
Abstract
Cervical cancer continues to pose a significant health challenge, especially in resource-limited settings, highlighting the need for the development of novel therapeutic agents. This study investigates the potential of 2,4-diphenyl indenol [1,2-b] pyridinol derivatives as inhibitors targeting the epidermal growth factor receptor (EGFR) through computational drug discovery methods. A genetic algorithm-multiple linear regression (GA-MLR) model was created, achieving strong predictive accuracy with R² = 0.9243, Q² = 0.8957, CCC = 0.9021, and MAE = 0.034. Molecular docking studies indicated that ligand 57 displayed the highest binding affinity of -29.2313 kcal/mol, followed by ligands 111 (-29.1459 kcal/mol) and 110 (-29.9082 kcal/mol), all of which stabilize key EGFR residues. Molecular dynamics (MD) simulations confirmed the stability of ligand 111, showing an improved binding free energy of -18.2235 kcal/mol. Additionally, pharmacokinetic analysis further validated their favorable ADMET (Absorption, Distribution, Metabolism, Excretion, and Toxicity) properties, supporting their potential as drug-like candidates. These findings establish a strong foundation for the development of EGFR-targeted therapies for cervical cancer.
Similar content being viewed by others


Introduction
Cervical cancer remains a significant global health challenge, particularly in low-resource settings where access to preventive measures and treatments is limited1. The primary cause of cervical cancer is persistent infection with high-risk human papillomavirus (HPV) genotypes, which can lead to the malignant transformation of cervical tissues2. HPV, consisting of over 200 known genotypes, primarily infects the epithelial tissues of the throat, mouth, and genital regions3. Although many HPV infections resolve spontaneously, high-risk genotypes such as HPV-16 and HPV-18 are strongly associated with the progression to cervical cancer4,5. This type of cancer predominantly affects the lower uterus and often originates from the squamous cells of the exocervix or the glandular cells of the endocervix6. It is closely linked to specific HPV DNA genotypes7. In the pursuit of effective treatments with minimal side effects, therapeutic strategies have increasingly focused on targeting the molecular drivers of cervical cancer. One key oncogenic factor is the HPV E6 protein, which disrupts cellular balance by inactivating tumor suppressor proteins such as p53, thereby promoting cellular transformation and proliferation8. Compounds designed to inhibit HPV E6 activity are promising candidates for therapy. In addition to HPV-targeted strategies, researchers are exploring the targeting of aberrantly expressed cellular proteins such as the epidermal growth factor receptor (EGFR). EGFR is frequently overexpressed or mutated in cervical cancer and plays a crucial role in tumor cell proliferation, survival, and metastasis9.
This study investigates novel 2,4-diphenyl indenol [1,2-b] pyridinol derivatives as potential EGFR with PDB ID [1M17] inhibitors. These derivatives were chosen for their unique chemical structures and anticipated favorable interaction with the active site of EGFR]10 &29]. The research employs an integrated computational approach that includes quantitative structure-activity relationship (QSAR) modeling, molecular docking, pharmacokinetics, and molecular dynamics (MD) simulations. These techniques help elucidate the structural and functional determinants influencing the biological activity of the derivatives. Molecular docking predicts binding affinity and orientation, while MD simulations provide insights into the stability and dynamics of ligand-receptor interactions. Pharmacokinetics analysis is conducted to ensure the identification of compounds with favorable ADMET (absorption, distribution, metabolism, excretion, and toxicity) profiles, supporting the potential of these compounds as safe and effective therapeutics. Although this study focuses on a specific cervical cancer cell line, the findings could have broader implications, informing research across other cancers driven by EGFR. By utilizing advanced computational methodologies, this work aims to deepen our understanding of cervical cancer biology, advance EGFR-targeted drug discovery, and accelerate the translation of computational predictions into clinical solutions.
Materials and methods
Data source
The dataset for this study was sourced from the literature cited in reference10. Lead compounds were selected based on their promising bioactivity profiles, as reported in prior studies.
Drawing chemical structures
Accurate representation of molecular structures is crucial for computational studies. Properly drawn structures ensure precise descriptor calculations and reliable QSAR modeling. The chemical structures of the compounds were drawn in 2D using ChemDraw software (version 16.0) to ensure accurate molecular representations for subsequent computational analyses11.
Geometry optimization
Geometry optimization refines molecular structures to their most stable conformations, which is essential for reliable docking and descriptor calculations. Molecular structures were optimized using Spartan 14 software (version 14.1v.14). Geometry optimization was performed using density functional theory (DFT) with the B3LYP functional and a 6-31G** basis set12. This step aimed to achieve the most energetically favorable conformations, providing a robust foundation for further analyses13.
Descriptor calculation
Molecular descriptors capture physicochemical, electronic, and topological properties that influence biological activity. These features are critical for the development of QSAR models. Physicochemical and structural descriptors were systematically calculated using PaDEL software (version 2013)14. PaDEL (version 2.21) was specifically chosen for its capability to compute a wide range of molecular descriptors essential for quantitative structure-activity relationship (QSAR) modeling15.
Data pretreatment
Data pretreatment ensures model accuracy by removing redundant, noisy, or highly correlated descriptors. Standardization improves the comparability of different molecular structures. Before model development, the dataset underwent thorough preprocessing to ensure consistency and reliability. This involved removing outliers, addressing missing values, and standardizing the dataset. Data preprocessing was conducted using VS-WPS software (version 12)16.
Data division
A well-balanced training and test set is essential for robust model validation and assessment of predictive performance. The dataset was divided into training and validation subsets using the Kennard-Stone algorithm, which was implemented with VS-WPS software (version 12). This method ensured an even distribution of chemical diversity across the subsets, enhancing the robustness of model training and validation17.
Model development
Developing a predictive QSAR model helps identify key molecular features that influence activity and guides rational drug design. QSAR models were developed using the QSAR-Co package (version 1.0.0), an offline software specifically designed for constructing robust QSAR models18. This software facilitated key tasks, including the construction of regression-based QSAR models, assessment of model robustness using internal metrics (Eqs. 2, 3, and 4), evaluation using external validation metrics (Eqs. 5, 6, and 7), visualization of model equations, and verification of model reliability through statistical validation Y-randomization11. The Multiple Linear Regression (MLR) validation tool was employed to evaluate the predictive capabilities of the developed QSAR models19. This comprehensive approach aimed to address challenges in QSAR model construction and ensure the reliability of predictions based on diverse experimental responses and bioactivity measures21.
To ensure the reliability of the developed quantitative structure-activity relationship (QSAR) model, we assessed multiple performance indicators and incorporated feedback from reviewers to enhance clarity and scientific rigor. The metrics evaluated included the cross-validated coefficient of determination (Q²), root mean squared error (RMSE), concordance correlation coefficient (CCC, as shown in Eq. 7), and mean absolute error (MAE, as shown in Eq. 6)11. The MAE measures the average absolute difference between predicted and observed values, indicating the model’s accuracy by assessing the magnitude of prediction errors19. The CCC, described in Eq. 7, evaluates the degree of agreement between observed and predicted values, accounting for both accuracy and precision17. The RMSE quantifies the average magnitude of errors, emphasizing the significance of outliers in the dataset and providing a robust understanding of prediction deviations. Cross-validation results are summarized by Q², which is derived from Eq. 2. This metric demonstrates the model’s ability to predict untested data, highlighting its generalization potential and relevance for unseen datasets20. Q² serves as a critical benchmark for assessing the robustness and predictive power of the QSAR model. We meticulously analyzed these comprehensive performance metrics to address potential concerns regarding multicollinearity, overfitting, and data variability. The results validate the QSAR model’s reliability and applicability for predicting chemical activity, providing a solid foundation for subsequent analyses and interpretations21.
VIF calculation
The Variance Inflation Factor (VIF), as described by Eq. 8, is a widely used metric for assessing multicollinearity in regression models. While high VIF values generally suggest potential multicollinearity issues, there are situations where elevated VIFs may be acceptable. This can occur, for example, when control variables are included or when interaction terms (such as products or powers of variables) are added to the model28,29,30. It is important to recognize that significant VIF values do not always indicate the presence of multicollinearity, especially when working with categorical variables that have sparse or limited occurrences. Therefore, a thorough evaluation that combines VIF with other diagnostic methods is often necessary to ensure accurate model interpretation and reliability.
To validate the robustness and stability of the constructed QSAR models, Y-randomization tests were performed. In this process, the dependent variable (biological activity) was randomly shuffled while keeping the independent descriptors unchanged, as described in Eq. 9. This method ensures that the randomized models generated do not have any logical relationship between the predictors and the response variable, making it a rigorous test for overfitting and model reliability29.
Through multiple iterations, it was anticipated that the randomized QSAR models would produce significantly low R² (R-squared) and Q² (cross-validated R-squared) values. These results confirm that the original QSAR model was not derived by chance and possesses genuine predictive power. Additionally, the metric cRp² (calibrated squared correlation) was calculated to assess the extent of correlation between observed and predicted activities across the randomized models. For the model to be considered stable and reliable, cRp² must exceed the threshold of 0.530.
Docking analysis
The molecular docking analysis was performed utilizing the ICM-Pro software, adhering to a systematic and precise workflow to ensure the accuracy of the results. Initially, both ligands and receptor proteins underwent thorough preparation, which included the optimization of their structural properties and the verification of format compatibility within the ICM-Pro software19. Special emphasis was placed on delineating the docking region to concentrate the analysis on the active site or other regions of interest28.
The docking procedure utilized robust algorithms and optimized parameters to generate a broad spectrum of ligand conformations and orientations within the binding pocket16. Subsequent to the docking, the results were meticulously analyzed to identify key molecular interactions, including hydrogen bonds, hydrophobic interactions, and pi-stacking interactions, while also calculating binding affinities. The analysis was further refined through validation steps that included cross-referencing docking scores with experimental data or literature values to ensure reliability and accuracy24.
To enhance the interpretability of the results, visualization tools embedded within ICM-Pro were employed, facilitating graphical representations of protein-ligand interactions. This systematic approach establishes a reliable framework for identifying promising drug candidates and contributes significantly to the domain of drug discovery and optimization.
Pharmacokinetics studies
The pharmacokinetic properties of the selected compounds were assessed using the SwissADME web tool (http://www.swissadme.ch/) and the PKCM (Pharmacokinetics of Chemical Mixtures) tool. The chemical structures of the compounds were input in both 2D and SMILES formats to ensure compatibility with the respective tools14,17.
SwissADME was utilized to predict lipophilicity (Log P values) and water solubility by evaluating the compounds’ polar surface area (PSA). These parameters provide insights into membrane permeability and solubility, which are critical for oral bioavailability. The drug-likeness of the compounds was evaluated based on Lipinski’s Rule of Five, and the Bioavailability Radar tool was employed to estimate their oral bioavailability profiles.
The PKCM tool complemented these findings by facilitating predictions of a wider array of pharmacokinetic parameters. By uploading the chemical structures in SMILES format, the assessment addressed absorption, distribution, metabolism, and excretion (ADME) properties, in addition to potential toxicological profiles17.
Binding free energy Estimation methodology
To validate the stability and binding affinity of the optimal molecular docking results under physiologically relevant conditions, molecular dynamics (MD) simulations were performed using the GROMACS 2019 software package along with the CHARMM force field22. The CHARMM General Force Field (CGenFF) was used to generate the molecular topology file and force field parameters for the ligand, ensuring compatibility and accuracy in the modeling process. The docked complexes were solvated in a cubic box of TIP3P water molecules, maintaining a minimum distance of 10 Å between the complex and the edges of the box to avoid boundary effects25. To neutralize the system’s charge, Na⁺ counterions were added. After setting up the system, an energy minimization process consisting of 50,000 steps was performed using the steepest descent integrator. This step effectively reduced any steric clashes and internal strain within the system25,26,27. Following energy minimization, equilibration steps were carried out to prepare the system for the production MD run. The system underwent a 0.1 ns NVT (constant volume and temperature) equilibration at 300 K, followed by a 1 ns NPT (constant pressure and temperature) equilibration at 1 bar pressure. Temperature regulation was achieved using the Berendsen thermostat, while pressure control utilized the Parrinello-Rahman barostat, ensuring a stable thermodynamic environment26. The equilibrated system was then subjected to a 100 ns MD simulation under constant temperature (300 K) and pressure (1 bar) to evaluate the dynamic behavior of the protein-ligand complex. Bond lengths were constrained using the LINCS algorithm to maintain structural integrity during the simulation. The Particle Mesh Ewald (PME) method was employed for accurate calculations of long-range electrostatic interactions, ensuring precise force field calculations. Binding free energy estimation was performed using the Molecular Mechanics/Poisson-Boltzmann Surface Area (MM/PBSA) approach. This methodology involved analyzing frames extracted from both the beginning and the end of the MD simulation trajectory, providing a robust estimation of binding free energy27. Furthermore, the methodology included cross-validation of the MM/PBSA results with literature-reported values or experimental data, wherever available, to reinforce the reliability of the binding free energy estimations. This comprehensive approach ensures a detailed understanding of the binding interactions, complements the docking results, and facilitates informed decisions in drug discovery.
Results
Model validation
The QSAR analysis produced significant findings, demonstrating the robustness and predictive accuracy of the GA-MLR (Genetic Algorithm for Multiple Linear Regression) model. The developed model can be expressed with the following equation:
pI{C50} = -189.5096 (± 11.2145) + 68.7114 (± 3.2601) SpMax3_Bhv + 29.8848 (± 0.7401) SIC1 + 28.0994 (± 1.447) SpMin3_Bhi.
This model quantitatively relates molecular descriptors to biological activity (pIC50), where pIC50 represents the negative logarithm of the IC50 value, serving as a measure of compound potency. The three descriptors, SpMax3_Bhv, SIC1, and SpMin3_Bhi, each contribute distinctly to the biological activity. SpMax3_Bhv (68.7114 ± 3.2601) quantifies the contribution of the heaviest non-hydrogen atoms in a molecule, influencing its ability to interact with the biological target. SIC1 (29.8848 ± 0.7401) captures the complexity of the molecular structure, reflecting the intricacies that affect activity. SpMin3_Bhi (28.0994 ± 1.447) describes interactions involving hydrogen atoms, emphasizing the impact of minimal interactions on the molecular properties and potency.The model was constructed using a dataset of 90 compounds, with molecular descriptors selected based on their statistical significance and relevance. Graphical representations, including scatter plots and residual analysis, illustrate the model’s performance, as shown in Figs. 1 and 2, and 3.
Internal validation metrics displayed in Table 1 show case the robustness and accuracy of the model. The coefficient of determination (R²) is 0.9784, indicating an excellent correlation between observed and predicted values. The adjusted R² of 0.9777 confirms the model’s reliability after accounting for the number of predictors. Additionally, the standard error of estimation (SEE) is notably low at 0.0693, demonstrating high precision in predictions. Leave-one-out cross-validation (LOO) further validates the model, resulting in a Q² value of 0.9757 and a standard deviation of prediction (SDEP) of 0.0719. The scaled average Rm² (0.9674) and scaled delta Rm² (0.0152) provide further evidence of the model’s strong predictive performance. The mean absolute error (MAE) of 0.0527 indicates minimal deviation between predicted and observed values.
External validation, conducted using a test set of 38 compounds as shown in Table 1, further affirms the model’s reliability. The R² values—0.9604 for the full dataset and 0.9583 after excluding 5% of high-residual data—highlight the model’s ability to capture variability in the observed values. The Q²F1 and Q²F2 metrics exceed 0.95, confirming the model’s predictive capacity. The concordance correlation coefficient (CCC) is also high, with values of 0.9714 for the complete dataset and 0.9786 after data refinement, demonstrating strong agreement between observed and predicted values. Error-based metrics, such as the root mean squared error of prediction (RMSEP) and MAE, further improve upon excluding high-residual data, indicating enhanced prediction accuracy. These results underscore the robustness, reliability, and predictive performance of the GA-MLR model, making it a valuable tool for predicting anti-cervical cancer activity.

Graph of residual against descriptor.
The SIC1 residual plot in Fig. 1 indicates that the linear regression model fits the data well. The residuals exhibit random scatter around zero, suggesting that there are no systematic patterns in the model’s errors. The spread of the residuals remains consistent across predicted values, indicating that the error variance is uniform. Additionally, the residuals appear to follow a normal distribution, which further supports the model’s reliability.

Graph of residual against descriptor.
The residual plot In Fig. 2 for the SpMin3_Bhi demonstrates that the model fits the data well and meets the essential assumption of constant variance. Further analysis will improve the accuracy and robustness of the model.

Residual plot against SpMax3 descriptor.
The residual plot for the SpMax3_Bhv variable shows several positive features. The residuals are randomly scattered around the horizontal line at zero, indicating that the model’s errors are random and do not exhibit any systematic pattern.
Results of VIF analysis
The Variance Inflation Factor (VIF) analysis in Table 2 reveals varying levels of multicollinearity among the predictor variables in the regression model. One variable has a high VIF score of 5.29, indicating a strong correlation with other variables, which may compromise the model’s stability and interpretability. Another variable shows moderate multicollinearity with a VIF of 4.34, while a third variable exhibits low multicollinearity with a VIF of 1.90. These findings raise potential concerns regarding the reliability of the regression analysis, especially for the variables with higher VIF values.
Results of Y-randomization
The Y-randomization analysis, as shown in SM Table 2, confirms that the high R² and Q²-LOO values observed in the original model are not due to chance or random variation. The significantly lower R² and negative Q²-LOO values from the randomly generated models further validate the original model. These results indicate that the model’s performance is not a consequence of random data distribution but rather reflects a genuine and meaningful relationship between the predictor variables and the response variable. Therefore, we can confidently conclude that the original model is robust and valid, demonstrating strong predictive capability for the given dataset.
Docking results
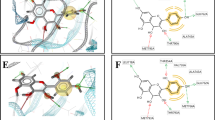
The study focused on analyzing the interactions between ligands and specific target protein residues, as outlined in Table 3. The results demonstrated strong interactions with key residues, including ARG460, PHE464, and VAL482, as shown in Fig. 4 (ID 57). Various types of bonding were observed, such as Pi-donor hydrogen bonds, conventional hydrogen bonds, and Pi-Pi T-shaped bonds. Notably, Ligand 1 exhibited an exceptionally high binding affinity for the target proteins, with a binding score of -29.2313 kcal/mol. In contrast, Fig. 5 (ID 110) revealed a more distinct interaction profile characterized by normal hydrogen bonds, Pi-Sigma interactions, and Pi-Pi stacked interactions with residues GLU183, THR179, and GLY142. Although the bond distance was 5.23 Å, the binding scores for these interactions indicated varying levels of affinity, suggesting differences in the strengths of these interactions. Additionally, Fig. 6 (ID 111) illustrated significant interactions with residues GLN520, PHE519, and PHE464. These interactions involved conventional hydrogen bonds, Pi-Pi stacked interactions, Pi-Pi T-shaped bonds, and Pi-Alkyl bonding types. Impressively, Ligand 3 demonstrated a strong binding affinity, with a binding score of -29.9082 kcal/mol, comparable to that of Ligand 1. These promising findings highlight the potential of these ligands, especially Ligand 3, as therapeutic candidates for cervical cancer treatment. The observed binding interactions and high binding scores suggest that these ligands may play a significant role in the development of novel treatment strategies for cervical cancer.

2D and 3D crystals receptor and ligand 57 interaction.

2D and 3D crystals receptor and ligand 110 interaction.

2D and 3D crystal receptor and 111 ligand interaction.
Dynamics simulation results analysis
The dynamic behavior of the protein-ligand complex and gain insights into the binding mechanism, we conducted 100 ns molecular dynamics simulations. The stability of the protein-ligand complex was assessed by monitoring the Root Mean Square Deviation (RMSD) of the protein backbone atoms from the initial structure. The RMSD plot (Fig. 7a) showed a stable trajectory with minimal fluctuations, indicating that the protein-ligand complex remained stable throughout the simulation. The Root Mean Square Fluctuation (RMSF) analysis (Fig. 7d) revealed regions of high flexibility within the protein structure, suggesting potential conformational changes. These regions were further investigated to identify their potential role in ligand binding and protein function. The Radius of Gyration (Rg), which reflects the compactness of the protein structure, showed a slight decrease throughout the simulation (Figure e), indicating a subtle compaction of the protein. This observation is consistent with the decrease in Solvent Accessible Surface Area (SASA) (Fig. 7b), suggesting a reduction in the protein’s exposure to the solvent. The number of hydrogen bonds formed between the ligand and the protein was monitored throughout the simulation. The analysis revealed that an average of [-1.24255] hydrogen bonds were formed, with key interactions observed between [ASN and Ligand-OH]. The dynamics of hydrogen bond formation and breakage were further explored to identify crucial residues involved in ligand binding and stability. The PCA was performed to identify the major collective motions of the protein-ligand complex. The first two principal components accounted for [70%] of the total variance, indicating that the protein-ligand complex undergoes significant conformational changes along these directions. Analysis of the principal component eigenvectors revealed that the major motions involved.
The binding affinity of the ligand, Molecular Mechanics/Generalized Born Surface Area (MM/GBSA) calculations were performed at both the initial (0 ns) and final (100 ns) time points of the simulation. The results showed a decrease in the binding free energy (ΔG_bind) from − 16.5939 kcal/mol at 0 ns to -18.2235 kcal/mol at 100 ns (Table 1). This decrease indicates an increase in binding affinity over the course of the simulation, suggesting a stable and favorable interaction between the ligand and the protein. The major contributors to the binding free energy were found to be van der Waals interactions (ΔG.vdW.) and Coulombic interactions (ΔGCoulomb). The positive values for polar solvation energy (ΔG_SolvGB) and covalent energy (ΔG_Covalent) suggest that these interactions may not be favorable for binding. Density Functional Theory (DFT) calculations were performed on the ligand to investigate its electronic properties and potential for further optimization. These findings provide valuable insights into the ligand’s electronic structure and its potential for interaction with the protein.

Dynamic simulation of protein ligand complex.
The plots presented in Fig. 7 from the molecular dynamics (MD) simulation provide essential insights into the structural and dynamic properties of the protein. The Root Mean Square Deviation (RMSD) plot indicates the protein’s stability over time, as evidenced by its plateau, suggesting that the protein structure remains stable throughout the simulation. The Root Mean Square Fluctuation (RMSF) plot highlights the flexible regions within the protein, which are likely important for its dynamics and interactions with ligands or other molecules.
Pharmacokinetics studies results
Molecules 1 and 2 share similarities in structural weight and characteristics, both predicted to have poor solubility, as indicated by the solubility prediction models presented in Table 4. In contrast, Molecule 3 demonstrates higher solubility, making it more favorable in terms of solubility compared to the weakly soluble Molecules 1 and 2. The pharmacokinetic and physicochemical characteristics of these molecules—including lipophilicity, blood-brain barrier (BBB) penetration potential, and interactions with various cytochrome enzymes—are evaluated. These characteristics are essential for determining the compounds’ overall drug-likeness, potential adverse effects, and bioavailability, all of which are critical factors in drug development and discovery. Although the bioavailability scores (0.55) of all three compounds are identical, Molecule 3 stands out for having fewer violations of the Lipinski, Ghose, Veber, Egan, and Muegge rule sets. This suggests that Molecule 3 is more compliant with established drug-likeness criteria. Collectively, these findings provide valuable insights into the solubility, absorption, and drug-likeness of the compounds, which are crucial factors to consider when evaluating their potential as pharmaceutical candidates (Table 5).
Conclusion
This study provides a thorough evaluation of 2,4-diphenyl indeno[1,2-b]pyridinol derivatives as potential therapeutic agents targeting the epidermal growth factor receptor (EGFR) to combat cervical cancer. The robust GA-MLR model developed in this research demonstrates a strong predictive power for pIC50 values, highlighting its utility in identifying promising candidates with high therapeutic potential. Docking studies indicate stable and high-affinity interactions between the ligands and the EGFR active site, with hydrophobic forces and hydrogen bonding playing crucial roles in stabilizing the ligand-receptor complexes. These findings emphasize the molecular basis of binding specificity and affinity, offering important insights into the design of effective inhibitors. Pharmacokinetic analyses provide valuable information regarding the drug-likeness, solubility, and bioavailability of the studied compounds, suggesting their potential suitability for pharmaceutical development. Additionally, molecular dynamics simulations further validate the stability of ligand-receptor interactions under dynamic conditions, confirming the reliability of the computational predictions. Overall, this study connects experimental findings with advanced computational techniques, significantly enhancing our understanding of cervical cancer biology and its therapeutic targeting. The findings pave the way for the development of novel, targeted treatments, representing a meaningful advancement in the global effort to combat cervical cancer. This work underscores the integration of in silico and in vitro approaches as a powerful strategy in drug discovery and development.
Data availability
Data Availability StatementThe datasets generated during and/or analyses during the current study are available from the corresponding author on reasonable request. And some are in the supplementary material.
References
Dong, J. X. et al. Long non-coding RNAs on the stage of cervical cancer (review). Oncol. Rep. 38 (4). https://doi.org/10.3892/or.2017.5905 (2017).
Choi, S., Ismail, A., Pappas-Gogos, G. & Boussios, S. HPV and cervical cancer: A review of epidemiology and screening uptake in the UK. Pathogens 12 (2). https://doi.org/10.3390/pathogens12020298 (2023).
Hull, R. et al. Cervical cancer in low and middle.income countries (review). Oncol. Lett. 20 (3). https://doi.org/10.3892/ol.2020.11754 (2020).
Kusakabe, M., Taguchi, A., Sone, K., Mori, M. & Osuga, Y. Carcinogenesis and management of human papillomavirus-associated cervical cancer. Int. J. Clin. Oncol. 28 https://doi.org/10.1007/s10147-023-02337-7 (2023). no. 8.
Okunade, K. S. Human papillomavirus and cervical cancer. J. Obstet. Gynaecol. 40 https://doi.org/10.1080/01443615.2019.1634030 (2020). no. 5.
Lee, J. E., Chung, Y., Rhee, S. & Kim, T. H. Untold story of human cervical cancers: HPV-negative cervical cancer. BMB Rep. 55 (9). https://doi.org/10.5483/BMBRep.2022.55.9.042 (2022).
Eun, T. J. & Perkins, R. B. Screening for cervical cancer. Med. Clin. N. Am. 104 https://doi.org/10.1016/j.mcna.2020.08.006 (2020). no. 6.
Mulato-Briones, B. et al. Cultivable microbiome approach applied to cervical cancer exploration. Cancers 16 (2). https://doi.org/10.3390/cancers16020314 (2024).
Kadayat, T. M. et al. Discovery and biological evaluations of halogenated 2,4-diphenyl indeno[1,2- B]pyridinol derivatives as potent topoisomerase IIα-targeted chemotherapeutic agents for breast cancer. J. Med. Chem. 62 (17). https://doi.org/10.1021/acs.jmedchem.9b00970 (2019).
Ibrahim, M. T. & Uzairu, A. Theoretical validation of some third-generation epidermal growth factor receptor (EGFR) inhibitors as non-small cell lung cancer (NSCLC) drugs. Egypt. J. Basic. Appl. Sci. 10 (1). https://doi.org/10.1080/2314808X.2023.2187516 (2023).
Isa, A. S. et al. Halogenated 2, 4-diphenyl Indeno [1, 2-B] pyridinol derivatives as potential inhibitors of the androgen receptor (PDB ID: 58TE): A study of QSAR modeling, molecular docking, and pharmacokinetics for prostate cancer treatment. Biomedical Anal. 1 (3), 240–269 (2024).
Ejeh, S., Uzairu, A., Shallangwa, G. A., Abechi, S. E. & Ibrahim, M. T. In silico identification of some novel ketoamides as potential pan-genotypic HCV NS3/4A protease inhibitors with drug-likeness, pharmacokinetic ADME profiles and synthetic accessibility predictions. Adv. J. Chem. Sect. A. 5 (3). https://doi.org/10.22034/ajca.2022.329332.1302 (2022).
Abdullahi, M., Uzairu, A., Shallangwa, G. A., Mamza, P. A. & Ibrahim, M. T. Computational modelling of some phenolic diterpenoid compounds as anti-influenza A virus agents. Sci. Afr. 19 https://doi.org/10.1016/j.sciaf.2022.e01462 (2023).
Isa, S., Uzairu, A., Umar, U. M., Ibrahim, M. T. & Umar, A. B. QSAR, docking and pharmacokinetic studies of 2,4-diphenyl indenol [1,2-B] pyridinol derivatives targeting breast cancer receptors, J. Chem. Lett. (2024). https://doi.org/10.22034/JCHEMLETT.2024.424800.1146
Abdulfatai, U., Uba, S., Umar, B. A. & Ibrahim, M. T. Molecular design and docking analysis of the inhibitory activities of some Α_substituted acetamido-N-benzylacetamide as anticonvulsant agents. SN Appl. Sci. 1 (5). https://doi.org/10.1007/s42452-019-0512-6 (2019).
Umar, B. et al. Investigation of novel imidazole analogues with terminal sulphonamides as potential V600E-BRAF inhibitors through computational approaches. Chem. Afr. 6 (6). https://doi.org/10.1007/s42250-023-00687-3 (2023).
Studies, P. Potential anti-colon cancer agents: Molecular modelling, docking, pharmacokinetics studies and molecular dynamic simulations. J. Taibah Univ. Med. Sci.
Ejeh, S. et al. Chemical bioinformatics study of Nonadec-7-ene-4-carboxylic acid derivatives via molecular docking, and molecular dynamic simulations to identify novel lead inhibitors of hepatitis C virus NS3/4a protease. Sci. Afr. 20 https://doi.org/10.1016/j.sciaf.2023.e01591 (2023).
Abdullahi, S. H., Uzairu, A., Ibrahim, M. T. & Umar, A. B. Chemo-informatics activity prediction, ligand based drug design, molecular docking and pharmacokinetics studies of some series of 4, 6-diaryl-2-pyrimidinamine derivatives as anti-cancer agents. Bull. Natl. Res. Centre. 45 (1). https://doi.org/10.1186/s42269-021-00631-w (2021).
Tabti, K. et al. In silico design of novel PIN1 inhibitors by combined of 3D-QSAR, molecular docking, molecular dynamic simulation and ADMET studies. J. Mol. Struct. 1253 https://doi.org/10.1016/j.molstruc.2021.132291 (2022).
Ouabane, M. et al. Structure-odor relationship in pyrazines and derivatives: A physicochemical study using 3D-QSPR, HQSPR, Monte Carlo, molecular docking, ADME-Tox and molecular dynamics. Arab. J. Chem. 16, 11 (2023). https://doi.org/10.1016/j.arabjc.2023.105207
Tabti, K., Sbai, A., Maghat, H., Lakhlifi, T. & Bouachrine, M. Computational exploration of the structural requirements of Triazole derivatives as Colchicine binding site inhibitors. ChemistrySelect 8 (26). https://doi.org/10.1002/slct.202301707 (2023).
El Masaoudy, Y. et al. In silico design of new pyrimidine-2,4-dione derivatives as promising inhibitors for HIV reverse transcriptase-associated RNase H using 2D-QSAR modeling and (ADME/Tox) properties. Moroccan J. Chem. 11 (2). https://doi.org/10.48317/IMIST.PRSM/morjchem-v11i2.35455 (2023).
Tabti, K., Sbai, A., Maghat, H., Lakhlifi, T. & Bouachrine, M. Computational assessment of the reactivity and pharmaceutical potential of novel Triazole derivatives: an approach combining DFT calculations, molecular dynamics simulations, and molecular Docking. Arab. J. Chem. 17 (1), 105376. https://doi.org/10.1016/j.arabjc.2023.105376 (2024).
Tabti, K., Sbai, A., Maghat, H., Lakhlifi, T. & Bouachrine, M. Computational assessment of the reactivity and pharmaceutical potential of novel Triazole derivatives: an approach combining DFT calculations, molecular dynamics simulations, and molecular docking. Arab. J. Chem. 17 (1). https://doi.org/10.1016/j.arabjc.2023.105376 (2024).
Faris, A. et al. In silico computational drug discovery: A Monte Carlo approach for developing a novel JAK3 inhibitors. J. Biomol. Struct. Dyn.. 1–23. https://doi.org/10.1080/07391102.2023.2270709
Saravanan, V., Chagaleti, B. K., Packiapalavesam, S. D. & Kathiravan, M. Ligand based pharmacophore modelling and integrated computational approaches in the quest for small molecule inhibitors against hCA IX. RSC Adv. 14 (5), 3346–3358 (2024).
Sigismund, S., Avanzato, D. & Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 12 (1), 3–20 (2018).
Sony, A. S. & Suresh, X. Molecular Modeling of Some Benzodiazole Derivatives with EGFR Protein 1M17.
Isa, A. S., Ibrahim, A. K. & Mukhtar, A. M. In silco analysis, anti-proliferative activity modeling, molecular docking and pharmacokinetic properties prediction of some tetrahydropyrazole-quinazoline derivatives as cancer therapeutic agents. Med. Med. Chem. 3. (2025).
Acknowledgements
The authors gratefully acknowledged the technical effort of Prof. Adamu Uzairu of the Department of Chemistry, Ahamdu Bello University Zaria.
Author information
Authors and Affiliations
Contributions
Authors’ contributionsASI designed and wrote the manuscript, AU, UMU, MTI ABU supervised and carried out the statistical analysis and KT perform Simulation dynamics AMM review the manuscript. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Isa, A.S., Uzairu, A., Umar, U.M. et al. In silico exploration of novel EGFR-targeting compounds: integrative molecular modeling, docking, pharmacokinetics, and MD simulations for advancing anti-cervical cancer therapeutics. Sci Rep 15, 7334 (2025). https://doi.org/10.1038/s41598-025-91135-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-91135-4
Keywords
This article is cited by
-
Unraveling the molecular mechanisms of paclitaxel in high-grade serous ovarian cancer through network pharmacology
Scientific Reports (2025)
-
Integrated QSAR, docking, pharmacokinetics, and molecular dynamics approaches for designing IRAK4 inhibitors against MYD88^L265P-driven diffuse large B-cell lymphoma
Discover Chemistry (2025)
