
Abstract
Fucosyltransferase 2 (FUT2) gene has been extensively reported to play its role in potential gut microbiota changes and norovirus susceptibility. The normal activity of FUT2 has been found to be disrupted by non-synonymous single nucleotide polymorphisms (nsSNPs) in its gene. To explore the possible mutational changes and their deleterious effects, we employed state-of-the-art computational strategies. Firstly, nine widely-used bioinformatics tools were utilized for initial screening of possibly deleterious nsSNPs. Subsequently, the structural and functional effects of screened nsSNPs on FUT2 were evaluated by utilizing relevant computational tools. Following this, the two shortlisted nsSNPs, including G149S (rs200543547) and V196G (rs367923363), were further validated by their molecular docking with norovirus capsid protein, VP1. As compared to wild-type, the higher stability and lower binding energy scores of the both the mutants indicated their stable binding with VP1, which ultimately leads to norovirus implications. These docking results were further verified by a comprehensive computational approach, molecular dynamic simulation, which gave results in the form of lower RMSD, RMSF, RoG, and hydrogen bond values of both the mutants, depicted in relevant graphs. Overall, this research explores and validated the two FUT2 nsSNPs (G146S and V196G), which may possibly linked with the norovirus susceptibility and gut microbiota changes. Moreover, our findings highlights the value of computational strategies in mutational analysis and welcomes any further experimental validation.
Similar content being viewed by others

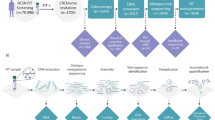

Introduction
A family of viruses known as noroviruses, which affect people of all ages, are the primary cause of sudden-onset inflammation of the stomach and intestines worldwide1. Communities, cruise ships, and healthcare institutions are just a few of the places where these extremely contagious diseases cause outbreaks. Some people have more severe symptoms and a longer sickness, even though the majority of people recover from norovirus infections without any problems. Most recently, scientists have focused on hereditary variables that influence the severity of the illness and norovirus infection. Located on chromosome 19q13.3, nsSNPs in Fucosyltransferase 2 (FUT2) gene is being considered to be associated with Norovirus infection. The roles of these nsSNPs (non-synonymous SNPs) in amino acid substitution, which can affect protein structure or function neutrally or negatively, are especially noteworthy2. The enzyme α(1,2) fucosyltransferase, which adds fucose to glycoproteins and glycolipids on the surfaces of epithelial cells, is encoded by the gene FUT23. The H antigen is produced as a result of this glycosylation process and functions as a precursor to the ABO blood group antigens. But the significance of FUT2 goes beyond blood type determination; it has been connected to a range of biological processes, such as interactions with the gut microbiota and viral infections. Individuals who have non-functional FUT2 alleles, also called “non-secretors”, are unable to secrete ABH antigens in body fluids such as tears, saliva, and mucosal surfaces, nor can they express the H antigen4. Interestingly, it has been shown that non-secretors are more vulnerable to norovirus infections5. There is a belief that the lack of H antigens on non-secretor mucosal surfaces influences the attachment of viruses and their subsequent entry into host cells6. Because FUT2 gene variants alter how viral surface proteins interact with host cell receptors, they may make a person more susceptible to contracting a norovirus7.
Further evidence indicates that the composition and diversity of the gut microbiota are altered by mutations in the FUT2 gene8. Because it controls a number of physiological processes such nutrition metabolism, immune system modulation, and pathogen defense, the gut microbiota is essential for human health9. Studies have shown that non-secretors with FUT2 gene mutations have different gut microbiota patterns than secretors10. An increased risk of several diseases, such as infectious diseases, metabolic disorders, and inflammatory bowel disease, has been linked to these changes in the gut microbial ecology11. Preventing norovirus infections benefits public health in several aspects. Reductions in the number of people with painful gastrointestinal symptoms, less demand for healthcare resources, more economic productivity, and improved protection for susceptible groups are all brought about by decreased viral dissemination12. One very effective way to look at genetic changes and how they can affect a person’s susceptibility to norovirus infection is to use FUT2 SNP analysis. By identifying certain harmful single nucleotide polymorphisms (SNPs), we can significantly reduce the risk of catching a norovirus infection13.
As different studies have discussed about the role of FUT2 alleles (non-secretors) in changed susceptibility to norovirus infection and gut microbiota profiles, the specific mechanism through which FUT2 gene mutations impact microbial community composition and how these changes contribute to norovirus susceptibility remain unclear14. Moreover, the association between norovirus susceptibility and FUT2 polymorphism have been studied extensively but there is still lake of comprehensive insights into FUT2 genetic mutations, which can provide clearer image of the mechanism. By considering these limitations, we aimed to explore and investigate the effects of FUT2 gene mutations on the composition of the gut microbiota and norovirus susceptibility. For this purpose, a pipeline of widely-used in-silico strategies including SIFT, Polyphene-2, MutPred, I-Mutant, DeepREx-WS, Molecular Operating Environment (MOE) and Molecular dynamic simulation, were employed15. By specifically concentrating on the deleterious nsSNPs, we delved deeper into their diverse structural and functional effect on FUT2. The ultimate purpose of our study is to explore potentially damaging nsSNPs in FUT2 gene, linked with various diseases. As the resulted nsSNPs are novel and have not been researched in any of the previous studies, this research also welcomes further experimental and clinical trials.
Methodology
This in-silico mutational investigation was conducted using a pipeline of different bioinformatics tools and strategies, which are depicted in Fig. 1. Every tool and web server, that was deployed, used GRCh38 as the reference human genome during every step16.

A framework illustrating the process for identifying the possibly damaging nsSNPs.
Acquiring genetic mutations
Complete details for all FUT2 SNPs, including location, global minor allele frequencies (MAFs), and residual changes, were accessed through the NCBI’s dbSNP (https://www.ncbi.nlm.nih.gov/snp/)17. A total of 5306 SNPs of FUT2 gene were retrieved. From overall acquired nsSNPs, only 372 nonsynonymous single nucleotide polymorphisms (nsSNPs), which are the most important in disease-causing, were found. Subsequently, we also employed the Ensembl database (https://www.ensembl.org/)18 to retrieve and compare nsSNPs, which determined 423 SNPs as missense. Extensive filtration was performed to eliminate duplicates and improve the overall list of nsSNPs in order to guarantee accuracy.
Selection of deleterious nsSNPs
To determine the possible consequences of single-nucleotide polymorphisms (nsSNPs) obtained from the dbSNP database, we utilized six bioinformatics tools including SIFT (Sorting Intolerant from Tolerant) (https://sift.bii.a-star.edu.sg/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), CADD (Combined Annotation Dependent Depletion) (https://cadd.gs.washington.edu/), Revel (Rare Exome Variant Ensemble Learner) (https://sites.google.com/site/revelgenomics/), MutationAssessor (http://mutationassessor.org/r3/), and MetaLR (http://sites.google.com/site/jpopgen/dbNSFP). SIFT19 and Polyphene-220 are widely utilized to assess the effect of amino acid change on the respective protein structure. The overall accuracy of SIFT and Polyophene-2 has been reported as 84%21 and 67%22, respectively. Likewise, CADD integrates different annotations to score variants on the basis of their deleteriousness. Different studies have reported CADD as 85% accurate23. Moreover, Revel is a tool used to predict the pathogenicity of nsSNPs, which has reported to show high accuracy in the form of AUC (an operating characteristic curve)24. Furthermore, MutationAssessor categorizes SNPs based on their structural and conservational impacts, and it has found to be 79% accurate in different studies25. Finally, MetaLR is a widely-employed tool to sort nsSNPs into benign and deleterious in the form of different scores, where scores of 0.5 or less considered as benign and vice versa. A study reported that MetaLR predicts lower but more accurate number of nsSNPs (as compared to other tools like DANN and FATHMM)26. We utilized all the tools simultaneously to predict the FUT2 nsSNPs and chose only those nsSNPs which were predicted deleteriously consistently in all of the mentioned tools. This robust method helped us to rely on its increased efficiency. To further validate the deleterious effect of the shortlisted mutations, SNP&GO (https://snps-and-go.biocomp.unibo.it/snps-and-go/)27, PANTHER (https://www.pantherdb.org/)28, and PredictSNP (https://loschmidt.chemi.muni.cz/predictsnp/)29 were employed, each providing complementary insights into the functional impact of amino acid substitutions. Using sequence-based characteristics and Gene Ontology (GO) annotations, SNP&GO predicts if a mutation is associated with a disease27, whereas PANTHER (Protein ANalysis THrough Evolutionary Relationships) classifies mutations based on evolutionary conservation and functional annotation, identifying whether a given substitution is likely to disrupt protein function28. On the other hand, the consensus-based tool, PredictSNP, combines predictions from several well-known techniques, such as MAPP, PhD-SNP, PolyPhen-1, PolyPhen-2, SIFT, and SNAP, to provide a very accurate classification of mutations as either neutral or harmful29.
Determining structural and functional impact
We used a web-based program, named MutPred 1.2 (http://mutpred.mutdb.org/)30, to look into how the amino acid changes (nsSNPs) will impact protein structure and function. This server predicts multiple structural and functional impacts including the alterations to the transmembrane protein, ordered interface, catalytic site, relative solvent accessibility, allosteric site, GPI-anchor amidation, N-linked glycosylation, metal binding, and strand. P values less than 0.05 were used to categorize mutations as having normal confidence, and p values less than 0.01 as having high confidence.
Protein’s stability evaluation
We utilized a web-based tool, I-Mutant 2.0 (https://folding.biofold.org/i-mutant/i-mutant2.0.html), to investigate the potential effects of the shortlisted damaging nsSNPs on the stability of the FUT2 protein31. This tool predicts changes in protein stability implicated by mutations using in-built machine learning based algorithm. After simulating the protein at the physiological condition of pH 7.0 and 25 °C, we used I-Mutant 2.0 to evaluate the nsSNPs. A “reliability index” (RI) between 0 and 10 was given by the program, where higher values denote greater stability. The aim of this RI index is basically the identification of deleterious nsSNPs. To further support the I-Mutant 2.0 results, additional computational tools including MUpro (https://mupro.proteomics.ics.uci.edu/)32, mCSM (https://biosig.lab.uq.edu.au/mcsm/)33 and DDMut (https://biosig.lab.uq.edu.au/ddmut/)34, were employed. MUpro predicts the impact of mutations on protein stability using support vector machines and neural networks32, while mCSM is a graph-based tool analyzing interatomic interactions to assess mutation-induced stability changes and functional effects33. Similarly, DDMut assesses a protein’s structural and sequence-based characteristics to predict how a mutation will destabilize it34.
Conservation analysis of nsSNPs
Understanding evolution is important to check whether mutations can cause health issues in humans or not35. Using the DeepREx-WS (https://deeprex.biocomp.unibo.it/), each amino acid in FUT2 protein sequence was checked for evolutionary conservation36. This web-based program scans protein sequences and predicts several properties, including conservation, using deep learning. Furthermore, it uses a deep learning based methodology, involving deep neural networks, to analyze protein sequences.
Structural modelling of FUT2 and mutants
Robetta Modelling server (https://robetta.bakerlab.org/)37 was employed to investigate further the potential effects of the most important mutations (nsSNPs) on the three-dimensional structure of the FUT2 protein. By utilizing the Rosetta package of this tool, the three-dimensional structures of all the shortlisted twelve mutants were obtained. For comparison, a 3D model of the wild-type FUT2 protein was also obtained. Subsequently, we used TM-align (https://zhanggroup.org/TM-align/) to compare each mutant’s structure with that of the wild-type protein. Information on structural superposition, TM-score, and root mean square deviation (RMSD) were acquired by this analysis. The average difference between the positions of corresponding atoms in two subsequent structures is measured by RMSD. It also indicates the higher structural divergence of given mutants, as compared to wild type38. Conversely, the TM-score is a numerical value ranging from 0 to 1, where 1 denotes the highest level of structural similarity. From these analyses, two mutants with, the largest structural deviations (higher RMSD) from the wild-type, were selected based on the preliminary analysis. The proposed mutants were, then, remodeled using highly accurate AlphaFold 2 (https://alphafold.ebi.ac.uk/)39 structure prediction tool. Pymol software (https://www.pymol.org/) was utilized to analyze the modelled protein structures in an interactive manner, facilitating an in-depth analysis of their structural characteristics and possible functional uses40.
Docking analysis
Following the proposal of two possibly deleterious mutants, we performed molecular docking to gain a better understanding of the possible interactions of these mutants (along with the wild type) with the Norovirus capsid protein. The capsid protein is the virus’s outer shell, and it contains areas that bind to host cell receptors41. For this aim, we firstly obtained the 3D structure of the norovirus GII.4 strain capsid protein from Protein Data Bank (https://www.rcsb.org/) (PDB ID: 6OUU)42. GII.4 is the most common genotype of human noroviruses, accounting for the bulk of norovirus outbreaks and illnesses globally. As a result of its predominance, the GII.4 strain is being explored for the development of possible therapeutic interventions43. As a pre-docking step, polar hydrogens were introduced into the structures, and their energies were minimized using the molecular operation system (MOE) visualization tool44. Subsequently, we used the state-of-the-art online protein–protein docking server, ClusPro v2.0 (https://cluspro.bu.edu/), which predicts protein interactions using energy calculations45. By utilizing this server, we accurately docked proposed FUT2 nsSNPs and norovirus capsid proteins by modeling possible interactions. Finally, we utilized the PDBsum server (https://www.ebi.ac.uk/thornton-srv/databases/pdbsum/) to delve deeper into these interactions46. The exact amino acid residues, linkages, and forces revealed by this server allowed us to better understand the various communication channels between FUT2 and norovirus.
Molecular dynamic simulation analysis
Molecular dynamic simulations are generally used to estimate the stability of a protein–protein complex and the mobility of individual atoms47. In our research, MD simulation was performed collectively on both the FUT2 nsSNPs (G149S, V196G) and norovirus capsid protein (VP1). To further compare the results, wild type FUT2 was also simulated with VP1. Initially, the system was prepared with the ff19SB force field and the Amber22 package48. The built-in program, tleap, was used to create and manage the complex problems in each of the systems. Each system was neutralized with Na+ or Cl− counter ions. Coordinate files and topology were employed to minimize the complexity of each neutralized system and cut its energy49. During the pre-processing stage, we refined collisions and conflicts within the protein structure using conjugate gradient and the steepest descent methods as a two-section energy minimization procedure50. In the first half, we applied protein restraints to lower the energy of water molecules over 2500 times. This comprised of 1000 steps for the steepest descent and 1500 steps for the conjugate gradient. During the second stage, we eliminated all restraints and lowered the overall energy of the complex to 2500 steps. This included the first 1000 steps of the steepest descent and the next 1500 steps of the conjugate gradient. The reduced complexes were, then, heated for 50 ps at 300 K. The system pressure was monitored using a Berendsen barostat51, while the temperature was controlled using a Langevin thermostat52. The AMBER22 SHAKE algorithm was used to enhance covalent bonding profile53. After 1000 ps of equilibration, the complex system was compressed using an NPT ensemble54. AMBER22’s GPU version (PMEMD.cuda) was used to run MD simulations on three complexes55. Each of the complexes was simulated for 300 ns, and PTRAJ and CPPTRAJ were utilized to evaluate the resulting trajectory56. The degree of degenerative alterations in protein–protein complex and dynamic behavior was measured using specific metrics such as root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (RoG), and hydrogen bond analysis. To explore proteins’ therapeutic potential, binding stability must be assessed, which influences the extent to which proteins interact with one another. Furthermore, binding stability is required for molecular optimization of novel proteins so that potential targets may be accurately evaluated. The binding stability was examined by the simulating trajectories and computing the RMSD as a function of time57. Following this, RMSF analysis was also done on individual amino acids to provide a better understanding of the stability of VP1 active site residues during protein–protein interactions58. To gain better understanding of the dynamics and binding and unbinding processes that took place during the simulation, we have assessed the Structural Compactness of each complex in an equilibrium scenario. To do this, we estimated the radius of gyration (Rg) as a function of time59. Macromolecular interactions, i.e. the bond between two or more protein molecules, are characterized by a number of properties, the most important of which is hydrogen-hydrophobic interaction at the interface60. Determining the total number of hydrogen bonds formed in each system was done using specific criteria in order to assess the systems at the atomic level. The requirements specified a distance of 0.35 nm between the donor and acceptor and an angle of 30° between the hydrogen donor and acceptor. Once both conditions were met, a hydrogen bond was considered to be formed61.
Estimation of post-translational modification (PTM) sites
In order to gain understanding of the possible functional implications of the proposed nsSNPs, we investigated a variety of post-translational modifications (PTMs) that can potentially affect the normal functioning of FUT2 function. Utilizing GPS-MSP (https://msp.biocuckoo.org/), possible methylation sites on the FUT2 protein were predicted. Likewise, the phosphorylation sites for serine, tyrosine, and threonine residues were found using GPS 6.0 (http://gps.biocuckoo.org/online.php) and NetPhos 3.1 (https://services.healthtech.dtu.dk/services/NetPhos-3.1/). Although neural network, ensembles with a threshold of 0.5, were used by NetPhos 3.162, GPS 6.0 has been reported to be more accurate and trustworthy in various studies63. Using RUBI (http://old.protein.bio.unipd.it/rubi/) and GPS-Uber (http://gpsuber.biocuckoo.cn/) tools, possible ubiquitination sites were found, with specific lysine residues. In order to predict ubiquitination for lysine residues, RUBI used a balanced threshold64. Similarly, the FUT2 protein’s glycosylation sites were found using NetOglyc4.0 (https://services.healthtech.dtu.dk/services/NetOGlyc-4.0)65, which identifies the possible functional differences resulting from nsSNPs by comparing their glycosylation patterns.
Building the phylogenetic relationship for FUT2 protein
To gain further insights into the phylogenetic relationship of human FUT2 protein (NCBI Accession: NP_000502.4) with the homologous proteins from the other relevant species, we retrieved and compared sequences from eight different species including Gorilla gorilla (XP_055226057.1), Pan paniscus (XP_008964946.1), Hylobates moloch (XP_058281478.1), Pan troglodytes (NP_001009120.1), Symphalangus syndactylus (XP_055091865.1), Nomascus leucogenys (XP_012365269.2), Pongo pygmaeus (XP_054319062.1), and Pongo abelii (XP_054396844.2)66. A web-based tool, ClustalW, was employed to align all of the sequences67. Finally, a phylogenetic tree was created using Neighbor-Joining (NJ) method, in MEGA software68. Additionally, iTOL v6 online program was utilized to better represent the resulted tree69.
FUT2 gene–gene interactions
Using GeneMANIA (https://genemania.org/)70 and STRING (https://string-db.org/cgi)71, we looked into how the identified nsSNPs can affect the FUT2 protein and how it can be interacted with other genes. To predict gene–gene correlations, GeneMANIA integrates information from multiple sources, such as co-expression, common pathways, and physical interactions. It offers a network map that highlights potential connections between FUT2 and relevant genes. In contrast, STRING focuses on protein–protein interactions and uses several data sources, including co-occurrence, co-expression, and experimental evidence, to identify the most important genes that interact with FUT2. The interactions are usually measured on a range of 0–1, where higher scores indicate stronger interactions.
Results
Acquired nsSNPs
A total of 5306 FUT2 SNPs including 372 nonsynonymous SNPs, 48 in the 5′UTR, 1081 in the 3′UTR, 162 coding synonymous, 2753 in the intron region, and the other SNPs (splice sites = 3, frameshift = 41, nonsense = 37) were retrieved from the dbSNP. From all of the retrieved SNPs, only nsSNPs were chosen, as they are more likely to be deleterious. Subsequently, 423 nsSNPs were acquired from Ensembl. Upon eliminating duplicates, these nsSNPs were refined to 362. An illustration of the retrieved SNPs is depicted in Fig. 2.

A bar graph representing the percentage of all the acquired FUT2 SNPs, where sky-blue color represents SNPs at 3′ UTR, orange color represents SNPs at 5′ UTR, yellow color represents nsSNPs, green color represents coding synonymous SNPs, blue color represents SNPs in intron regions and purple color represents other kind of SNPs (i.e. non-sense SNPs, and frameshift SNPs).
Screening of deleterious nsSNPs
Different in-silico tools including SIFT, Polyphene-2, CADD, Revel, MetaLR, and Mutation Assessor were employed to screen the deleterious mutations. Among these tools, SIFT categorizes replacements as “tolerated” if the score (TI = Total Index) is greater than 0.05 or as “deleterious” if it is less than 0.0519,72. SIFT results indicated that 193 nsSNPs were associated with deleterious impacts. The probability of damage from a replacement is indicated by the PolyPhen-2 score; values close to one suggest a higher probability of damage73. According to Polyphene-2, 215 nsSNPs were predicted to be harmful. Likewise, CADD uses a prediction score to classify SNPs as harmful or benign74, which identified 23 nsSNPs as potentially pathogenic. Since scores in Revel range from 0 to 1, higher-scoring mutations are probably more harmful75. A rigorous prediction using Revel predicted 194 nsSNPs to be diseased. Additionally, MetaLR, which assigns ratings between 0 and 1 with higher values denoting a higher likelihood of harm, was used. The MetaLR review revealed the 358 nsSNPs as deleterious. Lastly, Mutation Assessor assessed 263 nsSNPs as moderately damaging. The results of the tool are depicted in Fig. 3. Finally, 22 nsSNPs that were predicted to be deleterious using each of the six tools, were selected. Due to their deleterious prediction from each of the six tools, these 22 nsSNPs were selected for further exploration.

Represents the predicted nsSNPs percentage from eight bioinformatics tools. The number of nsSNPs, revealed by each tool, including SIFT (193), Polyphene-2 (215), CADD (23), Revel (194), MetaLR (358), and MutationAssessor (263), are depicted in the form of a pie chart.
The 22 shortlisted nsSNPs were further validated for their deleterious effect using three different tools including SNP&GO, Panther and PredictSNP. SNP&GO evaluation revealed that only nine mutations have damaging effect on FUT2 structure whereas Panther provided 19 mutations as deleterious. PredictSNP showed that all of the already shortlisted mutations have deleterious effects. Among all of the employed tools, only nine mutations including T65M, G149S, V196G, V200A, R202Q, G215R, V240M, R250P, and G301R were found deleterious as shown in Table 1.
MutPred prediction of structural and functional impacts
To investigate the structural and functional impact of 9 shortlisted nsSNPs, MutPred was utilized. This server represented the data based on the previously defined attributes, including p-values, in the form of likelihood scores (Table 2). Except V240M, all the other 8 nsSNPs were revealed to affect protein structure or function based on a threshold larger than 0.6. These 8 nsSNPs were shortlisted for further analyses.
FUT2 stability evaluation
Each of the shortlisted nsSNPs was dealt separately using I-Mutant tool, and their stability was predicted using RI values ranging from 0 to 10 (Table 3). All the 8 shortlisted nsSNPs showed declining stability from I-Mutant evaluation. Further insight using mCSM, Mupro and DDMut revealed that almost all the nsSNPs (except T65M and G301R) were found to destabilize the FUT2 structure. Considering their higher deleterious effects on FUT2 protein’s stability, these 6 nsSNPs were chosen for further processing.
Evolutionary conservation of nsSNPs
DeepRex web server provided information on each amino acid in FUT2, with 43.44% of residues as exposed and 56.56% of residues as buried (Table 4). The conservation threshold was automatically set at 0.17 (i.e. conserved < 0.17, highly conserved ≥ 0.17). G215 was the only residue that DeepREx-WS predicted would be less conserved and buried, excluding it for further analysis. Conversely, G149, V196, V200, and R202 were among the other highly conserved and buried residues. The residue, R250, was predicted to be highly functional, conserved and exposed. For each of the shortlisted nsSNPs, Table 3 shows the conservation scores. The function and structure of the FUT2 protein were predicted to be most deleteriously impacted by all nsSNPs located in highly conserved regions, according to these results.
Structural modeling of FUT2 and its mutants
To create 3D structures of mutant proteins, each nsSNP’s substitution in the FUT2 protein sequence was carried out separately and their 3D structures were modelled using Robetta online server. For each mutant model, the RMSD and TM scores were calculated using TM-Align. In wild type FUT2 and its mutant models, the average distance between the α-carbon backbones was measured by RMSD, whereas topological similarity was assessed by TM-score. Greater structural divergence between the mutant and the wild type was reflected in greater RMSD values. A 2 Å threshold was set up. At 2.28 Å and 2.24 Å, respectively, the mutants G149S (rs200543547) and V196G (rs367923363) had the highest RMSD values. R202Q (1.27 Å RMSD), V200A (1.91 Å RMSD), and R250P (1.78 Å RMSD), were among the other nsSNPs with less deviations. The RMSD values and the TM scores are presented in Table 5. Finally, the two nsSNPs (G149S, V196G) with the highest RMSD values from wild type FUT2 were remodeled using alpha-fold2. Using Pymol, the superimposed structures of the proposed two mutants with wild-type FUT2 were illustrated graphically (Fig. 4).

Represents the superimposed FUT2 mutants’ structures with their wild type (A) Wild_type FUT2 (blue), along with its superimposed mutant G149S (green) (B) wild_type FUT2 (blue), along with its superimposed mutant V196G (red), and (C) 3D structure of wild_type protein, FUT2.
The proposed mutants, along with the wild type FUT2, were subsequently examined using MolProbity and a web-based SAVES server. MolProbity produced reliable results for both of the two predicted modeled proteins and its wild-type FUT2. For the mutant and wild-type FUT2 genes, the highest ERRAT scores were wild_type = 92.18, G149S = 90.84, and V196G = 89.10 were observed.
Protein–protein docking analysis
The final two highly deleterious mutants were docked with the norovirus capsid protein (VP1) to determine their binding affinity and mode of interaction using ClusPro 2.0, providing ten distinct models for each mutant-receptor combination. Among these models, one best model from each complex was chosen. The G149S complex with the norovirus capsid protein showed the lowest energy (− 1508.4) and the most cluster members (53), indicating a stable favorable behavior. This suggests that the G149S mutation may affect norovirus susceptibility. Following this, the V196G-complex also demonstrated promising binding energy (− 1488.4) with 46 cluster members, as compared to the wild type-complex, which had a lower binding affinity (− 1344.6) and a lower cluster member count (44). These findings (Table 6) suggested that a mutation in the FUT2 protein might speed up the spread of norovirus infection.
Further analysis with PDBsum disclosed a network of connections between these complexes. We evaluated results based on three interactions: non-bonded contacts, which contribute to overall attraction; salt bridges, which are specific interactions involving the charged atoms that strengthen the binding; and hydrogen bonds, which form precise connections between molecules, similar to tiny bridges. The G149S mutant showed the highest interactions with the receptor, having 21 hydrogen bonds, 251 non-bonded contacts, and two salt bridges. Following this, the V196G mutant also demonstrated dependable connections by generating 14 hydrogen bonds, 171 non-bonded contacts, and one salt bridge. Finally, wild-type FUT2, complexed with norovirus capsid protein, offered the least interactions, creating 9 hydrogen bonds, 172 non-bonded contacts, and 1 salt bridge (Fig. 5). The overall results demonstrated that G149S mutant interacts significantly with the norovirus capsid protein, followed by the mutant V196G and the wild type, which can ultimately lead to the norovirus susceptibility.

(A) Surface representation of the wild type FUT2 (black color) interacting with the core residues of VP1 receptor (grey color) (A) Surface representation of the mutant G149S (red color) interacting with the core residues of VP1 receptor (grey color) (A) Surface representation of the mutant V196G (green color) interacting with the core residues of VP1 receptor (grey color).
Molecular dynamic simulation analysis
Compared to the wild type, the G149S, and V196G mutants formed more stable interactions with norovirus capsid protein (PDB ID: 6OUU). The time-dependent alterations of bound protein–protein complexes up to 300 ns were investigated using comprehensive MD simulation. Regarding stability, the wild-type complex with VP1 exhibited comparably greater fluctuations in the RMSD and RMSF metrics compared to both of the mutants, demonstrating a less stable conformation during the simulations. These deviations indicate that the wild-type has least interactions with the VP1, suggesting the less chances of norovirus implication in wild-type FUT2. Conversely, the G149S and V196G mutations exhibited enhanced binding interactions with VP1 due to structural changes, contributing to their increased stability in the protein–protein complex, which ultimately caused norovirus.
Root mean square deviation (RMSD) analysis
We evaluated and validated the stability of each optimized hit in a simulated environment using RMSD. An insight into the two proposed mutant complexes with VP1 revealed that they behave much more consistently with the receptor, as compared to the wild-type. The wild type complex with VP1 was more unstable compared to other mutants, with an RMSD of up to 22 Å over 300 ns (Fig. 6A). The RMSD increased during the simulation. For the first 90 ns, the RMSD climbed to 15 Å, then fluctuated, and finally reached up to 22 Å until 300 ns. This suggests a highly unstable complex between the wild type and capsid protein of norovirus (VP1), with very little opportunities of interaction between them. The G149S-VP1 complex was significantly more stable than the wild type, with an average RMSD of less than 10 Å throughout the simulation. For the first 40 ns, the complex had an initial growth with an average of 3–10. After then, it remained steady for the remainder of the simulation, lasting 300 ns, demonstrating a very stable complex with VP1 (Fig. 6B). Finally, the V196G-VP1 complex exhibited comparably lower deviations than the wild type, throughout the simulation. Figure 6C shows that the complex first increased up to 9 Å, experienced some deviations, and then stabilized at 9 Å RMSD after 240 ns. The overall RMSD results demonstrated that both the mutants G149S and V196G were comparably more stable than the wild type when interacting with the Norovirus capsid protein. As a result, we may conclude that wild-type FUT2 mutations can cause norovirus infection.

(A) Representing root mean square deviation of wild type-VP1 complex (B) representing Root mean square deviation of G149S-VP1 complex (C) Representing Root Mean Square Deviation of V196G-VP1 complex.
Root mean square fluctuation (RMSF) analysis
Each simulated system, including wild type and the mutants, exhibited a unique average RMSF. The wild-type molecule with the VP1 receptor showed the greatest RMSF values, reaching up to 32 Å during the simulation. The largest variations suggested a highly unstable interaction between wild-type FUT2 and the VP1 receptor (Fig. 7A). The G149S-VP1 complex, on the other hand, had the fewest changes, indicating very high stability. The RMSF for all residues was below 8 Å, with the exception of 491–496, which had RMSF approaching 12 Å (Fig. 7B). Similarly, the V196G-VP1 complex exhibited a more stable RMSF than the wild type. Throughout the 300 ns simulation, the average RMSF remained between 3 and 6 Å, with the exception of a few residues within 502–510, which displayed substantial variations (Fig. 7C). The overall RMSF results suggested that almost all residues in both mutants are more stable with the VP1 receptor than wild-type FUT2. This led to the conclusion that mutants had a greater affinity for Norovirus capsid protein.

(A) Representing root mean square fluctuation of wild type-VP1 complex (B) representing root mean square fluctuation of G149S-VP1 complex (C) representing root mean square fluctuation of V196G-VP1 complex.
Radius of gyration (RoG) analysis
The Rg values for both the wild type and the VP1 complex represented the lowest compactness and interacted between 70.3 and 70.45 Å during the simulation (Fig. 8A). The G149S–VP1 complex had a compactness between 51.5 and 51.6 Å, indicating the highest interaction between the mutant and receptor (Fig. 8B). V196G–VP1 also had a comparable low Rg of 56.7 Å, indicating a stronger binding than the wild type (Fig. 8C). Overall results of the RoG showed that the wild type has the least compactness when compared to the mutants. This suggests that these mutations interact with VP1 receptors with a comparable high binding affinity and thus may be a source of norovirus.

(A) Representing radius of gyration of wild type-VP1 complex (B) representing radius of gyration of G149S-VP1 complex (C) representing radius of gyration of V196G-VP1 complex.
Hydrogen bond analysis
Protein–protein complexes heavily depend on hydrogen bonding to maintain their secondary structure. Figure 9 shows a time-dependent study of hydrogen bonding, demonstrating that the two mutant complexes with VP1 displayed strong hydrogen bonding networks compared to the wild type FUT2. The wild-type FUT2-VP1 complex maintained around 6–9 hydrogen bonds on average throughout the simulation, with only rare instances where the count reached 11. In contrast, the G149S-VP1 mutant exhibited a significantly higher hydrogen bonding index, reaching up to 20, with a consistent increase compared to the wild-type complex. Similarly, the V196G-VP1 complex demonstrated an increased number of hydrogen bonds, rising up to 17 throughout the simulation. These results suggest that both the mutants exhibited comparably stronger interactions to the VP1 receptor than the wild type, and are able to effectively attach and infect norovirus. This information could assist in determining important protein interactions involving the capsid protein of norovirus.

(A) Representing H-bonds of wild type-VP1 complex (B) Representing H-bonds of G149S-VP1 complex, and (C) Representing H-bonds of V196G-VP1 complex.
Predicted PTMs (post‐transcriptional modifications)
Methylation
GPS-MSP 3.0 predicted no FUT2 sites to be methylated.
Phosphorylation
As demonstrated in Fig. 10, FUT2 phosphorylation locations were predicted using GPS 6.0 and NetPhos 3.1. 26 residues (Ser:11, Thr:09, TyrL:06) were predicted to be phosphorylated, according to NetPhos 3.1. Likewise, ten residues (Ser:03, Thr:05, Tyr:02) were identified by GPS 6.0 as possibly phosphorylated.

(A) Phosphorylation graph of FUT2 residues (B) Ubiquitination graph for FUT2 residues (C) Phosphorylation graph of FUT2, predicted by GPS server.
Ubiquitination
The RUBI and GPS-Uber servers were utilized to create the ubiquity forecast. 5 out of 10 lysines at positions 53, 180, 214, 321, and 342 was predicted to be ubiquitinated based on GPS-Uber algorithm. Among the ten lysine residues, RUBI predicted that one would be ubiquitinated. There was no predicted residue in a highly conserved or harmful nsSNP area. 10.0% of all proteins were thought to be ubiquitinated.
Glycosylation
Using NetOGlyc4.0, the most likely glycosylation sites were evaluated. At positions 51, 58, 2, 18, and 20, the wild-type FUT2 protein was found to be glycosylated, with scores of 0.55, 0.60, 0.54, 0.66, and 0.54, respectively. These locations are anticipated to be glycosylated.
Allelic frequency and clinical significance of proposed mutants
To get deep insights into the allelic frequency of the proposed mutants, we employed a genome aggregation database, named gnomAD76. Our comprehension of the prevalence of proposed mutations in different geographic regions may be increased by the information on several populations that this large database provides. After the analysis, V196G was found as the most frequent mutation, occurred in a total of 14 genomes and 196 exomes all over the world. Likewise, G149S was also found to be highly prominent in overall 14 genomes and 140 exomes in different regions like America, Europe and South Asia (Table 7).
To further check the clinical significance of the two proposed nsSNPs, we employed a database named ClinVar 77, which gives results on the basis of already utilized data from different researches and reported cases. From ClinVar results, both of the proposed mutants were found to be classified as highly significant for clinical profiles. Finally, Project HOPE (https://www3.cmbi.umcn.nl/hope/)78 server was employed to provide additional insights into the structural and functional consequences of mutations. This server revealed that both the mutations were located within a stretch of residues called Lumenal that was repeated in the protein. The mutation into another residue might disturb this repeat and consequently any function this repeat might have. Both the wild type (G149S) and mutational residues (V196G) included the most flexible residue, glycine. The mutation, involving glycine, can disrupt the required rigidity of protein and abolish its function. Furthermore, the higher MetaRNN scores for G149S (0.75) and V196G (0.92) indicated that the mutations are more likely to be pathogenic. Moreover, the mutated residue S at position 149 was bigger than the original residue G, which can lead to bumps. Conversely, at position 196, the mutated residue G was found smaller in size as compared to the wild residue V that might lead to the loss of interactions. The changed hydrophobicity of wild type and the mutant at position 196 also suggested that the hydrophobic interactions, either in the core of the protein or on the surface, might be lost.
Phylogenetic relationship for the proposed nsSNPs in FUT2
By performing the phylogenetic analysis of human FUT2 protein with the homologous proteins from other eight species, we focused on the residues of our proposed nsSNPs (i.e. G149 and V196). The alignment results of all the relevant species demonstrated that the residues G149 and V196 were highly conserved among all the species. To represent the evolutionary relationship among species, a phylogenetic tree was depicted (Fig. 11).

Represents a phylogenetic tree in circular manner, demonstrating the evolutionary relationship of FUT2 protein among various species.
FUT2 gene–gene interaction
Numerous genes, including as TCN1, KLF5, SI, FUT6, FUT3, GCNT1, MYOC, GPx2, MLN, CPA2, and CD82, were found to be expressed in tandem with FUT2. Likewise, it is co-localized with TCN1, KLF5, TACR2, FUT6, GCNT1, RPL12, MYOC, CTRB1, GPx2, CEACAM3, CA9, ALDH3A1, DSG3, CD82, and CLPS. Moreover, FUT2 and FUT1 also found to share protein domains. Each gene received a cumulative score based on STRING predictions. Figure 12 presents the GeneMANIA and STRING findings.

Gene interactions predicted by GeneMANIA and STRING are shown in (a) and (b), respectively.
Discussion
The function of the fucosyltransferase 2 (FUT2) gene in possible alterations in the gut microbiota and susceptibility to noroviruses has been widely documented79. It has been discovered that non-synonymous single nucleotide polymorphisms (nsSNPs) in the FUT2 gene affect the protein’s normal function. Previous studies have reported that the FUT2 secretors had been at significantly greater risk for both symptomatic and asymptomatic norovirus infections. This is consistent with our findings, which suggest that specific nsSNPs can further modulate this susceptibility. Previous epidemiological research have associated FUT2 polymorphisms with various degrees of norovirus susceptibility in different groups, which further supports our in-silico results. Moreover, researchers have become more interested in the correlation between the FUT2 gene and susceptibility to norovirus in various populations, but their researches were mostly based on epidemiological evidence or they analyzed a very limited number of genetic variants. Regardless of the increased interest to get insights into FUT2 interactions with norovirus susceptibility, the absence of a thorough examination has been restricting the understanding of potential variables contributing to this process. The main purpose of our study was to prepare a pipeline of various bioinformatics tools and databases and to apply that pipeline for the identification and exploration of potential highly deleterious FUT2 nsSNPs, which might be linked with norovirus sensitivity and gut microbiota changes. For this purpose, we initially acquired all of the 5306 FUT2 SNPs from dbSNP and Ensembl database. After extensively refined those SNPs for duplicates, only 362 non-synonymous SNPs were processed further because of their higher likeness of being deleterious. These nsSNPs were subsequently screened to explore their damaging effect using nine different widely-used bioinformatics tools including SIFT, polyphene-2, revel, metaLR, MutationAssessor, Panther, SNP&GO, PredictSNP and CADD. All of these tools have been reported to be highly specific and accurate. Out of overall 362 nsSNPs, only 9 were found to be deleterious as the consensus of all the tools, which were further checked for their possible impact on FUT2 structure and function using MutPred 1.2. The 8 out of 9 nsSNPs were found to have negative impact on overall FUT2 structure and function. These 12 nsSNPs were further checked for their impact on FUT2 stability using different tools including I-Mutant, mCSM, Mupro and DDMut, which showed that all of these 7 nsSNPs resulted in decreasing overall stability of FUT2. To further check whether these shortlisted 6 nsSNPs are conserved or not, we did the conservation analysis. Only one residue, G215R, showed comparatively less conservation than the other 5 residues, making it unreliable for further processing. By considering the fact that mutations in highly conserved residues can cause the structure to be less stabled, we took the remaining 5 nsSNPs as possibly deleterious and processed further. These 5 nsSNPs were induced one by one in FUT2 sequence and their three dimensional structures were predicted using an online web-server named Robetta. Subsequently, these mutants’ structures were compared with their wild type FUT2 structure using an online program, named TM-align. This web-based tool provided us results in the form of TM-Score and root mean square deviations of mutants’ structures from wild type structure. From all of the five mutants, only two mutants (G149S, and V196G) were chosen for further processing, based on their higher RMSD results (G149S = 2.28, V196G = 2.24) and TM scores (G149S = 0.91, V196G = 0.87). Following the shortlisting of two highly deleterious mutants, we delved deeper into their impact on norovirus susceptibility. For this purpose, we docked these proposed two mutants with the capsid protein of norovirus (VP1) using ClusPro. Previous researches have demonstrated the role of FUT2 in host-virus interaction through H-antigen. Conversely, in this study, the selection of VP1 as a receptor for FUT2 mutants was based its critical role in mediating the host-virus interaction as well. Moreover, it plays a crucial role in host cell recognition and binding during the initial stages80. The docking results of these two mutants and their wild type with VP1 demonstrated that both the mutants showed comparably higher stability and binding affinity with VP1 than the wild type, which supported the role of these mutations in norovirus susceptibility. Following the molecular docking, the atmoic level characterization and validation of mutants stability with VP1 was analyzed by comprehensive molecular dynamic simulation strategy. Compared to the wild type FUT2, both the mutants (G149S, V196G) showed less deviations and higher stability in the form of RMSD, RMSF, RoG and hydrogen bonds, which further validated their high binding interaction with VP1, ultimately playing role in norovirus sensitivity. Subsequently, these mutations were checked whether they can be resulted in the phosphorylation, ubiquitination or methylation of any residue. The results provided that these nsSNPs can cause at least 10 residues to be phosphorylated and about 5 out of 10 lysine to be ubiquitinated. To further check the allelic frequency of these two mutations, we employed gnomAD database, which provided G149S to be found in 154 and V196G to be found in 210 different genomes and exomes all over the world (more frequently in America, Europe and South Asia). Likewise, ClinVar assessment demonstrated that both of the proposed nsSNPs are highly significant. Finally, interaction of the FUT2 gene with other relevant genes further provided into their co-occurrence, mostly with FUT1 gene. In light of previous study, our results have significantly demonstrated the role of FUT2 in norovirus-related illnesses and offer vital information for further investigation. Our results offer strong support for the involvement of FUT2 gene alterations in the pathophysiology of norovirus infection. Previous studies have established that FUT2 secretors are at a greater risk of norovirus infection79,81, and our results demonstrate how specific nsSNPs, G149S and V196G, may further modulate this susceptibility by increasing the binding affinity of FUT2 to the norovirus capsid protein VP1. This is consistent with the understanding that host-virus interactions are influenced by FUT2’s role in producing H-antigens. Although there is no direct experimental evidence for these particular nsSNPs, the findings that they increase VP1 binding provide a logical extension given the known association between FUT2 genotype and the risk of norovirus infection. For instance, research has demonstrated that non-secretors, who lack a functional FUT2 enzyme, are resistant to some strains of norovirus43,81. Our results suggest a potential mechanism by which specific mutations in the FUT2 gene could alter its interaction with norovirus, leading to altered susceptibility. Although our study was comprehensive and utilized an effective strategy to enlist the possibly deleterious FUT2 nsSNPs, but it also faced some limitations as it highly relied on in-silico approaches. The built-in constraints of molecular dynamic simulation strategy was also one of the limitations as it might not efficiently included the complexity of in-vivo experiments. Furthermore, the accuracy of predictions might be limited by the training and algorithms used by the employed tools. Moreover, the interactions between FUT2 and VP1 may not be accurately represented by the energy functions and force fields utilized in docking and MD simulations since they are approximations of the real-world forces. To overcome these challenges, we welcome any further experimental validation and in-vivo trials, which we will be supposed do in our future research. Overall, this study identified and explored the two novel highly deleterious mutations including G149S (rs200543547), and V196G (rs367923363), which are needed to be further analyzed in future researches.
Conclusion
Non-synonymous SNPs in FUT2 gene have been reported to be associated with the norovirus susceptibility and gut microbiota composition. Exploration of these nsSNPs is crucial to delve deeper into their possible mechanism in the relevant diseases. In this research, we employed a comprehensive bioinformatics pipeline incorporating different computational tools and applied to all the reported nsSNPs of FUT2 gene. This strategy resulted in the identification of two nsSNPs, G149S and V196G, as deleterious in respective diseases, which were further validated through molecular docking and simulation approaches. These proposed nsSNPs showed higher stability and binding affinity with norovirus capsid protein, VP1, demonstrating their potential involvement in causing norovirus. Our strategy highlights the value of computational strategies in mutational analysis and appreciates further clinical validation of the resulting nsSNPs.
Data availability
Data are available from the corresponding author upon reasonable request.
References
Ahmed, S. M. et al. Global prevalence of norovirus in cases of gastroenteritis: A systematic review and meta-analysis. Lancet Infect. Dis. 14(8), 725–730 (2014).
Capriotti, E. & Altman, R. B. Improving the prediction of disease-related variants using protein three-dimensional structure. BMC Bioinform. 12(4), 1–11 (2011).
Kelly, R. J. et al. Sequence and expression of a candidate for the human secretor blood group Α (1, 2) fucosyltransferase gene (Fut2): Homozygosity for an enzyme-inactivating nonsense mutation commonly correlates with the non-secretor phenotype (∗). J. Biol. Chem. 270(9), 4640–4649 (1995).
Koda, Y. et al. Molecular basis for secretor type alpha(1,2)-fucosyltransferase gene deficiency in a Japanese population: A fusion gene generated by unequal crossover responsible for the enzyme deficiency. Am. J. Hum. Genet. 59(2), 343–350 (1996).
Hutson, A. M., Atmar, R. L. & Estes, M. K. Norovirus disease: Changing epidemiology and host susceptibility factors. Trends Microbiol. 12(6), 279–287 (2004).
Lindesmith, L. et al. Human susceptibility and resistance to Norwalk virus infection. Nat. Med. 9(5), 548–553 (2003).
Marionneau, S. et al. ABH and Lewis histo-blood group antigens, a model for the meaning of oligosaccharide diversity in the face of a changing world. Biochimie 83(7), 565–573 (2001).
Wacklin, P. et al. Faecal microbiota composition in adults is associated with the FUT2 gene determining the secretor status. PLoS ONE 9(4), e94863 (2014).
Sender, R., Fuchs, S. & Milo, R. Revised estimates for the number of human and bacteria cells in the body. PLoS Biol. 14(8), e1002533 (2016).
Wacklin, P. et al. Secretor genotype (FUT2 gene) is strongly associated with the composition of Bifidobacteria in the human intestine. PLoS ONE 6(5), e20113 (2011).
David, L. A. et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 505(7484), 559–563 (2014).
Lopman, B. A. et al. The vast and varied global burden of norovirus: prospects for prevention and control. PLoS Med. 13(4), e1001999 (2016).
Kindberg, E. & Svensson, L. Genetic basis of host resistance to norovirus infection. Future Virol. 4(4), 369–382 (2009).
Hong, X. et al. Association of fucosyltransferase 2 gene with norovirus infection: A systematic review and meta-analysis. Infect. Genet. Evolut. 96, 105091 (2021).
Iqbal, M. W. et al. Analysis of damaging non-synonymous SNPs in GPx1 gene associated with the progression of diverse cancers through a comprehensive in silico approach. Sci. Rep. 14(1), 28690 (2024).
Aganezov, S. et al. A complete reference genome improves analysis of human genetic variation. Science 376(6588), eabl3533 (2022).
Sherry, S. T. et al. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 29(1), 308–311 (2001).
Chen, Y. et al. Ensembl variation resources. BMC Genom. 11, 1–16 (2010).
Kumar, P., Henikoff, S. & Ng, P. C. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat. Protoc. 4(7), 1073–1081 (2009).
Adzhubei, I., Jordan, D. M. & Sunyaev, S. R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 76(1), 7–20 (2013).
Sim, N.-L. et al. SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 40(W1), W452–W457 (2012).
Ernst, C. et al. Performance of in silico prediction tools for the classification of rare BRCA1/2 missense variants in clinical diagnostics. BMC Med. Genom. 11, 1–10 (2018).
Schubach, M. et al. CADD v1.7: Using protein language models, regulatory CNNs and other nucleotide-level scores to improve genome-wide variant predictions. Nucleic Acids Res. 52(D1), D1143–D1154 (2024).
Ioannidis, N. M. et al. REVEL: An ensemble method for predicting the pathogenicity of rare missense variants. Am. J. Hum. Genet. 99(4), 877–885 (2016).
Reva, B., Antipin, Y. & Sander, C. Predicting the functional impact of protein mutations: Application to cancer genomics. Nucleic Acids Res. 39(17), e118–e118 (2011).
Dash, R. et al. Dynamic insights into the effects of nonsynonymous polymorphisms (nsSNPs) on loss of TREM2 function. Sci. Rep. 12(1), 9378 (2022).
Calabrese, R. et al. Functional annotations improve the predictive score of human disease-related mutations in proteins. Hum. Mutat. 30(8), 1237–1244 (2009).
Thomas, P. D. et al. PANTHER: Making genome-scale phylogenetics accessible to all. Protein Sci. 31(1), 8–22 (2022).
Bendl, J. et al. PredictSNP: Robust and accurate consensus classifier for prediction of disease-related mutations. PLoS Comput. Biol. 10(1), e1003440 (2014).
Pienaar, I. S., Howell, N. & Elson, J. L. MutPred mutational load analysis shows mildly deleterious mitochondrial DNA variants are not more prevalent in Alzheimer’s patients, but may be under-represented in healthy older individuals. Mitochondrion 34, 141–146 (2017).
Capriotti, E., Fariselli, P. & Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucleic Acids Res. 33(suppl_2), W306–W310 (2005).
Cheng, J., Randall, A. & Baldi, P. Prediction of protein stability changes for single-site mutations using support vector machines. Proteins Struct. Funct. Bioinform. 62(4), 1125–1132 (2006).
Pires, D. E., Ascher, D. B. & Blundell, T. L. mCSM: Predicting the effects of mutations in proteins using graph-based signatures. Bioinformatics 30(3), 335–342 (2014).
Zhou, Y. et al. DDMut: Predicting effects of mutations on protein stability using deep learning. Nucleic Acids Res. 51(W1), W122–W128 (2023).
Ramensky, V., Bork, P. & Sunyaev, S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 30(17), 3894–3900 (2002).
Manfredi, M. et al. DeepREx-WS: A web server for characterising protein-solvent interaction starting from sequence. Comput. Struct. Biotechnol. J. 19, 5791–5799 (2021).
Kim, D. E., Chivian, D. & Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 32(suppl_2), W526–W531 (2004).
Zhang, Y. & Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 33(7), 2302–2309 (2005).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596(7873), 583–589 (2021).
Yuan, S., Chan, H. S. & Hu, Z. Using PyMOL as a platform for computational drug design. Wiley Interdiscip. Rev. Comput. Mol. Sci. 7(2), e1298 (2017).
Chen, L. et al. Bioinformatics analysis of the epitope regions for norovirus capsid protein. BMC Bioinform. 14, 1–6 (2013).
Burley, S. K. et al. Protein Data Bank (PDB): The single global macromolecular structure archive. In Protein Crystallography: Methods and Protocols, 627–641 (2017).
Nordgren, J. & Svensson, L. Genetic susceptibility to human norovirus infection: An update. Viruses 11(3), 226 (2019).
Vilar, S., Cozza, G. & Moro, S. Medicinal chemistry and the molecular operating environment (MOE): Application of QSAR and molecular docking to drug discovery. Curr. Top. Med. Chem. 8(18), 1555–1572 (2008).
Kozakov, D. et al. The ClusPro web server for protein–protein docking. Nat. Protoc. 12(2), 255–278 (2017).
Laskowski, R. A. PDBsum: Summaries and analyses of PDB structures. Nucleic Acids Res. 29(1), 221–222 (2001).
Chapman, D. E., Steck, J. K. & Nerenberg, P. S. Optimizing protein–protein van der Waals interactions for the AMBER ff9x/ff12 force field. J. Chem. Theory Comput. 10(1), 273–281 (2014).
Tian, C. et al. ff19SB: Amino-acid-specific protein backbone parameters trained against quantum mechanics energy surfaces in solution. J. Chem. Theory Comput. 16(1), 528–552 (2019).
Salomon-Ferrer, R., Case, D. A. & Walker, R. C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 3(2), 198–210 (2013).
Kini, R. M. & Evans, H. J. Molecular modeling of proteins: A strategy for energy minimization by molecular mechanics in the AMBER force field. J. Biomol. Struct. Dyn. 9(3), 475–488 (1991).
Lin, Y. et al. Application of Berendsen barostat in dissipative particle dynamics for nonequilibrium dynamic simulation. J. Chem. Phys. 146(12), 124108 (2017).
Liu, J., Li, D. & Liu, X. A simple and accurate algorithm for path integral molecular dynamics with the Langevin thermostat. J. Chem. Phys. 145(2), 024103 (2016).
Forester, T. R. & Smith, W. SHAKE, rattle, and roll: efficient constraint algorithms for linked rigid bodies. J. Comput. Chem. 19(1), 102–111 (1998).
Stella, L. & Melchionna, S. Equilibration and sampling in molecular dynamics simulations of biomolecules. J. Chem. Phys. 109(23), 10115–10117 (1998).
Case, D. A. et al. AMBER 22 Reference Manual (2022).
Roe, D. R. & Cheatham, T. E. III. PTRAJ and CPPTRAJ: Software for processing and analysis of molecular dynamics trajectory data. J. Chem. Theory Comput. 9(7), 3084–3095 (2013).
Sargsyan, K., Grauffel, C. & Lim, C. How molecular size impacts RMSD applications in molecular dynamics simulations. J. Chem. Theory Comput. 13(4), 1518–1524 (2017).
Martínez, L. Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. PLoS ONE 10(3), e0119264 (2015).
Liu, P. et al. Lubricant shear thinning behavior correlated with variation of radius of gyration via molecular dynamics simulations. J. Chem. Phys. 147(8), 084904 (2017).
Chen, C. et al. Hydrogen bonding analysis of glycerol aqueous solutions: A molecular dynamics simulation study. J. Mol. Liq. 146(1–2), 23–28 (2009).
Park, I.-H. et al. Estimation of hydrogen-exchange protection factors from MD simulation based on amide hydrogen bonding analysis. J. Chem. Inf. Model. 55(9), 1914–1925 (2015).
Blom, N., Gammeltoft, S. & Brunak, S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J. Mol. Biol. 294(5), 1351–1362 (1999).
Xue, Y. et al. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteom. 7(9), 1598–1608 (2008).
Walsh, I., Di Domenico, T. & Tosatto, S. C. RUBI: Rapid proteomic-scale prediction of lysine ubiquitination and factors influencing predictor performance. Amino Acids 46, 853–862 (2014).
Steentoft, C. et al. Precision mapping of the human O-GalNAc glycoproteome through SimpleCell technology. EMBO J. 32(10), 1478–1488 (2013).
Halder, S. K. et al. Identification of the most damaging nsSNPs in the human CFL1 gene and their functional and structural impacts on cofilin-1 protein. Gene 819, 146206 (2022).
Thompson, J. D., Gibson, T. J. & Higgins, D. G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinform. 1, 1–22 (2003).
Hall, B. G. Building phylogenetic trees from molecular data with MEGA. Mol. Biol. Evol. 30(5), 1229–1235 (2013).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v6: Recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 52, W78–W82 (2024).
Franz, M. et al. GeneMANIA update 2018. Nucleic Acids Res. 46(W1), W60–W64 (2018).
Von Mering, C. et al. STRING 7—Recent developments in the integration and prediction of protein interactions. Nucleic Acids Res. 35(suppl_1), D358–D362 (2007).
Ng, P. C. & Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 31(13), 3812–3814 (2003).
Pavithran, H. & Kumavath, R. In silico analysis of nsSNPs in CYP19A1 gene affecting breast cancer associated aromatase enzyme. J. Genet. 100(2), 23 (2021).
Kircher, M. et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 46(3), 310–315 (2014).
Cubuk, C. et al. Clinical likelihood ratios and balanced accuracy for 44 in silico tools against multiple large-scale functional assays of cancer susceptibility genes. Genet. Med. 23(11), 2096–2104 (2021).
Koch, L. Exploring human genomic diversity with gnomAD. Nat. Rev. Genet. 21(8), 448–448 (2020).
Landrum, M. J. et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44(D1), D862–D868 (2016).
Venselaar, H. et al. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 11, 1–10 (2010).
Currier, R. L. et al. Innate susceptibility to norovirus infections influenced by FUT2 genotype in a United States pediatric population. Clin. Infect. Dis. 60(11), 1631–1638 (2015).
Lochridge, V. P. et al. Epitopes in the P2 domain of norovirus VP1 recognized by monoclonal antibodies that block cell interactions. J. Gen. Virol. 86(10), 2799–2806 (2005).
Prystajecky, N. et al. Personalized genetic testing and norovirus susceptibility. Can. J. Infect. Dis. Med. Microbiol. 25(4), 222–224 (2014).
Acknowledgements
This work was supported by the State Key Laboratory of Chemical Resources Engineering, Beijing University of Chemical Technology, Beijing 100029, China. The authors extend their appreciation to the Researchers Supporting Project number (RSP2025R197) King Saud University, Riyadh, Saud Arabia.
Author information
Authors and Affiliations
Contributions
Conceptualization, original draft writing, reviewing, and editing: Muhammad Waleed Iqbal, Muneer Ahmad, Muhammad Shahab, Xinxiao Sun. Formal analysis, investigations, funding acquisition, reviewing, and editing: Mudassar Mehmood Baig, Kun Yu, Guojun Zheng. Resources, data validation, data curation, and supervision: Qipeng Yuan, Turki M. Dawoud, Mohammed Bourhia, Fakhreldeen Dabiellil.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Iqbal, M.W., Ahmad, M., Shahab, M. et al. Exploring deleterious non-synonymous SNPs in FUT2 gene, and implications for norovirus susceptibility and gut microbiota composition. Sci Rep 15, 10395 (2025). https://doi.org/10.1038/s41598-025-92220-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-92220-4
Keywords
This article is cited by
-
Structure, function, and implications of fucosyltransferases in health and disease
Nature Communications (2025)
