ 50,000 total nuclei starting with between 10 and 15 mg of cryopreserved tissue. In all cases, the total number of nuclei per mg of tissue was sufficient for downstream sequencing application. Therefore, the percentage of good-quality nuclei was used as the main determinant for the best conditions (Fig. 2E–L). The selected conditions ranged from 6 strokes of pestle A (loose) for the brain to 10 strokes of pestle B (tight) for the prostate (Table 1). These results are in line with the higher stiffness of the prostate. It is worth noting that tissue aggregates were still visible suggesting that samples were not over solubilized (Fig. 2I–L). Therefore, determining the homogenization conditions empirically is a critical step for each tissue to be used with our method.

Different cryopreserved tissues require specific homogenization conditions. Homogenization conditions need to be optimized for each tissue. (A–D) Quantification of the number of nuclei per mg of tissue for each sample. Error bars represent the standard deviation for a biological triplicate (only duplicate for bladder due to tissue constraints). (E–H) Quality assessment of the extracted nuclei. Microscopy images were quantified manually. Nuclei were considered high-quality if: the membrane integrity was intact, the shape of the nuclei was consistent, and nuclei aggregates were low. The bars in red represent the optimal number of strokes required to obtain the best quality nuclei. Error bars represent the standard deviation for a biological triplicate. (I–L) Representative microscopy images for the optimal conditions highlighted in red (E–H). Nuclei were stained with 4′,6-diamidino-2-fenilindol (DAPI). Scale bars represent 100 and 50 µm respectively.
Sorting allows recovery of a pure suspension of quality nuclei
Obtaining high-quality single-nuclei data relies on preserving the nuclear integrity, which requires specific conditions and proper handling. Traditional protocols involving centrifugation and washing steps often result in the loss and damage of cells and nuclei. As an alternative, sorting intact nuclei before sequencing has proven effective9,43,44,45. The fluorescent DNA intercalator 7-aminoactinomycin D (7-AAD) has been shown to be safe for single-cell applications20,46,47. We utilized 7-AAD in combination with calibration beads of different sizes (8–16 µm) to set the gate for fluorescence-activated cell sorting (FACS) of nuclei (Fig. 3A). For each tissue, nuclei positive for 7-AAD and between 8 and 16 µm were selected (Fig. 3B–E). We obtained between 307 and 607 nuclei/µL (Table S2). A minimal concentration of 300 nuclei/µL was set as the lower threshold for tagging and sequencing to maximize the number of single nuclei profiled according to 10X Genomics guidelines. Overall, cell sorting minimally affected the recovery process while significantly reducing the presence of debris.

Nuclei are purified using fluorescence-activated cell sorting. Homogenized nuclei were subjected to fluorescence-activated nuclei sorting (FANS) to enrich high-quality nuclei while removing ambient RNA and debris. (A) Left—Scatter plot representing the nuclei distribution of an unlabeled sample based on the Side Scatter-Area (SSC-A) and 7-AAD intensity. The FANS gate (blue box) was set based on standard size beads ranging from 8 to 16 µm. Right – Histogram of the calibration beads using the forward scatter (FSC) signal. (B–E) Left—Scatter plots showing the distribution of nuclei for the brain (B), bladder (C), lung (D) and prostate (E) samples. Nuclei within the FANS gate were sorted and used for downstream analyses. Right – Distribution of the calibration beads and the nuclei (blue) using the FSC to approximate the size of nuclei for the different tissues.
Low-input cryopreserved tissues yield similar heterogeneity to single-cell atlases
Following purification, nuclei were tagged using the 10X Genomics Chromium droplet technology. Following library preparation, samples were sequenced at a minimal depth of 20,000 reads per cell. The quality of the data was assessed using established methods to ensure reliable and accurate data (see methods and Figure S1). We sequenced between 1550 and 7468 nuclei per sample (Figure S3). To compare the heterogeneity of the profiled samples with the tissue of origin, we integrated our results with publicly available single-cell atlases (Fig. 4 and Table S1). Low-input cryopreserved samples and single-cell atlases were normalized, and batch corrected to be combined into the same data set. For each tissue, our sequencing results overlapped with the cluster distribution of the single-cell atlases (Fig. 4A–D). Then, the different cell populations were identified and annotated using available signatures (Fig. 4E–H). For all tissues, combinations of tissue-specific subtypes in addition to immune and endothelial cells were recovered as expected for heterogenous tumors samples. For example, in the prostate tissue we identified all the expected cell populations including luminal, basal, immune and endothelial cells (Fig. 4H). Taken together, these results support the feasibility of our method using low-input cryopreserved tissues to generate representative single-nuclei datasets.

Low-input cryopreserved tissues yield similar heterogeneity to single-cell atlases. To evaluate the representativity of single-nuclei data starting with low-input material, we integrated our results with publicly available single-cell atlases. (A–D) Uniform manifold approximation and projection (UMAP) plots of the individual cells identified in each tissue (red) with a corresponding single-cell atlas (grey). Sources for the atlases are available in Table S1. Each dot represents a single nucleus or cell. (E–H) UMAP plots color-coded according to distinct cell types for the brain (9), the bladder (11), the lung (15) and the prostate (10). The cell annotations represent the cell populations found within the reference atlas for the specific tissue. Each dot represents a single nuclei or cell.
Discussion
Access to well-characterized samples in sufficient quantities is a major barrier for many single-cell experiments, as the high costs of these studies often limit the number of samples and biological replicates that can be processed. To address this issue, we propose a versatile method for extracting nuclei from cryopreserved human tissues available in tissue repositories, which requires minimal input material (Fig. 1). This method has been successfully applied to profile cells from brain, bladder, lung, and prostate tissues (Fig. 4), demonstrating its broad applicability. Notably, some tissues, such as the prostate, have historically posed challenges due to their highly fibrous nature, making them difficult to process for many applications. By focusing on nuclei rather than live single-cell suspensions, we eliminate the constraints associated with extracting viable whole cells. Our findings indicate that the solubilization of the tissues is the most variable and critical step (Fig. 2), necessitating careful optimization for each specific tissue type to ensure the extraction of high-quality nuclei prior to purification (Fig. 3). Once good-quality nuclei are obtained, the subsequent sequencing results are typically robust and reliable (Fig. 4). These results demonstrate that our method is applicable to a wide range of tissues with varying biophysical properties and can be potentially expanded to make the technology accessible to more tissues.
A significant limitation of current single-cell approaches is the substantial amount of tissue required. Samples with high clinical value for diagnostics are frequently available only in limited quantities, and cryopreserved samples, which cannot be replenished, are allocated with caution. Access to these samples is typically granted only when the potential for generating valuable information is sufficiently high. Our results demonstrate that using less than 15 mg of tissue, significantly less than the amounts required by standard reference atlases, yields the expected cellular heterogeneity for each tissue (Fig. 4). Notably, some cell types, such as epithelial cells in the lung are difficult to extract and often show biases in protocols that start with fresh tissues25,48. In this regard, extracting nuclei from cryopreserved tissues may offer a distinct advantage, as we profiled these cell populations that are sensitive to dissociation processes used for fresh samples. By using our method to extract nuclei from low-input cryopreserved tissues, we expand access to valuable samples. This methodology holds significant potential for advancing single-cell research, particularly in clinical and diagnostic contexts where sample availability is a critical constraint.
The development of methods to extract single-cell information from cryopreserved human tissues offers a significant advantage by enabling long-distance collaboration and the molecular analysis of retrospective studies. Our approach has successfully profiled cryopreserved samples stored for long periods (over 20 years), showcasing its versatility and robustness. Compared to other methods, which often require larger sample sizes and fresh tissues, our technique requires minimal tissue input while still delivering high-quality nuclei extraction and cellular heterogeneity across diverse tissues. Reducing the reliance on fresh tissues will enable the investigation of a broader array of tissue types and conditions, enhancing our understanding of tissue heterogeneity. Integrating single cell approaches with well-characterized tissues and long-term clinical follow-up will offer an effective strategy to tackle complex biomedical questions.
Limitations of study
While our method is applicable to at least four cancer tissue types: brain, bladder, lung, and prostate, it has not been extensively tested on additional normal or tumor tissues. The method was developed with a focus on optimizing conditions for low-input cryopreserved human tissues that range different grades stiffness, so we did not benchmark it against different protocols using the same starting material. Although our method is efficient, other conditions may be better suited for specific tissues. For example, protocols tailored for liver tissues often employ different enzymatic treatments to handle the higher metabolic activity26,49 while those designed for cardiac tissues typically use gentle mechanical dissociation to preserve delicate cell structures50. Additionally, we did not compare the cellular heterogeneity obtained from large versus small amounts of starting material, which limits our ability to conclude whether using a low input reduces the likelihood of identifying rare cell populations or introduces biases in cell proportions. Finally, our efforts were focused on transcriptomics, and we did not explore other single-cell approaches, such as those investigating chromatin features, which could provide complementary insights.
Conclusion
Overall, we present a functional, versatile, and robust method for isolating nuclei for single-nuclei transcriptomic analyses across multiple human cancer tissues, with the potential for adaptation to a broader range of applications and tissue types.
Change history
04 June 2025
The original online version of this Article was revised: The Data availability section in the original version of this Article was incorrect. It now reads: “The data generated for this publication is available on Sequence Read Archive51 under accession number PRJNA1137569. All publicly available sequencing datasets used in the manuscript are listed in Table S1.”
Abbreviations
- snRNA-seq:
-
Single-nuclei RNA sequencing
- 7-AAD:
-
DNA intercalator 7-aminoactinomycin D
- FACS:
-
Fluorescence-activated cell sorting
- UMIs:
-
Unique molecular identifiers
- DAPI:
-
4′,6-Diamidino-2-fenilindol
- FANS:
-
Fluorescence-activated nuclei sorting
- SSC-A:
-
Side Scatter-Area
- FSC:
-
Forward scatter
- UMAP:
-
Uniform manifold approximation and projection
References
Pan, L. et al. Single cell Atlas: a single-cell multi-omics human cell encyclopedia. Genome Biol. 25(1), 104 (2024).
Regev, A. et al. The human cell Atlas. eLife https://doi.org/10.7554/eLife.27041 (2017).
Jones, R. C. et al. The tabula sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science 376(6594), eabl4896 (2022).
Aran, D. & Single-Cell, R. N. A. Sequencing for studying human cancers. Annu. Rev. Biomed. Data Sci. 6, 1–22 (2023).
Fan, J., Slowikowski, K. & Zhang, F. Single-cell transcriptomics in cancer: Computational challenges and opportunities. Exp. Mol. Med. 52(9), 1452–1465 (2020).
Chappell, L., Russell, A. J. C. & Voet, T. Single-cell (Multi)omics technologies. Annu. Rev. Genomics Hum. Genet. 19, 15–41 (2018).
Elmentaite, R., Domínguez Conde, C., Yang, L. & Teichmann, S. A. Single-cell atlases: shared and tissue-specific cell types across human organs. Nat. Rev. Genet. 23(7), 395–410 (2022).
Slyper, M. et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med. 26(5), 792–802 (2020).
Nott, A., Schlachetzki, J. C. M., Fixsen, B. R. & Glass, C. K. Nuclei isolation of multiple brain cell types for omics interrogation. Nat. Protoc. 16(3), 1629–1646 (2021).
Nadelmann, E. R. et al. Isolation of nuclei from mammalian cells and tissues for single-nucleus molecular profiling. Curr. Protoc. 1(5), e132 (2021).
Bian, S. et al. Single-cell multiomics sequencing and analyses of human colorectal cancer. Science 362(6418), 1060–1063 (2018).
Machado, L., Relaix, F. & Mourikis, P. Stress relief: Emerging methods to mitigate dissociation-induced artefacts. Trends Cell Biol. 31(11), 888–897 (2021).
Denisenko, E. et al. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 21(1), 130 (2020).
van den Brink, S. C. et al. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods. 14(10), 935–936 (2017).
Adam, M., Potter, A. S. & Potter, S. S. Psychrophilic proteases dramatically reduce single-cell RNA-seq artifacts: A molecular atlas of kidney development. Development 144(19), 3625–3632 (2017).
Kim, N., Kang, H., Jo, A., Yoo, S. A. & Lee, H. O. Perspectives on single-nucleus RNA sequencing in different cell types and tissues. J. Pathol. Transl. Med. 57(1), 52–59 (2023).
Wolfien, M. D. R. & Galow, A. M. Single-Cell RNA sequencing procedures and data analysis. In Bioinformatics (ed. Nakaya, H. I.) (Exon Publications, 2021).
Maitra, M. et al. Extraction of nuclei from archived postmortem tissues for single-nucleus sequencing applications. Nat. Protoc. 16(6), 2788–2801 (2021).
Ding, J. et al. Systematic comparison of single-cell and single-nucleus RNA-sequencing methods. Nat. Biotechnol. 38(6), 737–746 (2020).
Truong, D. D. et al. Dissociation protocols used for sarcoma tissues bias the transcriptome observed in single-cell and single-nucleus RNA sequencing. BMC Cancer 23(1), 488 (2023).
Bakken, T. E. et al. Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLoS One. 13(12), e0209648 (2018).
Guillaumet-Adkins, A. et al. Single-cell transcriptome conservation in cryopreserved cells and tissues. Genome Biol. 18(1), 45 (2017).
Lake, B. B. et al. A comparative strategy for single-nucleus and single-cell transcriptomes confirms accuracy in predicted cell-type expression from nuclear RNA. Sci. Rep. 7(1), 6031 (2017).
Wu, H., Kirita, Y., Donnelly, E. L. & Humphreys, B. D. Advantages of single-nucleus over single-Cell RNA sequencing of adult kidney: rare cell types and novel cell states revealed in fibrosis. J. Am. Soc. Nephrol. 30(1), 23–32 (2019).
Renaut, S. et al. Single-cell and single-nucleus RNA-sequencing from paired normal-adenocarcinoma lung samples provide both common and discordant biological insights. PLoS Genet. 20(5), e1011301 (2024).
Andrews, T. S. et al. Single-cell, single-nucleus, and spatial RNA sequencing of the human liver identifies cholangiocyte and mesenchymal heterogeneity. Hepatol. Commun. 6(4), 821–840 (2022).
Soule, T. G. et al. A protocol for single nucleus RNA-seq from frozen skeletal muscle. Life Sci. Alliance https://doi.org/10.26508/lsa.202201806 (2023).
Whytock, K. L. et al. Isolation of nuclei from frozen human subcutaneous adipose tissue for full-length single-nuclei transcriptional profiling. STAR Protoc. 4(1), 102054 (2023).
Rousselle, T. V. et al. An optimized protocol for single nuclei isolation from clinical biopsies for RNA-seq. Sci. Rep. 12(1), 9851 (2022).
Mendelev, N. et al. Multi-omics profiling of single nuclei from frozen archived postmortem human pituitary tissue. STAR Protoc. 3(2), 101446 (2022).
Nguyen, Q. H., Pervolarakis, N., Nee, K. & Kessenbrock, K. Experimental considerations for single-Cell RNA sequencing approaches. Front. Cell Dev. Biol. 6, 108 (2018).
Hu, P. et al. Dissecting cell-type composition and activity-dependent transcriptional state in mammalian brains by massively parallel single-nucleus RNA-Seq. Mol. Cell. 68(5), 1006–15.e7 (2017).
Zheng, G. X. et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 8, 14049 (2017).
Young, M. D. & Behjati, S. SoupX removes ambient RNA contamination from droplet-based single-cell RNA sequencing data. Gigascience https://doi.org/10.1093/gigascience/giaa151 (2020).
Hao, Y. et al. Integrated analysis of multimodal single-cell data. Cell. 184(13), 3573–87.e29 (2021).
McGinnis, C. S., Murrow, L. M. & Gartner, Z. J. DoubletFinder: Doublet detection in single-cell RNA sequencing data using artificial nearest neighbors. Cell Syst. 8(4), 329–37.e4 (2019).
Abdulla, S., Aevermann, B., Assis, P., Badajoz, S., Bell, SM. & Bezzi, E., et al. CZ CELL×GENE Discover: A single-cell data platform for scalable exploration, analysis and modeling of aggregated data. bioRxiv. (2023):2023.10.30.563174.
Korsunsky, I. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat. Methods. 16(12), 1289–1296 (2019).
Wickham, H. ggplot2: Elegant graphics for data analysis (Springer-Verlag, 2016).
Singh, G. & Chanda, A. Mechanical properties of whole-body soft human tissues: A review. Biomed. Mater. https://doi.org/10.1088/1748-605X/ac2b7a (2021).
Butcher, D. T., Alliston, T. & Weaver, V. M. A tense situation: Forcing tumour progression. Nat. Rev. Cancer. 9(2), 108–122 (2009).
Xiong, Y. et al. MechanoBase: a comprehensive database for the mechanics of tissues and cells. Database (Oxford). https://doi.org/10.1093/database/baae040 (2024).
Li, L., Xu, B. & Liu, C. Sample enrichment for single-nucleus sequencing using concanavalin A-conjugated magnetic beads. STAR Protoc. 4(4), 102595 (2023).
Santos, M. D. et al. Extraction and sequencing of single nuclei from murine skeletal muscles. STAR Protoc. 2(3), 100694 (2021).
Gulko, A. et al. Protocol for flow cytometry-assisted single-nucleus RNA sequencing of human and mouse adipose tissue with sample multiplexing. STAR Protoc. 5(1), 102893 (2024).
Moraes Cabé, C. et al. High-quality brain and bone marrow nuclei preparation for single nuclei multiome assays. J. Vis. Exp. https://doi.org/10.3791/65715 (2023).
Gleitz, H. F. E., Snoeren, I. A. M., Fuchs, S. N. R., Leimkühler, N. B. & Schneider, R. K. Isolation of human bone marrow stromal cells from bone marrow biopsies for single-cell RNA sequencing. STAR Protoc. 2(2), 100538 (2021).
Koenitzer, J. R., Wu, H., Atkinson, J. J., Brody, S. L. & Humphreys, B. D. Single-nucleus RNA-sequencing profiling of mouse lung. Reduced dissociation bias and improved rare cell-type detection compared with single-cell RNA sequencing. Am. J. Respir. Cell Mol. Biol. 63(6), 739–747 (2020).
Payen, V. L. et al. Single-cell RNA sequencing of human liver reveals hepatic stellate cell heterogeneity. JHEP Rep. 3(3), 100278 (2021).
Chaffin, M. et al. Single-nucleus profiling of human dilated and hypertrophic cardiomyopathy. Nature 608(7921), 174–180 (2022).
Leinonen, R., Sugawara, H. & Shumway, M. The sequence read archive. Nucleic Acids Res. 39, D19-21 (2011).
Acknowledgements
We thank all lab members for insightful discussions and feedback. We are grateful to patients who agreed to dedicate their tissues for research projects. We would also like to thank all the people involved in collecting human samples and maintaining biobanks for research purposes. Your dedication and meticulous work are crucial to the advancement of our research. Lastly, we want to acknowledge and highlight the expert help provided by Stéphane Dubois from the Genomics platform of the Centre de Recherche du CHU de Québec – Université Laval. This work was supported by funds from the Natural Sciences and Engineering Research Council of Canada [#2019-06490 to S.B.] and the Canadian Institutes of Health Research [Grant # 451568 to S.B.].
Funding
This work was supported by funds from the Natural Sciences and Engineering Research Council of Canada [Grant #2019-06490 to S.B.] and the Canadian Institutes for Health Research [Grant #451568 to S.B].
Author information
Authors and Affiliations
Contributions
Conceptualization, C.S. and S.B.; Methodology, C.S., V.D., and S.B.; Software, C.S.; Formal Analysis, C.S.; Investigation, C.S., V.D. and F.K.; Resources, K.R., V.S.A., M.D., G.K., A.B., S.H., M.R., Y.B., Y.F., and V.F.; Data Curation, C.S.; Writing—Original Draft, C.S. and S.B.; Visualization, C.S. V.D.; Supervision, S.B.; Funding Acquisition, S.B. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
Prior to the collection of all human samples, the research protocols were reviewed and approved by the Institutional Review Boards (Comité d’éthique de la recherche du CHU de Québec-Université Laval and Comité d'éthique de la recherche de l’Institut universitaire de cardiologie et de pneumologie de Québec – Université Laval). Informed consent was obtained from all participants.
Consent of publication
During this work, the authors used ChatGPT (version July 13, 2024) to optimize the text and facilitate reading. After using this tool, the authors reviewed and edited the content as needed and take responsibility for the publication’s content.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Segovia, C., Desrosiers, V., Khadangi, F. et al. A versatile and efficient method to isolate nuclei from low-input cryopreserved tissues for single-nuclei transcriptomics. Sci Rep 15, 5581 (2025). https://doi.org/10.1038/s41598-025-90070-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-90070-8
