
Abstract
Recent advances in whole-genome sequencing (WGS) have increased the accessibility of this tool, offering substantial potential for pathogen surveillance, outbreak response, and diagnostics. However, the routine clinical adoption of WGS is hindered by factors such as high costs, technical complexity, and the requirement for bioinformatics expertise for data analysis. To address these challenges, we propose RapidONT, a workflow designed for cost-effective and accessible WGS-based pathogen analysis. RapidONT employs a mechanical shearing–based DNA extraction protocol, followed by library construction by using a multiplexing Oxford nanopore technologies (ONT) rapid barcoding kit. Flye software is used for de novo assembly without manual intervention, followed by basic assembly polishing using Medaka and Homopolish. The polished assemblies are then analyzed using the user-friendly web-based platform Pathogenwatch, which facilitates species identification, molecular typing, and antimicrobial resistance (AMR) prediction, all while requiring minimal bioinformatics expertise. The efficacy of RapidONT was evaluated using nine clinically relevant pathogens, encompassing a total of 90 gram-positive and gram-negative bacterial strains. The workflow demonstrated high accuracy in critical tasks such as multilocus sequence typing (MLST) and AMR identification, using only ONT R9.4.1 flowcell data. Notably, limitations were observed with Salmonella spp. and Neisseria gonorrhoeae. Furthermore, RapidONT enabled the generation of genomic information for 48 bacterial isolates by using a single flow cell, significantly reducing sequencing costs. This approach eliminates the need for extensive experimentation in obtaining crucial genomic information. This workflow facilitates broader WGS implementation in clinical pathogen analysis and diagnostics.
Similar content being viewed by others

Introduction
Clinical microbiology laboratories are essential for exploring microbial traits pertinent to clinical services, including the diagnosis and treatment of infectious disease as well as infection prevention and control1. Traditionally, the initial identification of microbes based on their observed color and shape through microscopy relies on staining techniques such as Gram staining2, followed by isolation in culture media3. However, recent advances in high-throughput sequencing have enabled the comparison of genomes from large numbers of bacterial isolates4,5. Whole-genome sequencing (WGS) offers unparalleled resolution compared with classical molecular typing methods such as multilocus sequence typing (MLST) and pulsed-field gel electrophoresis, rendering some of them redundant6,7,8. WGS can be used to effectively uncover molecular characteristics that are relevant to bacterial adaption, epidemiology, and drug resistance7,9 across a variety of species, including Acinetobacter baumannii6,10,11, Escherichia coli12, Klebsiella pneumoniae13,14,15, Neisseria gonorrhoeae16, Pseudomonas aeruginosa17, Salmonella Typhimurium18, and Staphylococcus aureus19.
Bacterial WGS relies on various de novo assembly strategies that are categorized by the read length employed, namely, short-read only, short- and long-read hybrid, and long-read only assemblies. Short-read technologies, such as Illumina sequencing, are widely used in WGS. However, this approach often yields fragmented genome assemblies20,21. Additionally, the high operational and capital costs associated with Illumina sequencing limit its integration into routine laboratory workflows. Generating a perfect bacterial genome using Oxford Nanopore Technologies (ONT) and Illumina data involves a long-read-based de novo assembly, followed by serial polishing with high-accuracy short reads to correct the error-prone contigs20. This process requires considerable human expertise and comes with the additional cost of Illumina sequencing, making it impractical for laboratory generation of complete bacterial genomes. Recent advancements have made ONT long-read-only sequencing a promising alternative for pure cultures. This approach generates high-quality genomes15,22 through ligation-based library preparation following careful DNA extraction to obtain long fragments. However, the optimal DNA extraction method for WGS can vary depending on the target organism. For bacteria that are difficult to lyse, additional enzymes or sonication may be required to achieve sufficient cell wall disintegration20,23. Furthermore, although ONT rapid barcoding kits facilitate swift library preparation, the impact of this rapid barcoding in combination with mechanical bead beating during DNA extraction on genome quality remains unclear and requires further investigation.
In this study, we propose the RapidONT workflow, a streamlined approach for bacterial WGS. RapidONT employs a universal DNA extraction protocol that uses beat beating for efficient cell disruption, regardless of the organism’s Gram staining characteristics. This protocol aligns with the prioritization of nine pathogens by the World Health Organization (WHO24). Subsequently, the ONT Rapid Barcoding Kit is used to facilitate rapid and simplified library construction, minimizing wet lab procedures. The de novo assembly process is straightforward, involves minimal manual intervention, and is followed by polishing to improve genome assembly quality. Following assembly, the web-based platform Pathogenwatch is used for species identification, molecular typing, and antimicrobial resistance (AMR) prediction. This platform requires only basic bioinformatics skills. In evaluation experiments, the results from the RapidONT workflow were compared against complete reference genomes.
Materials and methods
Bacterial isolates
The WHO 2017 global priority pathogens list categorizes 12 bacterial groups into three tiers: critical, high, and medium priority24. This study employed isolates representing 9 of these 12 priority groups, sourced from the Taiwan Surveillance of Antimicrobial Resistance (TSAR) program25, which maintains a collection of clinical isolates from hospitals across Taiwan. These isolates were collected as part of routine surveillance efforts to monitor antimicrobial resistance trends, and their use in this research was carried out in accordance with relevant guidelines and regulations. A total of 90 isolates, encompassing both gram-negative and gram-positive pathogens, were included in the analysis. Information on the isolation source and collection date is available in Table S1.
The analysis included the following gram-negative bacterial species: Acinetobacter baumannii complex (ABC) (10 isolates: 8 A. baumannii, 2 Acinetobacter pittii, and 1 Acinetobacter nosocomialis), Enterobacteriaceae (10 isolates: 8 Klebsiella pneumoniae, 1 Klebsiella quasipneumoniae, and 1 Escherichia coli), Haemophilus influenzae (10 isolates), Neisseria gonorrhoeae (10 isolates), Pseudomonas aeruginosa (10 isolates), and Salmonella spp. (10 isolates). The following gram-positive bacterial species were analyzed: Enterococcus faecium (10 isolates), Staphylococcus aureus (10 isolates), and Streptococcus pneumoniae (10 isolates).
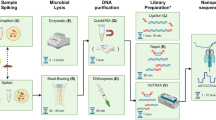
Gram stain–dependent and universal bacterial DNA extractions
For Gram stain–dependent DNA extraction approach, bacterial isolates were categorized based on their Gram stain characteristics. Subsequently, DNA extraction was performed using the DNeasy Blood & Tissue Kit (Qiagen GmbH, Cat. No. 69504, Hilden, Germany) following the manufacturer’s instructions. For gram-positive isolates, in the enzyme treatment step, lysostaphin at a concentration of 0.26 mg/mL was used instead of lysozyme for S. aureus. In the universal DNA extraction approach, the DNAs of all bacterial isolates, regardless of their Gram stain characteristics, were extracted using the DNeasy UltraClean Microbial Kit (Qiagen GmbH, Cat. No. 12224-50, Hilden, Germany) with automation using the QIAcube Connect machine (Qiagen GmbH, Cat. No. 9002864, Hilden, Germany) according to the manufacturer’s instructions. Bacterial lysis was achieved using a Precellys 24 tissue homogenizer (Bertin Technologies SAS, France). The homogenizer settings were optimized for efficient lysis: 6800 rpm for 30 s, followed by a 60-s pause, repeated over three cycles.
Illumina shotgun and ONT nanopore sequencing
Illumina shotgun sequencing
Illumina shotgun sequencing was conducted using the iSeq100 platform (Illumina). Library preparation involved the use of the Nextera DNA Flex kit and barcode kit (Illumina), according to established protocols. Sequencing was partially conducted by the Microbial Genomics Core Laboratories at the Institutes of Genomics and Bioinformatics, National Chung Hsing University.
ONT nanopore sequencing
For de novo whole-genome assembly, DNA extracted using the Gram stain-dependent methods was sequenced using various ONT kits, primarily the ligation sequencing kit and native barcoding kit, following previously described protocols11,26.
DNA extracted using the universal method was subjected to library construction with the ONT Rapid Barcoding Kit 96 (SQK-RBK110.96) following manufacturer’s instructions. However, the amount of DNA input was modified to 200 ng of DNA per sample, along with 1.3 µL of rapid barcode (RB01-96). The DNA library containing a maximum of 24 barcoded samples was loaded onto a MinION SpotON flowcell R9.4.1 (FLO-MIN106), and sequencing was executed using MinKNOW v22.08.9 with live basecalling, demultiplexing, and barcode trimming on Guppy v6.2.11 in high accuracy mode (HAC), targeting a minimum duration of 18 h.
Following the sequencing run, the flowcell was flushed using the flowcell wash kit (EXP-WSH004) as per ONT’s instructions. Before a second DNA library was loaded, the flowcell was primed as instructed in the SQK-RBK110.96 manual.
De novo genome assembly
For Nanopore sequencing reads derived from the Gram stain-dependent DNA extraction method, two distinct assembly strategies were implemented: long-read only and the short- and long-read hybrid assembly. The long-read only assemblies were generated using CCBGpipe26, whereas Unicycler v0.4.927 was employed in the hybrid method. To enhance the quality of the assembled genomes, a multistep polishing process was implemented. First, the long-read only assemblies were polished using ONT Medaka v1.4.3 twice and Homopolish v0.3.3 once28. This was followed by short-read polishing with Polypolish v0.5.029. The completeness of the genome assemblies was manually assessed using a combination of tools, including web-based BLASTN30, minimap2 v2.24-4112231, and Tablet v1.21.02.0832, to facilitate the integration of genome assemblies obtained using both methods. The strategy for genome assembly integration is detailed in the Supplementary Methods. The integrated complete genome assemblies were subjected to dual polishing by using the POLCA tool within MaSuRCA-4.1.033. This step generated circular, complete genomes that served as reference sequences for subsequent comparative analyses, including the evaluation of assemblies by using QUAST 5.2.034 and genomic characteristic predictions.
For Nanopore sequencing reads derived from the universal DNA extraction method, de novo assembly was conducted using Flye v2.9.235, resulting in draft genomes. These draft genomes were further polished using Medaka v1.9.1 with the r941_min_hac_g507 model and Homopolish v0.3.3. All software were run using 20 cores (Intel Xeon Gold 6230 2.10-GHz) with 1TB of memory. By using the complete genomes obtained using the conventional workflow as references, QUAST was employed to assess the various assemblies generated during the RapidONT workflow: Flye-assembled (draft), Medaka-only polished (Medaka) assemblies, Homopolish-only polished (Homopolish or homo for short), and Medaka followed by Homopolish polished (m + h, i.e., polish) assemblies.
Species identification, MLST, and AMR prediction
Following assembly and quality assessment, the complete reference genomes along with draft assemblies generated by Flye and the various polished versions (Medaka, Homopolish, and m + h) were uploaded to Pathogenwatch (https://pathogen.watch). This web-based platform facilitates rapid in silico characterization of bacterial genomes, including species identification, MLST, and AMR prediction36,37,38. For A. pittii and A. nosocomialis, mlst 2.23.0 was employed for MLST prediction39. AMR prediction for E. coli, H. influenzae, P. aeruginosa, and Salmonella enterica was performed using ResFinder 4.3.240.
Results
Complete genomes obtained through the conventional workflow, serving as references
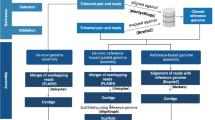
To evaluate the completeness of genome assemblies generated by the RapidONT workflow, complete reference genomes were established using a conventional workflow (Fig. 1). One S. aureus sample with a previously published complete genome sequence on NCBI (accession number: CP006838)41 was included, while DNA extraction for the remaining 89 samples performed based on their Gram staining characteristics. Hybrid assemblies (combining short and long reads) were generated using Unicycler, while long-read only assemblies were processed with CCBGpipe and subsequently polished through multiple steps (Table S2). After manual curation, circular genomes were successfully assembled for all 89 samples and further polished using Illumina reads with the POLCA tool, resulting in an average quality value (QV) of 93.03 (range, 46.42–100, Table S3). Detailed protocols are provided in Supplementary Methods. These high-quality complete genome assemblies represent a robust reference standard, serving as the baseline for evaluating and comparing genome assemblies obtained using the RapidONT workflow.

Comparison of bacterial whole-genome analysis: conventional versus RapidONT workflows. Conventional workflow: Obtaining complete genomes using the conventional workflow involves the use of different DNA extraction protocols and extensive manual interventions during the de novo assembly process. A total of 89 sequencing datasets, generated by both ONT and Illumina platforms, were de novo assembled using CCBGpipe and Unicycler, respectively. The CCBGpipe assemblies underwent a serial polishing process, and the polished contigs were used as references for completeness evaluation. Unique circular contigs generated by Unicycler, following plasmid validation, were integrated into the CCBGpipe results. The final complete circular genomes were polished twice with POLCA. The RapidONT workflow is a simplified approach. It uses a universal DNA extraction protocol, followed by de novo assembly and a basic assembly polishing process, minimizing the need for manual intervention.
Nanopore sequencing of 90 bacterial isolates by using two MinION flow cells
The RapidONT workflow comprises three key steps: universal DNA extraction, transposase-based rapid library construction, and de novo assembly using Flye (Fig. 1). To mimic real-world practices, DNA samples from various species were sequenced in a single run, eliminating the waiting time for sample collection for the same species. The universal DNA extraction method, regardless of Gram staining, consistently yields DNA that meets both quantity and quality requirements for direct use in ONT sequencing. The quality assessment data of extracted DNA are summarized in Table S1. Four sequencing runs were conducted using two MinION flow cells. The first flow cell generated passed sequencing data of 5.88 Gbp (Run1, barcode1-24) and 4.19 Gbp (Run2, barcode57-80) from 24 samples each, whereas the second flow cell produced 3.96 Gbp (Run3, barcode1-24) and 2.29 Gbp (Run4, barcode57-74) of passed sequences from 24 and 18 samples, respectively. To address potential carryover contamination despite a previous report indicating minimal risk from reusing a washed flow cell42, samples in the reused run were assigned distinct barcodes for efficient filtering of remnant sequences. Following overnight sequencing (~ 20 h), Flye generated draft assemblies for the targeted nine WHO priority pathogens, averaging 30 min per run (Supplementary Methods). These assemblies were polished once with Medaka and once with Homopolish, without requiring additional short-read data, to produce polished assemblies. The polishing process required less than 30 min per run. Post de novo assembly analysis was primarily performed using the web-based platform Pathogenwatch (Fig. 1). The streamlined RapidONT workflow produced genome assemblies that were subsequently compared with the complete reference genomes generated by the conventional workflow. This comparison allowed for the evaluation of assembly completeness and accuracy, demonstrating the effectiveness of this simplified process for rapid bacterial genome sequencing and analysis.
Sequencing performance and quality assessment of the RapidONT workflow
Out of the 90 samples, only one Salmonella spp. isolate with the lowest sequencing yield (8 Mbp) failed to produce a Flye assembly. However, during the polishing process, 12 additional Flye assemblies encountered difficulties due to numerous fragmented contigs and the inability to identify 20 homologous sequences by using Homopolish. These 13 samples (identified as hollow circles in Fig. 2A and crosses in Fig. 2B) included nine Salmonella spp. isolates, three S. aureus strains, and one P. aeruginosa strain. As depicted by the sequence summary in Fig. 2A, gram-negative strains generally yielded a greater number of sequence bases (average yield, 244 Mbp for 50 samples) than gram-positive strains (average yield, 69 Mbp for 20 S. aureus and S. pneumoniae samples), except for Salmonella spp. and E. faecium. The throughput ranged from the highest 497 Mbp in A. pittii to as low as 8 Mbp in Salmonella spp (Table S1). Among specific species, A. baumannii (average, 356 Mbp) and H. influenzae (average, 264 Mbp) exhibited significantly higher sequence yields (tested by Wilcoxon rank-sum test with p < 0.05), whereas Salmonella spp. (average, 50 Mbp), S. aureus (average, 53 Mbp) and S. pneumoniae (average, 86 Mbp) had significantly lower yields. In terms of read length, the average read length across all the 90 samples was 4,230 bp. However, significant differences were observed between species. P. aeruginosa (6,668 bp), H. influenzae (5,215 bp), and Enterobacteriaceae (5,069 bp) had significantly longer average read lengths, whereas Salmonella spp. (985 bp) and S. pneumoniae (2,838 bp) had shorter average read lengths than other species (Fig. 2A). These findings indicate that RapidONT effectively generated draft genome assemblies for eight pathogenic groups, except for Salmonella spp. Additionally, a minimum read length of 2,000 bp is recommended to ensure high-quality draft genome assemblies.

Sequencing and assembly metrics for different pathogen groups. (A) Sequencing data presenting key metrics such as total sequence bases, average read length, sequencing depth, and the percentage of the genome covered by at least fivefold sequencing reads. (B) Assembly metrics, including the number of contigs, length of the largest contig, assembly coverage rate, and assembly accuracy compared with the reference genome. Different colors indicate different species within each group: A. pittii is depicted in red, A. nosocomialis in light blue, K. quasipneumoniae in orange, and E. coli in purple. Hollow circles in (A) and crosses in (B) indicate samples that only have draft assemblies generated using Flye.
Bacterial genomes vary in size (e.g., > 7 Mbp for P. aeruginosa vs. < 2 Mbp for H. influenza). Therefore, sequence depth is a more reliable metric than sequence bases for determining sequencing needs. Through the normalization of sequence depth to genome size, we determined a minimum coverage of 20-fold to be necessary for genome assemblies with sufficient completeness (assembly coverage > 98.4%) and high accuracy (> 99.5%; Table S1). In all, 23 samples, including the 13 samples with only draft assemblies, fell below this 20-fold cutoff. Nevertheless, seven draft assemblies maintained adequate contiguity, with their N50 being equal to the maximum contig length (Table S1). The fivefold coverage rate reveals isolates of insufficient sequencing amount, particularly evident in fragmented assemblies (hollow circles in Fig. 2A). As expected, the 77 polished assemblies achieved a high average coverage rate (99.81%) and accuracy (99.94%). Notably, 31 samples achieved complete assembly coverage (100%), and 55 samples had accuracy exceeding 99.98%. However, the accuracy check revealed an outlier among the nine pathogens (Fig. 2B)—N. gonorrhoeae had a lower average accuracy of 99.64%. After N. gonorrhoeae was excluded, the average accuracy for the remaining species reached 99.98% (Table S1).
The assembly accuracy was also calculated based on the number of mismatches and indels reported in the QUAST results (Table S4). Accuracy was compared across three polishing strategies: Medaka-only polishing (Medaka), Homopolish-only polishing (Homopolish), and Medaka followed by Homopolish polishing (m + h). As shown in Fig. 3, the m + h polishing method was most effective in reducing both mismatches and indels across all nine pathogenic groups compared with the Medaka and Homopolish methods. Medaka effectively mitigated mismatch errors, especially for H. influenzae and P. aeruginosa, whereas Homopolish was better at handling indels, particularly in Enterobacteriaceae and P. aeruginosa assemblies. The m + h approach offered a balanced strategy for correcting both mismatches and indels across different bacterial genomes, resulting in highly accurate contigs. Therefore, the m + h polishing method was adopted in this study for RapidONT workflow.

Comparison of the effectiveness of various polishing methods in reducing mismatches and indels within draft assemblies for different pathogenic groups. Stacked bar graphs represent the total number of errors identified using QUAST across draft assemblies generated using Flye and polished assemblies processed using different methods, namely Medaka only (medaka), Homopolish only (homopolish), and Medaka followed by Homopolish (m + h), compared with the reference genomes.
Comparisons of species, MLST, and resistance analyses between the conventional and RapidONT workflows
Complete genomes, draft assemblies, and polished assemblies were uploaded to the Pathogenwatch platform for species identification, MLST typing, and predictions of AMR and virulence factors. The platform successfully identified two isolates of A. pittii and one of A. nosocomialis in the A. baumannii group, as well as one isolate each of Klebsiella quasipneumoniae and Escherichia coli in the Enterobacteriaceae group, and Salmonella enterica in the Salmonella spp. group across all genomes obtained from both workflows. While MLST and AMR predictions were available for most species (data in Table S5), the platform returned incomplete results for a few others. Specifically, MLST data were unavailable for A. pittii and A. nosocomialis, and AMR predictions were unavailable for E. coli, H. influenzae, P. aeruginosa, and S. enterica. To address these gaps, manual analysis was conducted to ensure comprehensive characterization. The MLST analysis of the 90 complete genomes detected an undetermined allele each in the following samples: N. gonorrhoeae (abcZ in VP84), S. aureus (aroE in VP77), and P. aeruginosa (acsA in VP27), as shown in Table S5. These findings suggest potential novel allelic variations not yet included in the current PubMLST reference database. For the 77 isolates with polished assemblies, we compared MLST predictions from both draft and polished assemblies against the reference genome with 536 alleles. Figure 4 presents the prediction agreement rates for MLST allele and AMR prediction (excluding one S. enterica sample with seven identical predictions). Furthermore, perfect predictions (100% agreement) were achieved using polished assemblies (m + h) generated using the RapidONT workflow for four pathogen groups: A. baumannii, H. influenzae, E. faecium, and S. pneumoniae (Fig. 4). However, three alleles in three K. pneumoniae strains (gapA in VP42, tonB in VP43, and gapA in VP66), one allele in P. aeruginosa (guaA in VP28), and four alleles in one S. aureus strain (gmk, pta, tpi, and yqiL in VP77) were not accurately predicted (Table S5), which may be attributed to the low sequence depth. The sequence depths of these five strains in sequential order were 10, 13, 51, 21, and 9 (Table S1). Notably, despite having sufficient sequence depths (60–141), N. gonorrhoeae displayed limited prediction success (2–3 alleles per isolate).

Agreement rates of genomic predictions of multilocus sequence typing alleles and antimicrobial resistance genes between reference genomes and various assemblies across different pathogenic groups.
For AMR predictions, only the number of resistant classes identified by Pathogenwatch or ResFinder was considered. The comparative analysis revealed that A. baumannii, H. influenzae, P. aeruginosa, S. aureus, and S. pneumoniae (in Fig. 4), along with the single S. enterica sample, exhibited perfect agreement (100%) between draft and polished assemblies compared with complete genomes. Although polishing increased the number of AMR predictions E. faecium (from 107 in Flye-assembled genomes to 119, 123, and 123 in Medaka, Homopolish, and m + h polished assemblies), two AMR predictions remained absent in two K. pneumoniae strains (i.e. VP42 and VP68 in Table S5). Similarly, N. gonorrhoeae exhibited a substantial improvement in AMR predictions after polishing (from 11 to 29), but seven strains still lacked a total of 12 AMR predictions compared with the reference genome. Interestingly, strains VP81, VP86, and VP89 achieved perfect prediction accuracy with polished assemblies. These findings suggest that the RapidONT workflow offers accurate AMR predictions from polished assemblies, comparable to those derived from complete genome obtained through the conventional workflow.
Discussion
Modern laboratories prioritize the development of rapid, scalable, and cost-effective workflows to minimize labor-intensive tasks. Despite rapid advances in WGS, its routine adoption has been hindered by challenges such as cost, potential inaccuracy, complex library construction processes, the need for a variety of DNA extraction protocols, and the need for expertise in postsequencing data analysis. The high capital expenditure associated with Illumina sequencing system often restricts WGS to well-funded research institutions and biotechnology companies. Conversely, ONT offers the MinION Mk1B starter pack for a very low price of US$1999, broadening access to whole-genome analysis, particularly in lower income regions43. Moreover, ONT actively invests in the development of rapid sequencing chemistries, simplifying library construction and enabling the generation of multiplexed sequencing libraries within an hour. This study presents a streamlined workflow designed to challenge the traditional “longer-is-better” paradigm in long-read sequencing. Although the universal bead-beating cell lysing protocol for DNA extraction and transposase-based library construction contribute to DNA fragmentation, the resulting average read length of 4,230 bp across 90 samples remains advantageous for de novo assembly compared with the shorter reads generated by Illumina systems. Additionally, the benefits of a universal DNA extraction protocol, coupled with the rapid turnaround time for library construction, outweigh the drawbacks associated with DNA fragmentation. This facilitates the broader adoption of WGS in various laboratory settings. We aimed to simplify experimental protocols by using a universal DNA extraction method and a rapid barcoding kit. To maximize the capacity of a single flow cell while mitigating potential barcode imbalance issues, we selected samples from nine WHO priority pathogen groups, with a maximum of 24 samples processes per run. To further reduce costs (below US$20 per sample), flushed flow cells were reused once, enabling the sequencing of a maximum of 48 samples per flow cell. The costs associated with the RapidONT workflow are outlined in Table S6. Notably, the Illumina website indicates that a price point below US$20 per sample is currently only achievable for targeted gene expression profiling (US$23) or 16S metagenomic sequencing (US$18) (https://sapac.illumina.com/science/technology/next-generation-sequencing/beginners/ngs-cost.html, accessed May 13, 2024). Furthermore, the average cost of bacterial genome sequencing by using Illumina technology typically ranges from US$50 to US$10044. Our findings demonstrate the effectiveness of the RapidONT workflow for a diverse range of human pathogens, with the exception of Salmonella spp., which had problems with short read length and low throughput, and N. gonorrhoeae, which had low assembly accuracy.
Our study findings indicate that a minimum average read length of 2000 bp and a minimum sequencing depth of 20-fold are crucial to obtaining accurate genomic information necessary for species identification, MLST, and AMR prediction in most bacterial pathogens. The employed universal DNA extraction method was particularly effective for gram-positive E. faecium, yielding a high number of long reads. However, the method was less effective for other gram-positive species, S. aureus and S. pneumoniae, resulting in a reduced quantity and shorter length of extracted DNA. Conversely, this method was successful for all tested gram-negative species, with the exception of Salmonella spp. This disparity highlights the need for future investigation into alternative bead-beating settings or DNA extraction kits to optimize DNA yield and quality across diverse bacterial taxa. For example, we have successfully integrated the DNeasy PowerLyzer Microbial Kit (Qiagen, Hilden, Germany) into the RapidONT workflow, enabling the sequencing of over 250 clinical isolates of the Enterobacter cloacae complex by using only six MinION flow cells. Furthermore, we are currently utilizing R10.4.1 flow cells to sequence over 100 K. pneumoniae isolates with the RapidONT workflow. While each flow cell process approximately 40 genomes of this size (~ 5 Mb), the resulting genome quality has shown significant improvement, achieving Q-scores of Q40 or higher.
This study employed Flye, Medaka, and Homopolish for de novo assembly and polishing of bacterial genomes, enabling accurate genomic predictions for MLST and AMR. However, a significant outlier was observed in the sequencing accuracy of N. gonorrhoeae. Although a relatively high sequencing depth (averaging 97) was achieved, the estimated prepolishing sequence accuracy for N. gonorrhoeae contigs assembled with CCBGpipe (ONT long-read only) was notably lower (99.7%) compared with other pathogens (99.9%) assembled using Polypolish (Table S2). This discrepancy persisted even after re-basecalling the N. gonorrhoeae samples in the super accuracy mode (SUP) and subsequent polishing with Medaka and Homopolish. This discrepancy suggests potential limitations extending beyond the basecalling and polishing methods. Whether the higher degree of sequence variability in N. gonorrhoeae is attributed to its higher levels of methylation45 or the inherent variability associated with its diploid nature46,47, which contributes to the observed basecalling errors, remains unclear. Although Illumina short-read alignments indicated a homozygous pattern, this approach might not fully capture the complexity introduced by methylation. Our analysis using the DeepMod2 tool48 revealed that these sequence variations were indeed located at methylated sites. This finding supports the hypothesis that methylation could play a role in the observed basecalling errors. These findings highlight the complex genomic structure of N. gonorrhoeae. The heightened levels of methylation, or the variability in methylation due to its diploid nature, might have influenced the accuracy of basecalling algorithms in this study.
Pathogenwatch provides a user-friendly interface for uploading genome assemblies and conducting various analyses, including species determination, MLST, cgMLST, and genomic predictions of AMR and virulence. While its specialized capabilities for specific bacterial species are a notable strength, some limitations do also exist. One such limitation is the targeted application of functionalities. For example, MLST analysis for Acinetobacter species, such as A. pittii and A. nosocomialis, was omitted despite using the same database as A. baumannii. Similarly, although carbapenemase-producing E. coli poses a global public health threat, Pathogenwatch lacks AMR predictions for this specific species. Other bioinformatics resources such as the Center for Genomic Epidemiology offer noncommercial, free online tools such as ResFinder for users with limited bioinformatics expertise. These resources bridge the gap between wet- and dry-lab practices in WGS applications. The development of a cohesive and user-friendly informatics system could enable public health organizations to more effectively share data, processes, and analyses, promoting reproducibility, accessibility, and verifiability in pathogen genomic studies while maintaining individual autonomy49.
Conclusion
The study introduces the RapidONT workflow, a streamlined approach for WGS of various bacterial pathogens that combines a universal DNA extraction protocol, ONT transposase-based rapid barcoding library construction, de novo assembly, and basic assembly polishing. This workflow is coupled with a user-friendly web-based platform for species identification, molecular typing, and AMR prediction. The minimum reference point requires a 20-fold sequencing depth with an average read length of at least 2,000 base pairs for accurate genomic characterization. This threshold allows for the termination of a sequence run and flow cell reuse after data acquisition, thereby reducing sequencing costs. The proposed RapidONT workflow streamlines experimental protocols and minimizes costs associated with WGS. The effectiveness of this approach in generating accurate genomic characteristics for crucial pathogen groups has been validated.
Data availability
The complete genome sequences of 89 isolates have been deposited in the National Center for Biotechnology Information (NCBI) under BioProject number PRJNA1000275 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1000275/). The Nanopore sequencing reads obtained through the RapidONT workflow have also been deposited in the NCBI Sequence Read Archive (SRP467862, https://trace.ncbi.nlm.nih.gov/Traces/study/?acc=SRP467862).
References
Samuel, L. P. et al. The need for dedicated microbiology leadership in the clinical microbiology laboratory. J. Clin. Microbiol. 59, e0154919. https://doi.org/10.1128/JCM.01549-19 (2021).
Tripathi, N. & Sapra, A. In StatPearls (2024).
Bonnet, M., Lagier, J. C., Raoult, D. & Khelaifia, S. Bacterial culture through selective and non-selective conditions: the evolution of culture media in clinical microbiology. New Microbes New Infect. 34, 100622. https://doi.org/10.1016/j.nmni.2019.100622 (2020).
Sheppard, S. K., Guttman, D. S. & Fitzgerald, J. R. Population genomics of bacterial host adaptation. Nat. Rev. Genet. 19, 549–565. https://doi.org/10.1038/s41576-018-0032-z (2018).
Shen, Y., Nie, J., Kuang, L., Zhang, J. & Li, H. DNA sequencing, genomes and genetic markers of microbes on fruits and vegetables. Microb. Biotechnol. 14, 323–362. https://doi.org/10.1111/1751-7915.13560 (2021).
Fida, M. et al. Acinetobacter baumannii genomic sequence-based core genome multilocus sequence typing using Ridom SeqSphere+ and antimicrobial susceptibility prediction in ARESdb. J. Clin. Microbiol. 60, e0053322. https://doi.org/10.1128/jcm.00533-22 (2022).
Uelze, L. et al. Typing methods based on whole genome sequencing data. One Health Outlook 2, 3. https://doi.org/10.1186/s42522-020-0010-1 (2020).
Baker, K. S. et al. Genomics for public health and international surveillance of antimicrobial resistance. Lancet Microbe https://doi.org/10.1016/S2666-5247(23)00283-5 (2023).
Sherry, N. L. et al. An ISO-certified genomics workflow for identification and surveillance of antimicrobial resistance. Nat. Commun. 14, 60. https://doi.org/10.1038/s41467-022-35713-4 (2023).
Hu, X. et al. Novel clinical mNGS-based machine learning model for rapid antimicrobial susceptibility testing of Acinetobacter baumannii. J. Clin. Microbiol. 61, e0180522. https://doi.org/10.1128/jcm.01805-22 (2023).
Chen, F. J. et al. Molecular epidemiology of emerging carbapenem resistance in Acinetobacter nosocomialis and Acinetobacter pittii in Taiwan, 2010 to 2014. Antimicrob. Agents Chemother. https://doi.org/10.1128/AAC.02007-18 (2019).
Humphries, R. M. et al. Machine-learning model for prediction of cefepime susceptibility in escherichia coli from whole-genome sequencing data. J. Clin. Microbiol. 61, e0143122. https://doi.org/10.1128/jcm.01431-22 (2023).
Hernandez-Garcia, M. et al. Impact of Ceftazidime-Avibactam treatment in the emergence of novel KPC variants in the ST307-Klebsiella pneumoniae high-risk clone and consequences for their routine detection. J. Clin. Microbiol. 60, e0224521. https://doi.org/10.1128/jcm.02245-21 (2022).
Chen, F. J. et al. Emergence of mcr-1, mcr-3 and mcr-8 in clinical Klebsiella pneumoniae isolates in Taiwan. Clin. Microbiol. Infect. 27, 305–307. https://doi.org/10.1016/j.cmi.2020.07.043 (2021).
Foster-Nyarko, E. et al. Nanopore-only assemblies for genomic surveillance of the global priority drug-resistant pathogen, Klebsiella pneumoniae. Microb. Genom. https://doi.org/10.1099/mgen.0.000936 (2023).
Ma, A. et al. Use of genome sequencing to resolve differences in gradient diffusion and agar dilution antimicrobial susceptibility testing performance of Neisseria gonorrhoeae isolates in Alberta, Canada. J. Clin. Microbiol. 61, e0060623. https://doi.org/10.1128/jcm.00606-23 (2023).
Liu, B. et al. Direct prediction of carbapenem resistance in Pseudomonas aeruginosa by whole genome sequencing and metagenomic sequencing. J. Clin. Microbiol. 61, e0061723. https://doi.org/10.1128/jcm.00617-23 (2023).
Fu, Y., M’Ikanatha, N. M. & Dudley, E. G. Whole-genome subtyping reveals population structure and host adaptation of salmonella typhimurium from wild birds. J. Clin. Microbiol. 61, e0184722. https://doi.org/10.1128/jcm.01847-22 (2023).
Giulieri, S. G. et al. Genomic exploration of sequential clinical isolates reveals a distinctive molecular signature of persistent Staphylococcus aureus bacteraemia. Genome Med. 10, 65. https://doi.org/10.1186/s13073-018-0574-x (2018).
Wick, R. R., Judd, L. M. & Holt, K. E. Assembling the perfect bacterial genome using Oxford Nanopore and Illumina sequencing. PLoS Comput. Biol. 19, e1010905. https://doi.org/10.1371/journal.pcbi.1010905 (2023).
Rayamajhi, N., Cheng, C. C. & Catchen, J. M. Evaluating Illumina-, Nanopore-, and PacBio-based genome assembly strategies with the bald notothen, Trematomus borchgrevinki. G3 (Bethesda) https://doi.org/10.1093/g3journal/jkac192 (2022).
Sereika, M. et al. Oxford Nanopore R10.4 long-read sequencing enables the generation of near-finished bacterial genomes from pure cultures and metagenomes without short-read or reference polishing. Nat. Methods 19, 823–826. https://doi.org/10.1038/s41592-022-01539-7 (2022).
Chen, N. & Yuan, X. A quick DNA extraction method for high throughput screening in gram-positive bacteria. Bio Protoc. 13, e4653. https://doi.org/10.21769/BioProtoc.4653 (2023).
Tacconelli, E. et al. Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327. https://doi.org/10.1016/S1473-3099(17)30753-3 (2018).
Ho, M. et al. Surveillance of antibiotic resistance in Taiwan, 1998. J. Microbiol. Immunol. Infect. 32, 239–249 (1999).
Liao, Y. C. et al. Completing circular bacterial genomes with assembly complexity by using a sampling strategy from a single MinION run with barcoding. Front. Microbiol. 10, 2068. https://doi.org/10.3389/fmicb.2019.02068 (2019).
Wick, R. R., Judd, L. M., Gorrie, C. L. & Holt, K. E. Unicycler: Resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13, e1005595. https://doi.org/10.1371/journal.pcbi.1005595 (2017).
Huang, Y. T., Liu, P. Y. & Shih, P. W. Homopolish: a method for the removal of systematic errors in nanopore sequencing by homologous polishing. Genome Biol. 22, 95. https://doi.org/10.1186/s13059-021-02282-6 (2021).
Wick, R. R. & Holt, K. E. Polypolish: Short-read polishing of long-read bacterial genome assemblies. PLoS Comput. Biol. 18, e1009802. https://doi.org/10.1371/journal.pcbi.1009802 (2022).
Camacho, C. et al. BLAST+: architecture and applications. BMC Bioinform. 10, 421. https://doi.org/10.1186/1471-2105-10-421 (2009).
Li, H. New strategies to improve minimap2 alignment accuracy. Bioinformatics 37, 4572–4574. https://doi.org/10.1093/bioinformatics/btab705 (2021).
Milne, I. et al. Using tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 14, 193–202. https://doi.org/10.1093/bib/bbs012 (2013).
Zimin, A. V. & Salzberg, S. L. The genome polishing tool POLCA makes fast and accurate corrections in genome assemblies. PLoS Comput. Biol. 16, e1007981. https://doi.org/10.1371/journal.pcbi.1007981 (2020).
Gurevich, A., Saveliev, V., Vyahhi, N. & Tesler, G. QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. https://doi.org/10.1093/bioinformatics/btt086 (2013).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. https://doi.org/10.1038/s41587-019-0072-8 (2019).
Argimon, S. et al. Rapid genomic characterization and global surveillance of Klebsiella using Pathogenwatch. Clin. Infect. Dis. 73, S325–S335. https://doi.org/10.1093/cid/ciab784 (2021).
Argimon, S. et al. A global resource for genomic predictions of antimicrobial resistance and surveillance of Salmonella Typhi at pathogenwatch. Nat. Commun. 12, 2879. https://doi.org/10.1038/s41467-021-23091-2 (2021).
Sanchez-Buso, L. et al. A community-driven resource for genomic epidemiology and antimicrobial resistance prediction of Neisseria gonorrhoeae at Pathogenwatch. Genome Med. 13, 61. https://doi.org/10.1186/s13073-021-00858-2 (2021).
Seemann, T. mlst. https://github.com/tseemann/mlst (2024).
Bortolaia, V. et al. ResFinder 4.0 for predictions of phenotypes from genotypes. J. Antimicrob. Chemother. 75, 3491–3500. https://doi.org/10.1093/jac/dkaa345 (2020).
Chen, F. J., Lauderdale, T. L., Wang, L. S. & Huang, I. W. Complete genome sequence of Staphylococcus aureus Z172, a vancomycin-intermediate and daptomycin-nonsusceptible methicillin-resistant strain isolated in Taiwan. Genome Announc https://doi.org/10.1128/genomeA.01011-13 (2013).
Liao, Y. C. et al. Rapid and routine molecular typing using multiplex polymerase chain reaction and MinION sequencer. Front. Microbiol. 13, 875347. https://doi.org/10.3389/fmicb.2022.875347 (2022).
Gómez-González, P. J., Campino, S., Phelan, J. E. & Clark, T. G. Portable sequencing of Mycobacterium tuberculosis for clinical and epidemiological applications. Brief. Bioinform. https://doi.org/10.1093/bib/bbac256 (2022).
Bruzek, S., Vestal, G., Lasher, A., Lima, A. & Silbert, S. Bacterial whole genome sequencing on the illumina iSeq 100 for clinical and public health laboratories. J. Mol. Diagn. 22, 1419–1429. https://doi.org/10.1016/j.jmoldx.2020.09.003 (2020).
Blow, M. J. et al. The epigenomic landscape of prokaryotes. PLoS Genet. 12, e1005854. https://doi.org/10.1371/journal.pgen.1005854 (2016).
Tobiason, D. M. & Seifert, H. S. The obligate human pathogen, Neisseria gonorrhoeae, is polyploid. PLoS Biol. 4, e185. https://doi.org/10.1371/journal.pbio.0040185 (2006).
Tobiason, D. M. & Seifert, H. S. Genomic content of Neisseria species. J. Bacteriol. 192, 2160–2168. https://doi.org/10.1128/JB.01593-09 (2010).
Ahsan, M. U., Gouru, A., Chan, J., Zhou, W. & Wang, K. A signal processing and deep learning framework for methylation detection using Oxford Nanopore sequencing. Nat. Commun. 15, 1448. https://doi.org/10.1038/s41467-024-45778-y (2024).
Black, A., MacCannell, D. R., Sibley, T. R. & Bedford, T. T. recommendations for supporting open pathogen genomic analysis in public health. Nat. Med. 26, 832–841. https://doi.org/10.1038/s41591-020-0935-z (2020).
Acknowledgements
We thank the National Core Facility for Biopharmaceuticals (NCFB, MOST 111-2740-B-400-002) and the National Center for High-performance Computing (NCHC) of National Applied Research Laboratories (NARLabs) of Taiwan for providing the computational resources. We also thank the Taiwan Surveillance of Antimicrobial Resistance (TSAR) program for providing the clinical isolates used in this study. These isolates were collected as part of routine surveillance efforts, and their use in this research was conducted in accordance with the program’s ethical guidelines. This work was supported by grants from the National Health Research Institutes (IV-111-PP-11 to HKS, IV-111-PP-23 to FJC, and PH-112-PP-05 to YCL) and the Ministry of Science and Technology (111-2314-B-400-033 to YCL).
Author information
Authors and Affiliations
Contributions
FJC and YCL conceptualized the study. HCW, YTC, and CHL conducted the experiments and sequencing. HWC, YTC, and YCL performed data analysis. ICW, HWC, and YCL curated the data. HCW, HKS, YCL and FJC investigated. SCK and TLL provided the bacterial isolates and associated data from the TSAR program. HCW and YCL drafted the manuscript. All authors contributed to critical manuscript review and editing.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
The bacterial isolates used in this study were provided by the Taiwan Surveillance of Antimicrobial Resistance (TSAR) program, which collects clinical isolates as part of routine surveillance activities in hospitals across Taiwan. The collection of these isolates was conducted under the ethical approvals granted to the TSAR program by the Research Ethics Committee of the National Health Research Institutes, Taiwan (EC1010602-E, EC1030406-E, and EC1050606-E). The use of these anonymized isolates in this research did not require additional ethical approval or informed consent, as no patient-identifiable information was accessed.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Wu, HC., Chiu, YT., Wu, IC. et al. Streamlining whole genome sequencing for clinical diagnostics with ONT technology. Sci Rep 15, 6270 (2025). https://doi.org/10.1038/s41598-025-90127-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-90127-8
