
Abstract
The discovery of unique microRNA (miR) patterns and their corresponding genes in sarcoma patients indicates their involvement in cancer development and suggests their potential use in medical management. MiRs were identified from The Cancer Genome Atlas (TCGA) dataset, with a Deep Neural Network (DNN) employed for novel miR identification. MiRDB facilitated target predictions. Functional enrichment analysis, identify critical pathways, protein-protein interaction network, and diseases/clinical data correlations were explored. COX regression, Kaplan-Meier analyses, and CombioROC was also utilized. The population consisted of 119 females and 142 males, and 1046 miRs were uncovered. Ten miRs was selected for further analysis using DNN. Upon analyzing for gene ontology, it was found that these genes showed enrichment in various activities. We identified a significant association between the overall survival rate of sarcoma patients and miRs levels. The combination of miR.3688 and miR.3936 achieved the greatest diagnostic standing. MiRs have the capability to screen sarcoma patients to identify undetected tumors, predict prognosis, and pinpoint prospective targets for treatment. Further large clinical trials are required to validate our findings.
Similar content being viewed by others


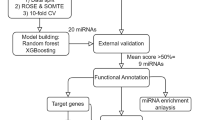
Introduction
Sarcomas are a broad category of malignancies originating from soft tissues and bone, comprising around 100 distinct pathological types1. Annual incidence of sarcomas is less than 6 cases per 100,000 persons. Sarcomas comprise up to 15% of pediatric malignancies, < 2% of all adult solid tumors, and < 1% of all adult malignancies1,2,3. Numerous sarcomas have their highest occurrence in adolescence and young adulthood, and sarcoma accounts for 8% of all cancers in this age group; thus, age-specific epidemiology lends credence to the idea that this is when the disease is most prevalent. This seamless incidence across ages has long blurred the conventional dividing line between pediatric and medical oncology care4.
Concerning the type of sarcoma, a 12-year analysis of the Surveillance, Epidemiology, and End Results (SEER) database in the US showed that the most prevalent histology was sarcoma-not otherwise specified (14.8%), and the primary location was soft tissue (43.4%)5. While certain environmental factors and genetic abnormalities have been associated with sarcoma development, most are sporadic and idiopathic. Familial adenomatous polyposis syndrome, neurofibromatosis-1, retinoblastoma, and Li-Fraumeni syndrome are some hereditary diseases associated with sarcoma6. Surgery, radiation, and chemotherapy are the mainstay in treating sarcomas7. Nonetheless, the use of conventional therapies such as chemotherapy or anti-angiogenic agents typically leads to a survival duration ranging from 12 to 18 months3.
To enhance the identification of accessible diagnostic, prognostic, and therapeutic targets, the utilization of DNA, RNA, and proteins is increasing. Research shows that only 2% of the human genome codes for proteins, while over 90% is actively transcribed. Noncoding RNAs are associated with most transcribed sequences8. One type of noncoding RNA that plays a role in many biological processes through posttranscriptional alterations is known as microRNAs (miRs)9,10,11,12,13,14. It is not well-known how miRs are expressed in sarcomas. However, prior research has demonstrated that several sarcoma subtypes could profit significantly from a deeper understanding of miR expression patterns for diagnostic, prognostic, and therapeutic reasons15. Notably, miRs’ oncogene or tumor suppressor roles in cancer rely on the mRNAs they interact with15. The findings of distinct miR patterns and associated genes in sarcoma patients show a role in oncogenesis and may help patient management.
Significant enhancements in diagnostic and prognostic accuracy in cancers have been owing largely to breakthroughs in big data processing tools. The big data approach commonly uses bioinformatics and artificial intelligence to analyze data from a set of people to predict an event for the general population. In contrast, the small data paradigm predicts data at an individual level16,17. Moreover, structured data sources can be easily managed and aggregated, dimensionality and noise reduced, and accuracy raised with these methods. Displays and summaries give interpretable, relevant information readily adaptable in clinical situations to make decisions, decrease expenses, and produce better outcomes18,19.
To the best of our knowledge, we pioneered the use of bioinformatics combined with deep learning models to identify miRs with differential expression in a large dataset of sarcoma cases, filling a gap in the literature that has currently ignored this combination in these rare patients. Additionally, sarcoma-related biological pathways, coexpressions of relevant genes, and diagnostic and prognostic markers aimed to uncover.
Results
Patient demographics
Our population comprised 119 females and 142 males. Of them, 162 (62.1%) were alive, and 99 (37.9%) were dead. Among the present 261 samples, 1046 miRs were found. The average age at the initial pathologic diagnosis was 60.87 ± 14.65, and the average number of days to the last follow-up was 863.44 ± 1099.92. Table 1 displays demographic and clinicopathological characteristics.
Identification of dysregulate expression miRs
The data was normalized and filtered to prepare the miR data for machine learning and A Deep Neural Network (DNN). Then, the essential features were extracted using a correlation coefficient threshold > 0.5. Finally, using the particular criteria, 70 miRs (tumor vs. normal) were found. A DNN with an accuracy of 79.15, an MSE of 2.21, an RMSE of < 0.01, an R2 of 0.99, and an AUC of 1.0 was used to examine the essential miRs. After that, 20 important miRs were found, and 10 of them with co-efficiencies greater than 0.90 were chosen for additional DNN study. The coefficients of miR.3688, miR.1292, and miR.3936 were 1.00, while miR.3678 had a coefficient of 0.96. MiR.588 and miR.3140 showed coefficients of 0.95, miR.1305, miR.3199-1, and miR.1255a had coefficients of 0.94, and miR.19b-2 had a coefficient of 0.93.
Target gene prediction of miR markers
The biological targets of 20 recognized miRs from DNN were predicted using the online analytical tool miRDB. Ninety-nine selected genes were used for subsequent analysis.
Gene ontology analyses of miRs, functional annotation, and pathway enrichment analyses
The deployment of ten critical genes for gene ontology (GO) was performed using R software. As Supplementary Table S1 online summarizes, genes exhibited enrichment with cellular signaling pathways, central nervous system development, and metabolic processes. Genes were significantly enriched in activities such as binding to nucleic acids, proteins, and ions through channels, facilitating the transmembrane transfer, and acting as receptors for benzodiazepines, insulin-like growth factors, and pre/post-synaptic signals. These cellular components are found or activated mainly through the CNS development process in the nucleus and cytoplasm (Fig. 1).

Pathway enrichment analyses.
PPI network construction
String analyzed and visualized the gene interaction, which received a 0.9 score. The outcomes showed a significant link between the 25 genes. As shown in Fig. 2, there are six association pathways between (I) STXBP5L, NRXN1, SYT1, and GPM6A; (II) ELAVL2, STRBP, PUM2, CPEB2, ATXN1, ELAVL1, CTBP1, AR, LATS2, TEAO1, MAPK1, APBB1IP, and PTPRE; (III) EBF2 and YOD1; (IV) BICD2 and HOOK3; (V) EDNRB and NMU; and (VI) GCNT1 and GALNT3 genes.

PPI network construction.
Investigation of the correlations of clinical/demographic features with sarcoma
Data on sarcoma patients revealed a significant moderate positive link between race and ethnicity (correlation = 0.3). (Fig. 3)

Investigation of the correlations of Clinical/Demographic features with Sarcoma. X indicates insignificant relationship.
Identifying prognostic markers of miRs
Kaplan-Meier survival analysis was used to assess the impact of the prognostic signature of important miRs in sarcoma. The findings indicated that the overall survival of patients with sarcoma was considerably increased by downregulating miR.3150, miR.3680, miR. 1229, and miR. 1265. On the other hand, overall survival was markedly reduced by downregulating miR.3144 and miR.3941. The details are presented in Table 2.
Identifying diagnostic markers of miRs
The combination of miR.3688 and miR.3936 attained the highest rank, as indicated by its AUC of 0.766, sensitivity of 0.90, and specificity of 0.50. The second and third potential diagnostic markers were corresponding combinations of miR.1255a, miR.1292, and miR.3936 (AUC: 0.90, sensitivity: 0.86, specificity: 1.0), and miR.1255a, miR.1292, and miR.3678 (AUC: 0.86, sensitivity: 0.83, specificity: 1.0). Furthermore, the generalized linear model (GLM) analysis determined that the combination of miR.3688 and miR.3936 exhibited the best Akaike information criterion (AIC) value. Figure 4; Table 3 illustrate additional potential diagnostic markers that can be identified using the receiver operating characteristic (ROC) and GLM analysis.

Identifying diagnostic markers of miRs.
External validation
The expression levels of candidate miRs in sarcoma were verified using 12 experiments in the dbDEMC including EXP00111(GSE18546), EXP00157(GSE31045), EXP00189(GSE28423), EXP00282(GSE45364), EXP00340(GSE65071), EXP00467(GSE55625), EXP00527(GSE106817), EXP00536(GSE113486), EXP00564(GSE135518), EXP00607(GSE113740), EXP00619(GSE112264), EXP00629(E_MTAB_3273).
Discussion
In this study, bioinformatic analysis and DNN were combined to identify novel biomarkers of sarcoma. Our data showed the potential value of the combination of miR.3688 and miR.3936 in sarcoma diagnosis. Furthermore, the result of the survival analysis revealed that the downregulating miR.3150, miR.3680, miR.1229, and miR.1265 was associated with an increased survival rate. Otherwise, upregulating miR.3144 and miR.3941 were associated with a lower survival rate. Additionally, 25 genes in six different pathways were found to be associated with miRs in our investigation. Overall, we uncovered that miRs could have a positive impact on identifying and managing sarcoma patients.
A first-year mortality rate of 10–15% had been reported for sarcoma cases, likely highlighting diagnostic delays20. Most studies diagnosed approximately 30% of cases with metastatic conditions1. Unfortunately, recurring and/or metastatic conditions probably caused the deaths of 30% or more of the patients within five years20. In other words, delays in accurate diagnosis, inefficient or inadequate therapy or prognosis, fewer chances to participate in clinical trials, and less availability of innovative drugs all contribute to a greater mortality rate for patients with rare cancers compared to those with common cancers21. Since delays in diagnosis result in increased tumor size, metastasis, complications, and decreased survival rates, early diagnosis is essential for sarcoma patients to minimize the necessity for extensive surgery and increase their chances of survival.
As stated above, the prognosis of sarcomas is significantly influenced by the size of the tumor and the extent of metastasis at the time of diagnosis. Delaying the diagnosis of sarcoma can also affect fertility preservation, induce psychological distress, lead to patient dissatisfaction, and impede treatment adherence1,22. To understand better, a growing number of cancer datasets are being processed, and studies are exploring the potential positive impacts of bioinformatics and artificial intelligence in healthcare and uncovering the complicated biology of cancers. For example, noteworthy and compelling discoveries derived from this combination have been observed in glioblastoma, non-alcoholic fatty liver disease, colorectal, pancreatic, uterine, and cervical cancers23,24,25,26,27,28.
Priorly, studies of bioinformatics on possible therapeutic, prognostic, or diagnostic factors in sarcoma have found the biomarkers, including combined CNB2, PRC1, SCM229, methylation sites of cg07814289, cg09494609, cg1414402530, mRNAs of APOL1, EFEMP1, LYZ, MEDAG, MYH11, RARRES1, TNFAIP2, TNFSF10, ZNF385A, Long noncoding RNAs of CTD-2228K2.7, HOTAIRM1, NCF1C31, BCL7B expression7, expression of CENPF, KIF11, KIF23, TTK, MKI67, TOP2A, CDC45, MELK, AURKB, BUB132, Long noncoding RNA of MALAT1/miR-508-3p/CCNA2 as a regulatory axis33, the CD48 antigen (CD48), putative P2Y purinoceptor 10 (P2RY10), RAS protein activator like-3 (RASAL3)34, and CDK1, CCNB1, CDC20, CCNB2, AURKB, MAD2L1, CENPE, KIF2C and PCNA35,36.
The application of DNN-based approaches as a supporting analytical framework in cancer research is gaining progress. Some recent papers have discussed the practical uses of DNN-based methods in cancer research, such as medication response prediction, cancer diagnosis, and prognosis. These papers also highlight the potential of this new analytical platform to realize the goal of precision and personalized medicine. These designs may not be widely used at this point, but they could help fill in some of the gaps in paradigmatic analysis that older inference frameworks are struggling with37. The DNN is an algorithm considered for analysis in the present study. Machine learning-based bioinformatics applications have benefited from DNN since it makes it possible to discover important features with high accuracy in prediction analysis38.
In light of machine learning, several investigations have been done on sarcoma diagnosis, staging, subtype differentiation, therapeutic targets, and prognosis. For instance, some were based on radiomics and digital patho/histopathology39,40,41, NETosis-related and overall gene patterns42, or Fourier transform infrared spectroscopy of tissues43. Genes such as PRKAR1B, INPP5A, GLI344, HMMR45, cells such as macrophages and cytotoxic, CD8 + T, natural killer (NK), mast, B, NK CD56bright, TEM, TCM, T helper 2 cells, FABP4, and NDRG146 were some potential identified biomarkers by machine learning.
While bioinformatics and machine learning shed light on sarcoma management in unison, working in combination could revolutionize oncology fields of diagnosis, molecular subtyping, histopathology, treatment targets, and prognosis. On the other hand, because of proper sensitivity, high specificity, and ease of access from various bodily fluids, miRs in circulation are a promising biomarker47. Some recent specific studies on miRs and sarcoma are summarized in Supplementary Table S2 online. Research points to miRs as a key player in cancer development and progression. A promising new approach to cancer management could be the identification of dysregulated miRs and their roles in cellular transformation, progression, benign and malignant states, and regulating many biological pathways48. In this regard, 21 essential miRs were identified in a pan-cancer study as being related to eight or more cancers and upregulated in over 60% of patients per cancer49. The therapeutic utility of miRs as biomarkers for the prediction, diagnosis, and prognosis of cancer treatment responses is the focus of numerous ongoing clinical trials50.
When comparing our newly discovered biomarkers with prior ones, it is evident that miR plays a crucial role in intercellular interaction and is released into physiological fluids. In several ways, miRs might enter the circulation and end up in easily accessible fluids, including saliva, blood, and urine. Damage to tissues or cells can trigger their release or be actively transported through mechanisms such as microvesicles, exosomes, or protein bonding51. Additionally, we found 25 genes in six different pathways associated with our miRs. Only Early B-Cell Factor-2 (Ebf2) was found to be associated with sarcoma and to play an essential regulatory role in osteosarcoma in earlier studies. One of them found overexpressed EBF2 impeded cell death and promoted osteosarcoma cell migration and invasion. The study showed that miRs that are anti-oncogenic in osteosarcoma might work by blocking the oncogenic transcription factor EBF252. Another research also indicated that decreased expression of a certain miR in osteosarcoma cells leads to the increased production of EBF2, hence facilitating the development of the disease53.
Moreover, in pan-cancer analysis, APBB1IP overexpression was connected with greater immune-cell infiltration, and the expression of APBB1IP in distinct cancers might be attributed to the tumor immune microenvironment54. Besides, the fusion gene HOOK3-FGFR1 and the gene ATXN1 were shown to be linked to the NF-kappaB pathway, which leads to the activation of proinflammatory cytokine genes and the initiation of an inflammatory reaction55,56. In parallel, several genes were discovered to be involved in cellular activities such as migration, invasion, apoptosis, and proliferation; these genes include PUM2, SYT1, and CTBP157,58,59, which significantly altered in several cancers, such as gastrointestinal, genitourinary, hematologic, lung, and skin60,61,62. For example, overexpressing SYT1 inhibits the development of colorectal cancer cell pseudopods, mediated by ERK/MAPK signaling, and therefore reduces migration, invasion, and metastasis59. Also, bladder cancer in vitro invasion is enhanced by overexpression of L1PA2-SYT163.
A large number of the genes that we have found also have roles in the central and peripheral nervous system’s synaptic network. Essential genes for synaptic health include STXBP5L, NRXN1, SYT1, and GPM6A. Neuropsychiatric and neurodevelopmental diseases were caused by these genes’ dysregulations64,65,66,67. For example, STXBP568, LATS269, ELAVL270, and CPEB271 are particular markers uncovered in glioma. Moreover, research has shown that cancer patients often have a loss of heterozygosity in the LATS gene. In addition to its critical function as a tumor suppressor, the LATS gene demonstrates advantages in preserving the organism’s genomic integrity and fitness72. In parallel with the effects of these genes on the nervous system, CPEB2 has also been implicated in breast tumors73, multiple myeloma74, and colorectal cancer75. Besides, the anti-tumor effectiveness of NRXN1-mediated antibody-drug combination treatment in small-cell lung cancer was lost when NRXN1 was knocked out in SHP77 cells76.
Numerous additional genes we found, previously have been identified as influential factors in the various cancers. The gene GPM6A, for instance, has been implicated in the development of lymphoid leukemia, sporadic non-functioning pituitary adenomas, and colorectal cancer77. Similarly, ELAVL1 has been associated with cervical cancer78, hepatocellular carcinoma79, prostate, breast, and gastric cancer80. YOD1 has been uncovered as a novel regulator of the Hippo pathway in liver cancer81 and as an oncogene in triple-negative breast cancer through its interaction with CDK182. Additionally, it has shown potential for managing pancreatic cancer83. BICD2 has been found to impact oral squamous cell carcinoma by affecting cell proliferation, migration, invasion, glutaminolysis, and apoptosis via modulation of the miR-149-5p/IGF2BP1 axis84. Also, HOOK3 has been implicated in the development of various malignancies, including prostate cancer, myelodysplastic syndrome, non-small-cell lung cancer, and papillary thyroid carcinoma55. Lastly, the gene EDNRB has been associated with lung adenocarcinoma85, triple‑negative breast cancer86 and has been identified as a potential treatment target for gastric cancer through its interaction with miR19b-3p at the posttranscriptional level87.
Furthermore, the gene NMU has emerged as a significant marker for tumor growth and/or progression in endometrial, renal, colorectal, and breast cancers88,89. In addition, the aberrant expression of GCNT1 has been linked to aggressive disease in various cancer types90, while GALNT3 has been implicated in tumor promotion in pancreatic and ovarian cancers91, as well as lung adenocarcinoma92 and intrahepatic cholangiocarcinoma93. Also, STRBP has been associated with triple-negative breast cancer94 and is involved in changed brain and testicular development in Down’s syndrome and cryptorchidism95. Moreover, Ataxin-1 has been linked to an increased risk of multiple sclerosis96 and cervical cancer97, while the androgen receptor (AR) is associated with androgen insensitivity syndrome and prostate cancer98. Besides, the MAPR family of genes has demonstrated a distinct expression pattern in breast cancer99. Lastly, it has been identified as playing a role in chemoresistant retinoblastoma100 and mammary hyperplasia with a higher incidence of tumors101.
Improving sarcoma management by targeting correct miRs is just one of several tumors that could benefit from the significant impact of miRs and related genes, which has been discussed. Altogether, the rapid progress in bioinformatics enables the exploration of the complex relationships between miR patterns and downstream oncogenic targets, activated signaling mechanisms, cancer diagnosis and survival, and medication response dynamics. Besides, integrating extensive miR datasets using artificial intelligence and machine learning can potentially convert these patterns into comprehensive personalized medicine tools that can enhance clinical decision-making102. Currently, there is a lack of studies that combine bioinformatics and deep learning to provide more reliable results on miRs.
Limitations
Big data analysis has many advantages, such as improved quality of health care services and cost savings, but certain limitations need consideration. One of the most significant barriers to applying big data techniques in health care is the distributed nature of medical data. Another one is the lack of human clinical study in our investigation. Moreover, unstandardized methods for RNA isolation, sampling, and reference controls make it difficult to compare data among studies, thereby requiring reference procedures to quantify circulating miRs47. Additionally, an in-depth knowledge of miR biosynthesis and extracellular trafficking pathways is needed to clarify the source of miRs in healthy people and patients47.
In conclusion, improving sarcoma patient care in the context of standard clinical practice presents challenges. An unmet need in the field of biomarker-driven personalized diagnosis and treatment is the identification of prognostic and predictive variables for these uncommon conditions103. Meanwhile, miRs reflect various pathological processes and can be encapsulated in proteins or exosomes, released into the bloodstream, and protected from the action of endogenous RNAse enzymes48,104. Therefore, miRs can be used to screen patients for possible undetected tumors, predict prognosis, and identify potential targets for treatment. However, additional clinical studies in larger patient populations are necessary to confirm the practical value of miRs in the diagnosis, prognosis, and treatment of sarcoma.
Materials and methods
Data collection
The Cancer Genome Atlas (TCGA) dataset (https://gdac.broadinstitute.org/) provided the miR-Seq data of sarcoma patients and clinical parameters, including sex, age, race, ethnicity, vital status, case numbers, person neoplasm cancer status, and metastatic diagnosis. Of 261 samples obtained from the TCGA, 1046 miRs were found.
Data preprocessing and the identification of miRs
DNN of machine learning was employed to find novel miRs. Filtering and normalization are crucial phases in data processing. In this regard, preprocessing steps such as filtering and normalization were assessed before applying machine learning to miR data. Initially, duplicate genes and samples were removed with R programming filtering code. Next, the R software’s Limma R packages are used to normalize miRs. The miRs were filtered according to specific criteria, such as log fold change |FC| > 1 and P-value < 0.05. All analysis and graphic visualization in this stage were done using the R program (version 4.2.3).
Identifying predictive markers
Using DNN for classification and correlation for feature selection, we performed a bioinformatic investigation to predict miRs as crucial markers in sarcoma.
Implementation and evaluation
Version 3.7 of the Python programming language was employed to implement machine learning. The packages that were used were Scikit-learn, Matplotlib, NumPy, and Pandas. The created models were independently assessed and optimized using 70/30 training and test data. The AUC, accuracy, F1 Score, R2 score, and confusion matrix were the five indicators used to measure the performance metrics of the approaches used to identify significant miRs.
Machine learning categorization’s true positives and negatives are assessed using an accuracy metric. It is the degree to which a measurement and its actual value are closest. An indicator for classifying unbalanced data into false positive and false negative categories is the F1 Score. The ROC is frequently used to evaluate a prediction model’s discriminative power. The area under the ROC is represented by the AUC curve thatindicates the algorithm’s ability to categorize each class accurately. The confusion matrix is a table that outlines the four classification categories True Negative (TN), True Positive (TP), False Negative (FN), and False Positive (FP) and specifies the purpose of the procedure. When evaluating performance models from the standpoint of feature selection, the R2 score, also known as the coefficient of determination, is typically utilized105,106,107.
Target gene prediction for predictive miR markers
Web-based miRNA-target prediction and functional annotations are provided by miRDB (http://mirdb.org). The final target genes were chosen using a cutoff criterion of a score higher than 90.
Functional and pathway enrichment analyses of miRs
Using the cluster Profiler tool in R with P-adjusted < 0.05, the functional enrichment analysis and important pathway identification of the miR signature were annotated and displayed. GO is a standard database that provide the basis for annotating and visualizing the determined miRs.
PPI network analysis
Using the biological database STRING (https://string-db.org/), the protein-protein interaction network of miR’s targets was illustrated108. The comprehension of cellular pathways and functional genomics is contingent upon these connections. More than 0.9 was deemed to be the statistically significant interaction score.
Correlation between demographic /clinical data with disease
The use of the ggcorrplot package and cor function in the R program allowed for the analysis of correlations between disease and clinical /demographic data, including sex, race, ethnicity, vital status, patient-healthy, person neoplasm cancer status, and metastatic diagnosis. A correlation of less than 0.3 was considered weak, 0.3 to 0.6 moderate, and more than 0.6 substantial.
Identifying prognostic markers
The Univariate COX regression and Kaplan-Meier survival curves were plotted for the top-selected miRs with the survival, survminer, ggplot2, and ezcox R packages. The entire data set was subject to screening criteria, which included a cutoff level of p < 0.05 and HR ≠ 1. SPSS version 20 was utilized for Kaplan-Meier survival analysis, and p < 0.05 was considered significant. We note that, clinicopathological variables were initially included in the Cox regression model to account for potential confounding factors. However, none of these variables were significantly associated with the outcome, and as such, they were not included in the final reported results.
Combined ROC curve
GLM with combined ROC curve analysis was used to determine the effectiveness of the diagnostic process and construct diagnostic models. The discrimination of individual or combination biomarkers was assessed by evaluating their sensitivity, specificity, cutoff value, positive predictive value, negative predictive value, and AUC. Package combioROC in R was used to implement the entire process.
GLM suggests its applicability to a wide range of outcome types, including binary outcomes, multi-class outcomes, count data, and quantitative outcomes109. It extends the framework of ordinary linear regression models to include non-normal response distributions and nonlinear relationships concerning the mean. GLM comprises three essential components: the random component, the systematic component, and the link function110.
Validation of candidate miRs in the dataset
The candidate miRNAs were validated using online web servers, namely dbDMEC (https://www.biosino.org/dbDEMC/index), which contain the demiRs in human cancers based on public repositories like ArrayExpress, Gene Expression Omnibus (GEO), Sequence Read Archive (SRA), and TCGA.
Data availability
The data used in this study were taken from the TCGA dataset, openly available in the URL: https://gdac.broadinstitute.org/.
References
David, B. B. L., Abdon Mello, C., Santos Thuler, L. C. & de Melo, A. C. Overview of adult sarcoma burden and clinical pathways in Brazil. JCO Global Oncol. 8, e2100387 (2022).
Miwa, S., Yamamoto, N., Tsuchiya, H. & Sarcoma molecular pathology, diagnostics, and therapeutics. Int. J. Mol. Sci. ;24(6). (2023).
Dajsakdipon, T., Siripoon, T., Ngamphaiboon, N., Ativitavas, T. & Dejthevaporn, T. Immunotherapy and biomarkers in Sarcoma. Curr. Treat. Options Oncol. 23 (3), 415–438 (2022).
Reed, D. R., Naghavi, A. & Binitie, O. Sarcoma as a model for adolescent and young adult care. J. Oncol. Pract. 15 (5), 239–247 (2019).
Gage, M. M. et al. Sarcomas in the United States: recent trends and a call for improved staging. Oncotarget 10 (25), 2462–2474 (2019).
Hui, J. Y. Epidemiology and etiology of sarcomas. Surg. Clin. North. Am. 96 (5), 901–914 (2016).
Yang, D. et al. BCL7B is a potential novel diagnosis and prognosis biomarker for sarcomas using bioinformatics analysis. Med. (Baltim). 100 (28), e26632 (2021).
Shabani, P. et al. Role of miR-142 in the pathogenesis of osteosarcoma and its potential as therapeutic approach. J. Cell. Biochem. 120 (4), 4783–4793 (2019).
Atoum, M. F., Alowaisy, D. & Deeb, A. A. Analysis of microRNA processing machinery gene DROSHA, DICER1, and XPO5 variants association with atherosclerosis: a case–control study. Biomed. Biotechnol. Res. J. (BBRJ) 8(4). (2024).
Makhlouf, S. J., Khabour, O. F., Rawashdeh, H. M. & Sakee, B. L. Polymorphisms in MicroRNA biogenesis genes and the risk of Preeclampsia in Jordan. Biomed. Biotechnol. Res. J. (BBRJ) 8(3). (2024).
Pande, A. Co-regulatory network of transcription factor and MicroRNA: a key player of gene regulation. Biomedical Biotechnol. Res. J. (BBRJ) 5(4). (2021).
Furci, L., Schena, E., Miotto, P. & Cirillo, D. M. MicroRNA induction in human macrophages associated with infection with ancient and modern TB strains. Int. J. Mycobacteriology 4(Suppl 1). (2015).
Furci, L., Schena, E., Miotto, P. & Cirillo, D. M. Alteration of human macrophages microRNA expression profile upon infection with Mycobacterium tuberculosis. Int. J. Mycobacteriology 2(3). (2013).
Alipoor, S. D., Adcock, I. M., Folkerts, G., Garssen, J. & Mortaz, E. A Bioinformatics analysis of exosomal MicroRNAs released following mycobacterial infection. Int. J. Mycobacteriol. 8(3). (2019).
Lim, H. J. & Yang, J. L. Regulatory roles and therapeutic potential of microRNA in sarcoma. Crit. Rev. Oncol. Hematol. 97, 118–130 (2016).
Nazari, E. et al. Decision fusion in healthcare and medicine: a narrative review. Mhealth 8, 8 (2022).
Mishra, S. K., Singh, A., Dubey, K. B., Paul, P. K. & Singh, V. Role of bioinformatics in data mining and big data analysis. In: (eds Singh, V. & Kumar, A.) Advances in Bioinformatics. Singapore: Springer Nature Singapore; 271–277. (2024).
Nazari, E. et al. A comprehensive overview of decision fusion technique in healthcare: a systematic scoping review. Iran. Red Crescent Med. J. 22 (10), e30 (2020).
Nazari, E., Biviji, R., Farzin, A. H., Asgari, P. & Tabesh, H. Advantages and challenges of information fusion technique for big data analysis: proposed framework. J. Biostatistics Epidemiol. (2021).
Tichanek, F., Försti, A., Hemminki, O., Hemminki, A. & Hemminki, K. Steady survival improvements in soft tissue and bone sarcoma in the nordic countries through 50 years. Cancer Epidemiol. 102449. (2023).
Soomers, V. L. M. N. et al. The route to diagnosis of sarcoma patients: results from an interview study in the Netherlands and the United Kingdom. PLoS One. 15 (12), e0243439 (2020).
Weaver, R., O’Connor, M., Carey Smith, R. & Halkett, G. K. B. The complexity of diagnosing sarcoma in a timely manner: perspectives of health professionals, patients, and carers in Australia. BMC Health Serv. Res. 20, 1–10 (2020).
Golbaghi, M. P., Valizadeh Laktarashi, H. & Nazari, E. Identification of C1QTNF2 and its combination with AASS as a novel biomarker of Uterine cancer: RNA-sequencing and machine learning analysis. Iran. Biomed. J. 28 (7), 382 (2024).
Khalili-Tanha, G., Khalili-Tanha, N., Farahani, M., Rezaei-Tavirani, M. & Nazari, E. The G protein-coupled receptor-related gene signatures for diagnosis and prognosis in Glioblastoma: a deep learning model using RNA-Seq Data. Asian Pac. J. Cancer Prev. 25 (12), 4201–4210 (2024).
Pourali, G. et al. Abstract 4930: identification of CBX7 and PCDHB18 as novel prognostic biomarkers of cervical cancer: RNA-sequencing and machine learning analysis. Cancer Res. 84 (6_Supplement), 4930 (2024).
Rouzbahani, A. K. et al. Machine learning algorithms and biomarkers identification for pancreatic cancer diagnosis using multi-omics data integration. Pathol. - Res. Pract. 263, 155602 (2024).
Nazari, E. et al. Bioinformatics analysis and machine learning approach applied to the identification of novel key genes involved in non-alcoholic fatty liver disease. Sci. Rep. 13 (1), 20489 (2023).
Asadnia, A. et al. The Prognostic Value of ASPHD1 and ZBTB12 in Colorectal Cancer: a machine learning-based Integrated bioinformatics approach. Cancers 15 (17), 4300 (2023).
Tu, B., Jia, Y. & Qian, J. Bioinformatics analysis identified five widely expressed genes associated with prognosis in sarcoma. Int. J. Gen. Med. :3711–3725. (2022).
Wang, X-W. et al. A 3-DNA methylation signature as a novel prognostic biomarker in patients with sarcoma by bioinformatics analysis. Medicine 100 (20), e26040 (2021).
Zou, D. et al. Bioinformatics analysis reveals the competing endogenous RNA (ceRNA) coexpression network in the tumor microenvironment and prognostic biomarkers in soft tissue sarcomas. Bioengineered 12 (1), 662–672 (2021).
Song, Y. et al. Identification of metastasis-associated biomarkers in synovial sarcoma using bioinformatics analysis. Front. Genet. ;11. (2020).
Hong, X., Liu, H., Chen, C., Lai, T. & Lin, J. Bioinformatics Identification and validation of aging–related molecular subtype and prognostic signature in sarcoma. Cancer Invest. 41 (5), 512–523 (2023).
Zhu, N. & Hou, J. Assessing immune infiltration and the tumor microenvironment for the diagnosis and prognosis of sarcoma. Cancer Cell Int. 20, 1–11 (2020).
Li, Q. et al. CDK1 and CCNB1 as potential diagnostic markers of rhabdomyosarcoma: validation following bioinformatics analysis. BMC Med. Genom. 12, 1–13 (2019).
Lu, S. et al. Bioinformatics Analysis and Validation identify CDK1 and MAD2L1 as prognostic markers of Rhabdomyosarcoma. Cancer Manage. Res. 12 (null), 12123–12136 (2020).
Wysocka, M., Wysocki, O., Zufferey, M., Landers, D. & Freitas, A. A systematic review of biologically-informed deep learning models for cancer: fundamental trends for encoding and interpreting oncology data. BMC Bioinform. 24 (1), 198 (2023).
Thareja, P. & Chhillar, R. S. (eds) Power of deep learning models in bioinformatics. innovations in data analytics; 2023 2023//; Singapore: Springer Nature Singapore.
Crombé, A., Roulleau-Dugage, M. & Italiano, A. The diagnosis, classification, and treatment of sarcoma in this era of artificial intelligence and immunotherapy. Cancer Commun. 42 (12), 1288–1313 (2022).
Xu, W., Hao, D., Hou, F., Zhang, D. & Wang, H. Soft tissue sarcoma: preoperative MRI-based radiomics and machine learning May be accurate predictors of histopathologic grade. Am. J. Roentgenol. 215 (4), 963–969 (2020).
Foersch, S. et al. Deep learning for diagnosis and survival prediction in soft tissue sarcoma. Ann. Oncol. 32 (9), 1178–1187 (2021).
Qi, L. et al. Deciphering the role of NETosis-related signatures in the prognosis and immunotherapy of soft-tissue sarcoma using machine learning. Front. Pharmacol. ;14. (2023).
Chaber, R. et al. Predicting ewing sarcoma treatment outcome using Infrared spectroscopy and machine learning. Molecules [Internet] 24(6). (2019).
Ren, J. et al. Identification of methylation signatures and rules for sarcoma subtypes by machine learning methods. Biomed. Res. Int. 2022 (1), 5297235 (2022).
van Ijzendoorn, D. G. P. et al. Machine learning analysis of gene expression data reveals novel diagnostic and prognostic biomarkers and identifies therapeutic targets for soft tissue sarcomas. PLoS Comput. Biol. 15 (2), e1006826 (2019).
Ren, E., Deng, Y., Yuan, W., Wu, Z. & Zhang, G. Xie Q-q. An immune-related gene signature for determining Ewing sarcoma prognosis based on machine learning. J. Cancer Res. Clin. Oncol. 147 (1), 153–165 (2021).
Ho, P. T. B., Clark, I. M. & Le, L. T. T. MicroRNA-based diagnosis and therapy. Int. J. Mol. Sci. ;23(13). (2022).
Mishra, S., Yadav, T. & Rani, V. Exploring miRNA based approaches in cancer diagnostics and therapeutics. Crit. Rev. Oncol. Hematol. 98, 12–23 (2016).
Hu, Y. et al. Identification of key differentially expressed MicroRNAs in cancer patients through pan-cancer analysis. Comput. Biol. Med. 103, 183–197 (2018).
Kim, T. & Croce, C. M. MicroRNA: trends in clinical trials of cancer diagnosis and therapy strategies. Exp. Mol. Med. 55 (7), 1314–1321 (2023).
Condrat, C. E. et al. miRNAs as biomarkers in Disease: latest findings regarding their role in diagnosis and prognosis. Cells ;9(2). (2020).
Li, M., Shen, Y., Wang, Q. & Zhou, X. MiR-204-5p promotes apoptosis and inhibits migration of osteosarcoma via targeting EBF2. Biochimie 158, 224–232 (2019).
Chen, G. et al. Potential regulatory effects of Mir-182-3p in Osteosarcoma via targeting EBF2. Biomed. Res. Int. 2019, 4897905 (2019).
Ge, Q. et al. Immunological role and prognostic value of APBB1IP in Pan-cancer analysis. J. Cancer. 12 (2), 595–610 (2021).
Yang, K. et al. HOOK3 suppresses proliferation and metastasis in gastric cancer via the SP1/VEGFA axis. Cell. Death Discovery. 10 (1), 33 (2024).
Lou, L. Q., Zhou, W. Q., Song, X. & Chen, Z. Elevation of hsa-mir-7-5p level mediated by CtBP1-p300-AP1 complex targets ATXN1 to trigger NF-κB-dependent inflammation response. J. Mol. Med. (Berl). 101 (3), 223–235 (2023).
Silva, I. L. Z., Kohata, A. A. & Shigunov, P. (eds) Modulation and Function of Pumilio Proteins in cancer. Seminars in cancer Biology (Elsevier, 2022).
Wu, Y. & Zhao, H. CTBP1 strengthens the cisplatin resistance of gastric cancer cells by upregulating RAD51 expression. Oncol. Lett. 22 (5), 810 (2021).
Shi, J. et al. Synaptotagmin 1 suppresses colorectal cancer metastasis by inhibiting ERK/MAPK signaling-mediated Tumor Cell Pseudopodial formation and Migration. Cancers (Basel) ;15(21). (2023).
Gong, Y. et al. PUMILIO proteins promote colorectal cancer growth via suppressing p21. Nat. Commun. 13 (1), 1627 (2022).
Liu, Z. & Lv, C. RNA binding protein PUM2 promotes hepatocellular carcinoma proliferation and apoptosis via binding to the 3’UTR of BTG3. Oncol. Lett. 24 (4), 346 (2022).
Blevins, M. A., Huang, M. & Zhao, R. The role of CtBP1 in oncogenic processes and its potential as a therapeutic target. Mol. Cancer Ther. 16 (6), 981–990 (2017).
Wang, Z. et al. Comprehensive identification of onco-exaptation events in bladder cancer cell lines revealed L1PA2-SYT1 as a prognosis-relevant event. Iscience ;26(12). (2023).
Kumar, R. et al. Homozygous mutation of STXBP5L explains an autosomal recessive infantile-onset neurodegenerative disorder. Hum. Mol. Genet. 24 (7), 2000–2010 (2015).
Cooper, J. N. et al. Landscape of NRXN1 gene variants in phenotypic manifestations of Autism Spectrum disorder: a systematic review. J. Clin. Med. 13 (7), 2067 (2024).
Riggs, E., Shakkour, Z., Anderson, C. L. & Carney, P. R. SYT1-Associated neurodevelopmental disorder: a narrative review. Child. (Basel) ;9(10). (2022).
Melland, H. et al. Expanding the genotype and phenotype spectrum of SYT1-associated neurodevelopmental disorder. Genet. Sci. 24 (4), 880–893 (2022).
Wang, J. et al. Aberrant hypermethylation induced downregulation of antisense lncRNA STXBP5-AS1 and its sense gene STXBP5 correlate with tumorigenesis of glioma. Life Sci. 278, 119590 (2021).
Guo, C. et al. LATS2 inhibits cell proliferation and metastasis through the Hippo signaling pathway in glioma. Oncol. Rep. 41 (5), 2753–2761 (2019).
Kim, Y. et al. ELAVL2 loss promotes aggressive mesenchymal transition in glioblastoma. Npj Precision Oncol. 8 (1), 79 (2024).
Zhao, G. et al. CPEB2 inhibit cell proliferation through upregulating p21 mRNA stability in glioma. Sci. Rep. 13 (1), 23103 (2023).
Akhtar, M. S. LATS in cancer repression: extracting its role in hippo pathway and beyond. Gene Rep. :101826. (2023).
Pascual, R. et al. The RNA binding protein CPEB2 regulates hormone sensing in mammary gland development and luminal breast cancer. Sci. Adv. 6 (20), eaax3868 (2020).
Zeng, P. et al. CPEB2 enhances cell growth and angiogenesis by upregulating ARPC5 mRNA stability in multiple myeloma. J. Orthop. Surg. Res. 18 (1), 384 (2023).
Lam, C. S. et al. Identification of microRNA 885-5p as a novel regulator of tumor metastasis by targeting CPEB2 in colorectal cancer. Oncotarget 8 (16), 26858–26870 (2017).
Yotsumoto, T. et al. NRXN1 as a novel potential target of antibody-drug conjugates for small cell lung cancer. Oncotarget 11 (39), 3590–3600 (2020).
Lacore, M. G. et al. The glycoprotein M6a is associated with Invasiveness and radioresistance of glioblastoma stem cells. Cells 11 (14), 2128 (2022).
Boudhiba, N. et al. Association between the ELAVL1 gene single nucleotide polymorphisms and the genetic susceptibility to cervical cancer by high resolution melting in a Tunisian population. Mol. Biol. Rep. 50 (3), 2559–2567 (2023).
Kanzaki, H. et al. The RNA-binding protein ELAVL1 regulates Hepatitis B virus replication and growth of hepatocellular carcinoma cells. Int. J. Mol. Sci. ;23(14). (2022).
Cai, Z. et al. ELAVL1 promotes prostate cancer progression by interacting with other m6A regulators. Front. Oncol. 12, 939784 (2022).
Kim, Y. & Jho, E. H. Deubiquitinase YOD1: the potent activator of YAP in hepatomegaly and liver cancer. BMB Rep. 50 (6), 281–282 (2017).
Han, Z. et al. Deubiquitylase YOD1 regulates CDK1 stability and drives triple-negative breast cancer tumorigenesis. J. Exp. Clin. Cancer Res. 42 (1), 228 (2023).
Zhang, Z. et al. YOD1 serves as a potential prognostic biomarker for pancreatic cancer. Cancer Cell Int. 22 (1), 203 (2022).
Qiu, L. et al. circBICD2 targets miR-149-5p/IGF2BP1 axis to regulate oral squamous cell carcinoma progression. J. Oral Pathol. Med. 50 (7), 668–680 (2021).
Wei, F., Ge, Y., Li, W., Wang, X. & Chen, B. Role of endothelin receptor type B (EDNRB) in lung adenocarcinoma. Thorac. Cancer. 11 (7), 1885–1890 (2020).
Liu, S., Zhang, J., Zhu, J., Jiao, D. & Liu, Z. Prognostic values of EDNRB in triple–negative breast cancer. Oncol. Lett. 20 (5), 149 (2020).
Hu, X., Liu, H. & Li, C. MiRNA-19b-3p downregulates the endothelin B receptor in gastric cancer cells to prevent angiogenesis and proliferation. Acta Biochim. Pol. 70 (2), 363–370 (2023).
Przygodzka, P., Soboska, K., Sochacka, E., Boncela, J. & Neuromedin, U. A small peptide in the big world of cancer. Cancers (Basel) ;11(9). (2019).
Przygodzka, P. et al. Neuromedin U secreted by colorectal cancer cells promotes a tumour-supporting microenvironment. Cell. Communication Signal. 20 (1), 193 (2022).
Hodgson, K. et al. The role of GCNT1 mediated O-glycosylation in aggressive prostate cancer. Sci. Rep. 13 (1), 17031 (2023).
Barkeer, S. et al. Novel role of O-glycosyltransferases GALNT3 and B3GNT3 in the self-renewal of pancreatic cancer stem cells. BMC Cancer. 18 (1), 1157 (2018).
Luo, D. et al. The EMT-Related genes GALNT3 and OAS1 are associated with immune cell infiltration and poor prognosis in lung adenocarcinoma. Front. Biosci. (Landmark Ed). 28 (10), 271 (2023).
Lixin, S., Wei, S., Haibin, S., Qingfu, L. & Tiemin, P. Mir-885-5p inhibits proliferation and metastasis by targeting IGF2BP1 and GALNT3 in human intrahepatic cholangiocarcinoma. Mol. Carcinog. 59 (12), 1371–1381 (2020).
Chen, S. Y. et al. Spermatid perinuclear RNA-binding protein promotes UBR5-mediated proteolysis of Dicer to accelerate triple-negative breast cancer progression. Cancer Lett. 586, 216672 (2024).
Salemi, M. et al. Expression of STRBP mRNA in patients with cryptorchidism and down’s syndrome. J. Endocrinol. Invest. 35 (1), 5–7 (2012).
Ma, Q., Oksenberg, J. R. & Didonna, A. Epigenetic control of ataxin-1 in multiple sclerosis. Ann. Clin. Transl Neurol. 9 (8), 1186–1194 (2022).
Kang, A. R., An, H. T., Ko, J., Choi, E. J. & Kang, S. Ataxin-1 is involved in tumorigenesis of cervical cancer cells via the EGFR-RAS-MAPK signaling pathway. Oncotarget 8 (55), 94606–94618 (2017).
Tan, M. H. E., Li, J., Xu, H. E., Melcher, K. & Yong, E. Androgen receptor: structure, role in prostate cancer and drug discovery. Acta Pharmacol. Sin. 36 (1), 3–23 (2015).
Kimura, I. et al. Functions of MAPR (membrane-associated progesterone receptor) family members as heme/steroid-binding proteins. Curr. Protein Pept. Sci. 13 (7), 687–696 (2012).
Mohren, L. et al. Role of protein tyrosine phosphatase receptor type E (PTPRE) in Chemoresistant retinoblastoma. Int. J. Mol. Sci. 25 (8), 4572 (2024).
Liang, J., Shi, J., Wang, N., Zhao, H. & Sun, J. Tuning the protein phosphorylation by receptor type protein tyrosine phosphatase Epsilon (PTPRE) in normal and Cancer cells. J. Cancer. 10 (1), 105–111 (2019).
Chen, H. microRNA-based cancer diagnosis and therapy. Int. J. Mol. Sci. [Internet] 25(1). (2024).
Czarnecka, A., Błoński, P., Chmiel, P. & Rutkowski, P. Novel biomarkers in bone sarcomas — diagnosis, treatment selection, and clinical trials. Oncol. Clin. Pract. ;0(0). (2024).
Chakrabortty, A., Patton, D. J., Smith, B. F. & Agarwal, P. miRNAs: potential as biomarkers and therapeutic targets for cancer. Genes [Internet] ; 14(7). (2023).
Fergus, P. & Chalmers, C. Performance evaluation metrics. applied deep learning: tools,techniques, and implementationp. 115–138 (Springer, 2022).
Dinga, R., Penninx, B. W., Veltman, D. J., Schmaal, L. & Marquand, A. F. Beyond accuracy: measures for assessing machine learning models, pitfalls and guidelines. BioRxiv :743138. (2019).
Varoquaux, G. & Colliot, O. Evaluating machine learning models and their diagnostic value. Mach. Learn. Brain Disorders. 601, 30 (2023).
Szklarczyk, D. et al. STRING v11: protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 47 (D1), D607–D13 (2019).
Song, L., Langfelder, P. & Horvath, S. Random generalized linear model: a highly accurate and interpretable ensemble predictor. BMC Bioinform. 14 (1), 5 (2013).
Mamun, A. & Paul, S. Model selection in generalized Linear models. Symmetry 15 (10), 1905 (2023).
Araki, Y. et al. A validation study for the utility of serum microRNA as a diagnostic and prognostic marker in patients with osteosarcoma. Oncol. Lett. 25 (6), 1–15 (2023).
Yoshida, K. et al. Downregulation of miR–10b–5p facilitates the proliferation of uterine leiomyosarcoma cells: a microRNA sequencing–based approach. Oncol. Rep. 49 (5), 86 (2023).
García-Heredia, J. M. et al. A new treatment for sarcoma extracted from combination of miRNA deregulation and gene association rules. Signal. Transduct. Target. Therapy. 8 (1), 231 (2023).
Xu, W., Huang, Y., Lei, Z. & Zhou, J. Mir-939-3p induces sarcoma proliferation and poor prognosis via suppressing BATF2. Front. Oncol. 14, 1346531 (2024).
Lee, S. M. Role of host microRNAs in Kaposi’s sarcoma herpesvirus tumorigenesis and lytic reactivation 2023.
Pillozzi, S. et al. 110P characterization of miRNA-mRNA network in soft tissue sarcoma. ESMO Open. ;8(1). (2023).
Karras, F. S. et al. Comparative analysis of miRNA expression in dedifferentiated and well-differentiated components of dedifferentiated chondrosarcoma. Pathol. - Res. Pract. 244, 154414 (2023).
Yang, F. et al. MicroRNA PC-3p-2869 regulates antler growth and inhibits proliferation and migration of human osteosarcoma and chondrosarcoma cells by targeting CDK8, EEF1A1, and NTN1. Int. J. Mol. Sci. [Internet] ; 24(13). (2023).
Li, M. H., Wu, Z. Y., Wang, Y., Chen, F. Z. & Liu, Y. Expression of miR-29 and STAT3 in osteosarcoma and its effect on proliferation regulation of osteosarcoma cells. Eur. Rev. Med. Pharmacol. Sci. 23 (17), 7275–7282 (2019).
Acknowledgements
None.
Funding
This study was supported by Shahid Beheshti University of Medical Sciences.
Author information
Authors and Affiliations
Contributions
Study conception and design: Elham Nazari, Shadi Shafaghi, Abdolreza Mohammadnia Acquisition of data: Maryam Hajimoradi Analysis and interpretation of data: Elham Nazai Drafting of manuscript: Rahem Rahmati, Fatemeh Zarimeidani, Farnaz Ahmadi, Hannaneh Yousefi-Koma Critical revision: Elham Nazari, Shadi Shafaghi, Rahem Rahmati.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics declarations
The data used in this study were taken from the TCGA dataset (http://tcga-data.nci.nih.gov/tcga/), and all considerations related to the code of ethics and patient satisfaction have been considered and announced on this site. Therefore, considering that experimental data was not used in this study, there was no need to obtain consent and code of ethics.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rahmati, R., Zarimeidani, F., Ahmadi, F. et al. Identification of novel diagnostic and prognostic microRNAs in sarcoma on TCGA dataset: bioinformatics and machine learning approach. Sci Rep 15, 7521 (2025). https://doi.org/10.1038/s41598-025-91007-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-91007-x
