
Abstract
The recent classification of the koala to endangered across most of its range has emphasised the urgent need for enhanced disease surveillance. Little is known about the distribution and clinical significance of phascolarctid gammaherpesvirus 2 (PhaHV-2) outside the Australian states of Victoria and South Australia. PhaHV-2 may have significant impacts on koala wellbeing, justifying investigation into its distribution and impact. To better characterise virus distribution and factors associated with infection, we developed a novel quantitative polymerase chain reaction assay with a high sensitivity, specificity and throughput capacity, to facilitate rapid detection and quantification of PhaHV-2. We applied this assay to 157 predominantly clinically-derived, urogenital swab samples. Results indicated that the distribution of PhaHV-2 extends throughout the endangered New South Wales koala populations. Frequency of detection of PhaHV-2 was highest in South Australia (25%) and lowest in northern Qld (0%) and was more likely in older koalas than younger koalas. We corroborate previous findings of an association with presence of PhaHV-1 and find no evidence to support sex as a predictor for viral presence in clinically-derived samples. The capacity to rapidly detect mucosal shedding of PhaHV-2 will enable rapid isolation of affected individuals and aid further research into the pathophysiological impacts of this virus in koalas and the influence of co-infections.
Similar content being viewed by others


Introduction
The conservation of the Australian endemic koala (Phascolarctos cinereus) faces many challenges, including disease, which serves as a significant obstacle to the persistence of koala populations1. Previously estimated at millions2, the national koala population was recently approximated to be 330,000 koalas3, with further population reductions expected in the state of New South Wales (NSW) particularly3,4,5,6,7. Following the 2019/20 bushfire season, the species’ listing was elevated to endangered in NSW, Queensland (Qld) and the Australian Capital Territory (ACT) in 2022 under the Environmental Protection and Biodiversity Conservation Act (EPBCA) based on longstanding and ongoing population decline as a result of habitat degradation, predation, motor vehicle trauma and disease8. The impact of infectious disease, the most prominent of which is chlamydial disease, is likely exacerbated by a range of environmental factors, as well as co-infection with one or more other infectious agents1. Chlamydia pecorum infection is prevalent among Australian koala populations7 and, as it is associated with infertility and ocular and urogenital disease9, there is a strong interest in identifying factors that may predispose koalas to this disease. Greater loads of koala retrovirus (KoRV) have been associated with chlamydial infection and disease, but causative relationships are less well known7,10,11,12,13,14. Two gammaherpesviruses have also been detected in koalas: phascolarctid herpesvirus 1 (PhaHV-1)15, and phascolarctid herpesvirus 2 (PhaHV-2)16. Gammaherpesviruses are generally lymphotropic and also invade epithelial cells17, and have been identified as important to disease outcomes for C. trachomatis in humans18. Understanding the dynamics of PhaHV-2 infection is crucial, considering its potential influence on disease progression and co-infection19. While PhaHV-2 is currently classified as low risk, the low certainty attached to this assessment emphasises the need for further evaluation of its role in disease; while the risk remains unquantified it cannot be effectively mitigated1.
Research has provided insight into the distribution and clinical significance of PhaHV-1 but comparatively little is known about PhaHV-2 20,21,22,23. PhaHV-1 has been significantly associated with both koala retrovirus (KoRV) and C. pecorum infection in koalas, indicating potential co-infection interactions20. The known distribution of PhaHV-1 was recently expanded beyond Victoria to NSW and Qld, and detection rate was significantly higher in archived samples from rehabilitated koalas, compared to free-living, suggesting increased viral shedding in koalas taken into care21. PhaHV-1 appears more prevalent among older age classes, suggesting horizontal transmission is likely20,21. Research into PhaHV-2 has provided preliminary insights into distribution and pathogenesis in Victorian (Vic) and South Australian (SA) koala populations only20,22,23. In those states, the detection of PhaHV-2 on urogenital and oropharyngeal swab samples has ranged from 0 to 0.9% in French Island, Vic; 40% in Mount Lofty, SA; up to 54.6% in Raymond Island, Vic20,22,23. PhaHV-2 has also been significantly associated with C. pecorum infection and reduced fertility in southern koalas20. Viral load is likely to depend on whether the virus detected is in a lytic or latent phase, and the site being sampled, which can cause variable results17. The latency sites of PhaHV-2 are undetermined, but the virus has been detected in the lymph nodes and spleen15,22 of infected koalas, suggesting they are potential sites of viral latency22. By developing a sensitive diagnostic assay, with the capacity to detect and quantify low viral loads, we attempt here to mitigate the influence of low-level and intermittent shedding on diagnostic accuracy17,24.
In this study we designed and optimised a qPCR assay with high analytical sensitivity and specificity. While our assay can quantify viral load, as our sampling strategy largely utilises archived clinical samples with limited metadata, we here focus on viral detection to extend our knowledge of the range of PhaHV-2 to the threatened koala populations of NSW. We have also investigated correlations between PhaHV-2 and koala age, sex, chlamydial infection status and PhaHV-1, using predominantly clinically-derived urogenital swab samples.
Results
Assay design and validation
Following primer design and optimisation, HV_230_Fwd and VK-PhaHV-2 Rev were selected as the optimal primer combination due to their agreement with the inclusion criteria listed in the Methods (see also Supplementary Table 1, Supplementary Fig. 1). All qPCRs for PhaHV-2 displayed congruous melt temperatures at 87–87.5 °C and standard curve efficiencies ranged from 88.7 to 110.1%. The average 95% LOD for the assay was quantified as 132 copies/reaction (equating to a Ct value between 35 and 36; Supplementary Table 2, Supplementary Fig. 2), based on two repeat standard curves. Specificity of the selected primers was corroborated using the NCBI BLAST function, with all bidirectional sequences sent for sequencing (n = 12) exclusively matching with PhaHV-2, with sequence identity ranging between 95.77 and 100%. No matches with PhaHV-1 were detected.
Determining PhaHV-2 presence
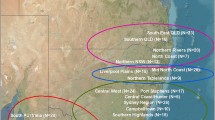
The urogenital swab samples used in this study are only appropriate for detecting active shedding of the virus at mucosal sites and not latent infections. Additionally, as our sampling strategy was largely opportunistic from archived samples, we cannot accurately determine viral prevalence and therefore report viral detection only. PhaHV-2 was detected in 6/14 localities at which samples were collected and all bioregions with the exception of the Central Qld region sampled from Connors Range (Fig. 1; Table 1). Frequency of detection of PhaHV-2 varied among the sample sets from the five biogeographic regions: Central Qld (0%), Northern NSW/Southern Qld (2.0%), Mid North NSW (19.2%), Central NSW (9.1%) and SA (25.0%) (Fig. 1) (Table 1). The overall frequency of detection of PhaHV-2 (9.6%) in our sample set was significantly lower than that previously found across populations in Victoria (Vaz et al., 2019), (15/157, 9.6%; vs. 149/702, 21.2%; X2 = 11.31, p value = 0.0008, df = 1, Odds ratio = 0.392, 95% CI = 0.22–0.69). The frequency of PhaHV-2 detected in our South Australian samples (25%) is similar to that previously found in South Australia (20.7%) (Kasimov et al., 2020).

Map of eastern Australia, displaying the geographical localities and sample sizes where urogenital swab samples were obtained between 2016–2022. Different biogeographical regions are represented by different colours: orange = Central Qld, pink = Southern Qld/Northern NSW; blue = Mid-north NSW; green = Central NSW; red = South Australia.
Age, sex, PhaHV-2 active shedding and co-infection status
Generalised linear regression model (GLM) results indicated PhaHV-2 is associated with PhaHV-1 presence, but not C. pecorum or bioregion (n = 157) (Table 2). The proportion of PhaHV-2 in PhaHV-1 positive samples (8/36, 22.2%) was significantly higher than the proportion of PhaHV-2 in PhaHV-1 negative samples (7/121, 5.8%; X2 = 8.68, p value = 0.0032, df = 1, Odds ratio = 4.653, 95% CI = 1.56–13.91). Sex was not a significant predictor for PhaHV-2 presence (n = 143) (Table 2). PhaHV-2 detection was more likely in older koalas than younger koalas (Age classes I-II, 0/36, 0% vs. Age classes III-V, 12/69, 17%; X2 = 7.07, p value = 0.0078, df = 1, Odds ratio = 0.063, 95% CI = 0.004–1.097; Tables 2 and 3). For 22 C. pecorum positive koalas, clinical signs were not associated with PhaHV-1, PhaHV-2, age or sex (Table 2). All data used in models is provided in Supplementary Table 3.
Discussion
In this study we designed and optimised a novel PhaHV-2 qPCR assay with a high analytical sensitivity and specificity, and throughput capacity. Using this assay, we discovered a more expansive distribution of PhaHV-2 to include Central NSW, Mid North NSW and Northern NSW. We explored relationships between koala age, sex, C. pecorum, PhaHV-1 and PhaHV-2 presence by testing 157 predominantly clinically-derived urogenital swab samples. We found no evidence of a sex-effect on active-shedding of PhaHV-2. In contrast to age groups of III or above (four years of age or above based on examination of dentition), PhaHV-2 was not detected in age class I or II, suggesting that transmission is, at least in part, horizontal. Detection of PhaHV-2 was associated with detection of PhaHV-1 but was not associated with the detection of C. pecorum (n = 157) or with clinical signs in a subset of koalas positive for C. pecorum (n = 22), but this investigation was restricted by incomplete metadata in both field and clinical records.
The overall detection of PhaHV-2 across all samples was 9.6% (15/157 samples). We applied the assay to samples across a wide geographical range, to extend knowledge of PhaHV-2 presence but because our sampling strategy was largely opportunistic, we cannot accurately determine prevalence. We detected PhaHV-2 throughout NSW localities: Central West NSW (1/11), Sydney region (1/23), Mid North Coast NSW (5/12), Port Stephens (3/9) and Northern NSW (1/12). We did not detect active shedding in the Southern Highlands, Campbelltown, Liverpool Plains, Northern Rivers of NSW and Qld but cannot declare absence in these areas. While the overall frequency of detection of PhaHV-2 in our study (9.6%) was lower than that previously found in Victorian populations20, we detected PhaHV-2 in 25% of South Australian samples which is similar to PhaHV-2 previously found in wild caught South Australian koalas (20.7%)22. The low detection rate across most of our samples could be associated with small sample sizes across populations in our study, or differing sensitivity of detection associated with the different sample types employed between studies or geographical differences in PhaHV-2 sequences. Vaz, et al.20 employed a sampling approach that encompassed a more extensive array of sample sites (nasal, oropharynx, urogenital and cloacal swab samples). Our study exclusively examined PhaHV-2 in urogenital swab samples, possibly not the optimal sample type for PhaHV detection as identified by Wright, et al.21 when investigating PhaHV-1. Vaz also tested 810 samples in Victoria20 whereas our approach tested 157 samples spread unevenly across multiple bioregions. Our assay has allowed the expansion of the known range of PhaHV-2, but to compare prevalence of infection, the optimal sample types for detecting lytic and latent infection need to be determined using a larger sample size and more sample types.
We did not detect PhaHV-2 in koalas of age class I or II suggesting that, unless there is an unusually long period between infection and mucosal shedding for this virus, transmission is unlikely to be exclusively vertical from mother to offspring. While Vaz, et al.20 and Kasimov, et al.22 both found no effect of age on PhaHV-2 detection, it is possible that the broader age groups used in those studies obscured findings specific to age classes I and II. If horizontal transmission is possible, this has significant implications for biosecurity and quarantine design, with isolation potentially required to prevent transmission in hospital or quarantine settings, but further work should be done to clarify impacts and transmission routes of PhaHV-2 before recommendations can be made.
We found no evidence to support sex as a predictor for viral presence in clinically-derived samples, in corroboration with previous literature20,22. Although Vaz, et al.20 did not find a statistical difference between PhaHV-2 presence in males versus females, a significant difference between females with and without pouch young was noted. Stalder, et al.23 observed males were significantly more likely to test positive for PhaHVs but did not differentiate PhaHV-1 and 2 in their analysis. Further investigation on both free-living and clinically-derived koala samples is required to determine any potential associations between sex and PhaHV-2 presence.
PhaHV-2 has been associated with detection of C. pecorum and wet bottom20, but we did not find an increased likelihood of clinical signs in koalas with both C. pecorum and PhaHV-2. From the already existing data associated with the samples used for this study, we did not find a significant association between PhaHV-1 and likelihood of clinical signs of C. pecorum infection, in agreement with co-infection analysis by Wright, et al.21, but not consistent with analysis performed by Vaz, et al.20 and Kasimov, et al.22. Our analyses were limited to only 22 samples as recorded information on clinical signs was inconsistent across samples, so associations between the severity in clinical signs and coinfection was not possible. Potential associations between coinfections with the PhaHVs and C. pecorum and clinical signs means careful sampling and standardised metadata record-keeping is required to bridge our understanding of potential coinfection dynamics. The relatively few samples with PhaHV-2 detected in our study may have impacted our ability to identify any associations with clinical signs. Future clinical studies can incorporate more samples, health data and viral load to investigate the impacts of PhaHV-2 on koala health.
Assay performance and partial validation (Stage I) was consistent with assay development specifications outlined by the World Organisation for Animal Health (WOAH)25. To better understand sites of PhaHV-2 active shedding and latency, the qPCR assay may be best combined with serological assays. Sequence variation between positive PhaHV-2 samples emphasises the importance of further sequencing as the assay is applied to future samples across a wider geographic area. Nonetheless, our assay has expanded knowledge of PhaHV-2 distribution and offers the capacity to swiftly quantify viral load to progress exploration of PhaHV-2 pathogenesis and the influence of co-infection on chlamydial infertility.
Methods
Assay design and validation
Due to limited reference sequence available (424 bp) of the published PhaHV-2 dpol gene16, we applied the conventional PCR protocol developed by Kasimov, et al.22 to generate more sequence to enable primer design. Conventional PCR was conducted on 25 clinically-derived urogenital swab samples, collected in Qld and NSW veterinary clinics, to identify conserved regions across a greater number of samples. The assay was performed in a 20 µL reaction volume, consisting of 10 µL MyTaq HS Red Mix (Bioline, 2023), 1 µL of each primer at a final concentration of 0.5 µM, 6 µL of water and 2 µL template DNA; run at 95 °C for 2 min of denaturation, followed by 34 cycles of denaturation at 95 °C for 5 s, annealing at 57 °C (modified from 64 °C following trials to determine optimal annealing temperature) for 15 s and extension at 72 °C for 10 s, followed by a final extension at 72 °C for 10 s in a Bio-RAD T100 thermocycler. Amplicons were then visualised using gel electrophoresis and those with appropriate lengths (n = 15) were submitted for bi-directional Sanger sequencing using forward and reverse primers (Macrogen, Seoul, South Korea). Sequences from the 15 samples were initially analysed using Molecular Evolutionary Genetics Analysis software (MEGA)26 to align with the dpol gene of PhaHV-2 (GenBank accession number: JQ996387.1) and low quality sequences were trimmed. Sequences were also trimmed using a quality limit of 0.05 and assembled using Qiagen CLC Main Workbench 24.0.1 to create consensus sequences with PhaHV-2 dpol as a reference sequence (Supplementary Fig. 1). The specificity of consensus sequences was confirmed using the National Centre for Biotechnology Information’s (NCBI) Basic Local Alignment Search Tool (BLAST) function27.
Variation between the sequences limited conserved sites appropriate for primer design (Supplementary Fig. 1). Regions conserved across all sequences were considered for potential qPCR primer binding sites. Inclusion criteria consisted of; guanine-cytosine (GC) content greater than 50%, low base repeatability, an amplicon length of approximately 100 bp and an absence of primer self-complementarity. Three forward (HV2_230_Fwd, HV2_175_Fwd and HV2_52 Fwd) and three reverse (VK-PhaHV-2 Rev, HV2_178_ Rev and HV2_315_Rev) primers (Supplementary Table 1) were trialled in seven combinations at various annealing temperatures, ranging from 54 to 57 °C. Five confirmed PhaHV-2 positive, urogenital swab samples from a previous study20 were used to test primer specificity and sensitivity. The VK-PhaHV-2 Rev primer (TTTCGAGCATCATGCGTCCT), originally developed by Kasimov, et al.22, was utilised due to its optimal primer binding site. All other primers were designed using primer3plus28 and Oligo-analyser29 was used to check for primer-dimers and self-dimers. The specificity of each primer was checked using the NCBI BLAST function. Our final selected primers were HV2_230_Fwd (GGTGACGTGCAATTCAGTGT) and VK-PhaHV-2 Rev.
The qPCR assay was performed in a 20 µL reaction volume, consisting of 2x SsoAdvanced Universal SYBR® Green Supermix (Bio-Rad, Australia), forward and reverse primers at a final concentration of 250 nM each, PCR grade water and 2 µL template DNA; run at 95 °C for 3 min of denaturation, followed by 39 cycles of denaturation at 95 °C for 10 s, annealing/extension at 57 °C for 30 s, and a final melt curve (65–90 °C) at 0.5 °C increments for 5 s. A standard curve and the limit of detection (LOD) were obtained using a synthetic positive control comprising the PhaHV-2 target insert sequence in a pMG-Amp vector (Macrogen, South Korea). The LOD and efficiency were calculated using a minimum of three replicates of 1 × 107 to 1 × 105 copies/µL, seven replicates of 1 × 102 to 50 copies/µL and ten replicates of 25, 10, 5 and 1 copies/µL (see Supplementary material). The average cycle threshold (Ct) for each plasmid dilution was calculated and the proportion of positive results per dilution was translated to a probit value (Supplementary Table 2)30. Probit values were then plotted (y-axis) against copies/reaction (x-axis) to determine the 95% (CI) LOD, and corresponding Ct (Supplementary Fig. 2)30.
To confirm assay specificity, several samples that produced positive or discordant results between replicates (n = 12) were sent for Sanger bidirectional sequencing (Macrogen, South Korea). Forward and reverse sequences for each sample were analysed and used to construct a consensus sequence. The consensus sequence was tested for percentage identity to the PhaHV-2 dpol gene using the NCBI BLAST function and to confirm there were no matches with PhaHV-1.
Study samples
We tested DNA previously extracted using the MagJET Genomic DNA Kit #K2721 (ThermoFisher Scientific, Australia) as described in Fernandez, et al.31 and the MagMAX™ CORE Nucleic Acid Purification Kit #A32700 (ThermoFisher Scientific, Australia) as described in Premachandra, et al.32 from a total of 171 urogenital swab samples. These samples comprised 156 samples from the University of Sydney’s Koala Health Hub archives, selected to cover a wide geographic range from various veterinary clinics and wildlife hospitals within NSW, SA and Qld; and 15 samples of previously extracted DNA of urogenital swabs from wild-caught koalas from a previous Qld study33. Clinical samples were collected opportunistically during routine veterinary examinations. All methods are reported in accordance with ARRIVE guidelines34 and were carried out in accordance with relevant guidelines and regulations and approved by the relevant institution (University of Sydney Ethics approval 2019–1547).
Study samples were tested in duplicate using the optimised qPCR protocol. Test qPCRs were performed against a standard curve of the plasmid at 10-fold dilutions (ranging from 1 to 107 copies/µL) to quantify viral load. Nuclease free water was used as a non-template control. Previously designed primers for koala ß-actin were employed as a DNA quality control35. A sample was deemed positive if duplicates successfully amplified PhaHV-2 and had a melt peak of 87-87.5° (see Results). Samples with discordant results, including off-target melt peaks or insufficient koala ß-actin amplification, were repeated at a 1:10 dilution to reduce potential inhibitor concentration36. If the viral status of a sample could not be resolved after repeating at 1:10 dilution, the sample was excluded from the investigation (n = 14/171 samples). Samples had previously been tested for presence of C. pecorum and PhaHV-1 using established protocols21,35,37 so these data were available to investigate coinfections with PhaHV-2.
Clinical samples were largely collected opportunistically from veterinary and rehabilitation clinics, so their precise location was not available. Sampled animals were categorised based on the genetically distinct bioregions previously identified by Johnson, et al.38; Central Qld, Northern NSW/Southern Qld, Mid North NSW, Central NSW and South Australia (SA) (Table 1; Fig. 1). The opportunistic nature of our sampling strategy is not suitable for calculating accurate prevalence of PhaHV-2, but rather extends knowledge of PhaHV-2 presence within sampled regions of NSW and Qld. The age of the koalas was estimated based on tooth wear classes I-VII using a previously-described methodology39 and ranged from classes I to VII, with 52 unknown (Table 3).
We employed generalized linear models (GLMs) to assess the relationships between PhaHV-2 detection to that of PhaHV-1, C. pecorum, age, sex and region. PhaHV-2 was selected as the binary outcome variable in R v4.2.0 40. As our sampling technique did not enable population-level evaluation of PhaHV-2 presence, sampling locations were grouped into five broad regions as outlined above. Data on sex and age was available for a subset of koalas (n = 143 and 105 respectively) so these variables were also included as possible predictors for PhaHV-2 presence. To investigate whether co-infection with PhaHVs impacts the presentation of clinical signs in koalas infected with C. pecorum, we studied an additional subset of koalas positive for C. pecorum and with data available on clinical signs consistent with chlamydial infection (n = 22, 12 of which displayed clinical signs). Reporting of ocular and urogenital clinical signs was variable, so this was reduced to a binary term. A secondary GLM was conducted using clinical data (binary; yes (presence of clinical signs) or no (absence of clinical signs)) as the outcome variable and PhaHV-2 detection, PhaHV-1 detection, age and sex as possible explanatory variables. GLMs were performed in R using the ‘LMe4’ package41. We followed the information theoretic approach, developed by Grueber, et al.42. The model is first standardized using the ‘arm’ package43,44, then the ‘MuMin package’45 to determine the top two AICc (small sample-size corrected Akaike information criterion) to generate the final model. The relative importance (RI) of each explanatory variable was determined by adding the Akaike weights from all the final models where the predictor was present, with an RI of 1 indicating a strong predictor. Univariate analyses were also performed to understand the nature of the associations, by calculating p values and 95% confidence intervals using odds ratios and chi-squared tests (X2) in 2 × 2 tables for significant predictors identified through GLM.
Data availability
Data is provided within the manuscript or supplementary information files.
References
Vitali, S. et al. National Koala Disease risk Analysis Report (University of Sydney, 2022).
Phillips, B. Koalas: the little Australians we’d all hate to lose (Australian Government Publishing Service, 1990).
Beyer, H. L. et al. Management of multiple threats achieves meaningful koala conservation outcomes. J. Appl. Ecol. 55, 1966–1975. https://doi.org/10.1111/1365-2664.13127 (2018).
Adams-Hosking, C. et al. Use of expert knowledge to elicit population trends for the koala (Phascolarctos cinereus). Divers. Distrib. 22, 249–262. https://doi.org/10.1111/ddi.12400 (2016).
McAlpine, C. et al. Conserving koalas: a review of the contrasting regional trends, outlooks and policy challenges. Biol. Conserv. 192, 226–236. https://doi.org/10.1016/j.biocon.2015.09.020 (2015).
Melzer, A., Carrick, F., Menkhorst, P., Lunney, D. & John, B. S. T. Overview, critical assessment, and conservation implications of koala distribution and abundance. Conserv. Biol. 14, 619–628. https://doi.org/10.1046/j.1523-1739.2000.99383.x (2000).
Quigley, B. L. & Timms, P. Helping koalas battle disease – recent advances in chlamydia and koala retrovirus (KoRV) disease understanding and treatment in koalas. FEMS Microbiol. Rev. 44, 583–605. https://doi.org/10.1093/femsre/fuaa024 (2020).
DCEEW, A. G. Koala listing under national environmental law, (2022). https://www.dcceew.gov.au/environment/biodiversity/threatened/species/koalas/listing-under-national-environmental-law
Robbins, A., Loader, J., Timms, P. & Hanger, J. Optimising the short and long-term clinical outcomes for koalas (Phascolarctos cinereus) during treatment for chlamydial infection and disease. PloS One. 13, e0209679–e0209679. https://doi.org/10.1371/journal.pone.0209679 (2018).
Xu, W. et al. Exogenous retrovirus isolated from koalas with malignant neoplasias in a US zoo. Proc. Natl. Acad. Sci. - PNAS. 110, 11547–11552. https://doi.org/10.1073/pnas.1304704110 (2013).
Kinney, M. E. & Pye, G. W. Koala retrovirus: a review. J. Zoo Wildl. Med. 47, 387–396. https://doi.org/10.1638/2015-0185.1 (2016).
McCallum, H., Kerlin, D. H., Ellis, W. & Carrick, F. Assessing the significance of endemic disease in conservation—koalas, chlamydia, and koala retrovirus as a case study. Conserv. Lett. 11, e12425–n. https://doi.org/10.1111/conl.12425 (2018).
Tarlinton, R., Higgins, D. & Maher, I. Koala retrovirus (KoRV) Fact Sheet 1–7 (Wildlife Health Australia 2019).
Blyton, M. D. J., Young, P. R., Moore, B. D. & Chappell, K. J. Geographic patterns of koala retrovirus genetic diversity, endogenization, and subtype distributions. Proceedings of the National Academy of Sciences - PNAS 119, e2122680119-e2122680119, (2022). https://doi.org/10.1073/pnas.2122680119
Vaz, P. et al. Detection of a novel gammaherpesvirus in koalas (Phascolarctos cinereus). J. Wildl. Dis. 47, 787–791 (2011).
Vaz, P. et al. Detection of a second novel gammaherpesvirus in a free-ranging koala (Phascolarctos cinereus). J. Wildl. Dis. 48, 226–229 (2012).
Cohen, J. I. Herpesvirus latency. J. Clin. Investig. 130, 3361–3369. https://doi.org/10.1172/JCI136225 (2020).
Prusty, B. K. et al. Chlamydia trachomatis infection induces replication of latent HHV-6. PloS One. 8, e61400–e61400. https://doi.org/10.1371/journal.pone.0061400 (2013).
Gulve, N. & Rudel, T. Chlamydia trachomatis and human herpesvirus 6 infections in ovarian cancer-casual or causal? PLoS Pathog. 15, e1008055–e1008055. https://doi.org/10.1371/journal.ppat.1008055 (2019).
Vaz, Legione, A. R., Harley, C. A., Devlin, J. M. & Fenwick, B. Detection and differentiation of two koala gammaherpesviruses by use of high-resolution melt (HRM) analysis reveals differences in viral prevalence and clinical associations in a large study of free-ranging koalas. J. Clin. Microbiol. 57 https://doi.org/10.1128/JCM.01478-18 (2019).
Wright, B. R. et al. Expanding the known distribution of phascolartid gammaherpesvirus 1 in koalas to populations across Queensland and New South Wales. Sci. Rep. 14, 1223. https://doi.org/10.1038/s41598-023-50496-4 (2024).
Kasimov, V. et al. Identification and prevalence of phascolarctid gammaherpesvirus types 1 and 2 in South Australian koala populations. Viruses 12, 948 (2020).
Stalder, K. et al. Prevalence and clinical significance of herpesvirus infection in populations of Australian marsupials. PLoS One. 10 https://doi.org/10.1371/journal.pone.0133807 (2015).
Michel, A. L. et al. Pathogen detection and disease diagnosis in wildlife: challenges and opportunities. OIE Revue Scientifique et Technique. 40, 105–118. https://doi.org/10.20506/rst.40.1.3211 (2021).
WOAH. in Manual of Diagnostic Tests and Vaccines for Terrestrial Animals 2022 Chap. 2.2.7. (2014).
MEGA11. Molecular Evolutionary Genetics Analysis Version 11 v. 38 (Oxford University Press, 2021).
Altschul, S. F., Gish, W., Miller, W., Meyers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410. https://doi.org/10.1006/jmbi.1990.9999 (1990).
Untergasser, A. et al. Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 35 https://doi.org/10.1093/nar/gkm306 (2007).
Owczarzy, R. et al. IDT SciTools: a suite for analysis and design of nucleic acid oligomers. Nucleic Acids Res. 36 https://doi.org/10.1093/nar/gkn198 (2008). W163-Q169, doi.
Pum, J. A practical guide to validation and verification of analytical methods in the clinical laboratory. Adv. Clin. Chem. 90, 215–281. https://doi.org/10.1016/bs.acc.2019.01.006 (2019).
Fernandez, C. M. et al. Genetic differences in Chlamydia pecorum between neighbouring sub-populations of koalas (Phascolarctos cinereus). Vet. Microbiol. 231, 264–270 (2019).
Premachandra, H. et al. Advancements in noninvasive koala monitoring through combining Chlamydia detection with a targeted koala genotyping assay. Sci. Rep. 14, 1–8 (2024).
Ellis, W. et al. Koalas of the Clarke Connors range. Australian Mammalogy. 45, 160–170. https://doi.org/10.1071/AM22026 (2023).
Percie du Sert. Reporting animal research: explanation and elaboration for the ARRIVE guidelines 2.0. PLoS Biol. 18, e3000411. https://doi.org/10.1371/journal.pbio.3000411 (2020).
Hulse, L. S. et al. Development and application of two multiplex real-time PCR assays for detection and speciation of bacterial pathogens in the koala. J. Vet. Diagn. Invest. 30, 523–529. https://doi.org/10.1177/1040638718770490 (2018).
Hoorfar, J. et al. Practical considerations in design of internal amplification controls for diagnostic PCR assays. J. Clin. Microbiol. 42, 1863–1868. https://doi.org/10.1128/JCM.42.5.1863-1868.2004 (2004).
Wright, B. R. et al. Development of diagnostic and point of care assays for a gammaherpesvirus infecting koalas. PLoS One. https://doi.org/10.1371/journal.pone.0286407 (2023).
Johnson, R. N. et al. Adaptation and conservation insights from the koala genome. Nat. Genet. 50, 1102–1111. https://doi.org/10.1038/s41588-018-0153-5 (2018).
Gordon, G. Estimation of the age of the koala, Phascolarctos cinereus (Marsupialia: Phascolarctidae), from tooth wear and growth. Australian Mammalogy. 14, 5. https://doi.org/10.1071/AM91001 (1991).
R Core Team. R: A Language and Environment for Statistical Computing_. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org, (2023).
Bates, D., Mächler, M., Bolker, B. & Walker, S. Fitting linear mixed-effects models using lme4. arXiv preprint arXiv:1406.5823 (2014).
Grueber, C. E., Nakagawa, S., Laws, R. J. & Jamieson, I. G. Multimodel inference in ecology and evolution: challenges and solutions. J. Evol. Biol. 24, 699–711. https://doi.org/10.1111/j.1420-9101.2010.02210.x (2011).
Gelman, A. Scaling regression inputs by dividing by two standard deviations. Stat. Med. 27, 2865–2873 (2008).
arm: Data analysis using regression and multilevel/hierarchical models. (2011).
MuMIn: multi-model inference. R package version 1. 0. 0. (2009).
Acknowledgements
The authors would like to thank Lyndal Hulse for providing samples from Queensland as well Port Macquarie Koala Hospital, Friends of the Koala, Port Stephens Koala Hospital, WIRES, UQ Koala Ecology Group and the many wildlife carers and veterinary clinics that submitted samples for diagnostics used in this study. This study was funded by the Australian Department of Agriculture, Water and the Environment Bushfire Recovery Multiregional Species Program. BRW and AC are supported by the NSW Wildlife Information Rescue and Education Service (WIRES).
Author information
Authors and Affiliations
Contributions
C.C. wrote the manuscript, conducted all laboratory work and data analysis; A.C. contributed to laboratory work and data analysis; Y.M. contributed to sampling and metadata collation, M. K. contributed to interpretation and provided samples; P. K. contributed to interpretation, primer design and provided samples; D.P.H. conceived and oversaw the project and contributed to interpretation of findings; B.R.W. conceived and oversaw the project, contributed to interpretation of findings, contributed to laboratory work and data analysis; all co-authors contributed to the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Church, C., Casteriano, A., Muir, Y.S. et al. New insights into the range and transmission dynamics of a koala gammaherpesvirus, phascolarctid gammaherpesvirus 2. Sci Rep 15, 6136 (2025). https://doi.org/10.1038/s41598-025-90626-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-90626-8
