
Abstract
Excess fat intake is associated with kidney toxicity and dysfunction. Because fatty acids can also be reversibly attached onto cysteine residues and modulate the function of several membrane-bound proteins, we studied the effect of high-fat diet (HFD) on the S-acylated proteome of mouse kidneys to uncover novel biochemical changes that might contribute to lipid-induced nephrotoxicity. We compared the S-acylated proteome of kidneys from mice fed a chow diet (CD) or a HFD. HFD caused albuminuria. The HFD intervention induced a large-scale repression of protein S-acylation as well as of the most abundant ceramides and sphingomyelin species, which are highly suggestive of a reduction in acyl-CoA availability. The HFD-induced S-acylation repression mostly affected proteins involved in endocytosis and intracellular transport. Notably, the kidneys of mice fed a HFD displayed a marked decrease in the total amount and in the S-acylated form of megalin, the main tubular protein retrieval system. Further in vitro experiments indicated that S-acylation inhibition results in a reduction of megalin protein level. We conclude that diet-induced derangement of fatty acid metabolism modifies the renal landscape of the S-acylated proteome during the early stages of the kidney injury, which might reduce the efficiency of protein reabsorption by the proximal tubule.
Similar content being viewed by others


Introduction
Chronic kidney disease (CKD) is a progressive deterioration in renal function that is defined by the presence of kidney damage or decreased kidney function for at least three months, independent of the underlying etiology1. Currently, the global prevalence of CKD is estimated to be 8–16% worldwide and continues to increase across diverse populations2,3, with diabetic nephropathy now being the leading cause4,5,6. From the initial inury, the progression to CKD represents the point of no return, whereby restoring damaged tissue to normal is practically impossible, and progressive organ dysfunction, high morbidity and high mortality are inevitable. The concomitant worldwide increase in the prevalence of obesity and type 2 diabetes suggests that excessive lipids are causative of insulin resistance, elevated blood glucose levels, and type 2 diabetes7,8,9. Meanwhile, mechanistic studies indicate that non-esterified fatty acids are highly toxic to the kidney cells, thereby directly contributing to the injury10,11,12. While lowering blood glucose and lipids has proven to be a valuable approach to indirectly managing the initial renal injury and to reducing the risk of progression to CKD13,14,15,16,17,18,19, there is a need to further characterize the molecular and biochemical changes underlying lipid-induced kidney injury, to identify new druggable targets for prevention of the injury or of its progression to CKD.
In the kidney, the bulk of fatty acids are usually committed to mitochondrial β-oxidation and ATP production to meet the high metabolic demand of the tubular epithelial cells. The remainder is primarily utilized for phospholipid and sphingolipid synthesis to sustain basal cellular functions20. In kidney disease, mitochondrial β-oxidation is repressed and anaerobic glycolysis becomes the main source of ATP, and the excess fatty acids are channelled into triacylglycerols and eventually into lipid droplets, with the primary goal of alleviating fatty acid-induced cell stress21,22. Indeed, kidney biopsies from patients with diabetic nephropathy show marked lipid droplet accumulation in both glomeruli and tubulointerstitium23. For cellular utilization, fatty acids must be firstly condensed with coenzyme A (CoA). Acyl-CoA sits at the crossroads of all catabolic and anabolic fatty acids-related pathways. Moreover, acyl-CoA, most typically palmitoyl-CoA, can be enzymatically attached onto cysteine residues of proteins via a labile thioester linkage24, a reversible post-translational modification termed S-acylation regulating trafficking, stability, and function of several membrane-bound proteins. S-acylated proteins control key physiological process involved in glucose and lipid metabolism, such as AMP kinase (AMPK), GLUT4, CD3625,26,27, and very low density lipoproteins (VLDL)28. Protein S-acylation is catalyzed by zinc-finger Asp-His-His-Cys (zDHHC) motif-containing acyl S-acyltransferases and reversed by acyl protein thioesterases (APTs)29. Recently, it has been shown that S-acylation of β-catenin is crucial in the promotion of the fibrotic process in the kidney30,31. To better understand the role of S-acylation in lipid-induced nephrotoxicity, we characterized the renal S-acylated proteome of mice with high-fat diet (HFD)-induced kidney dysfunction.
Results
High-fat diet (HFD) leads to hyperglycemia and elevated albuminuria
To test the effect of excess fat intake on protein S-acylation in kidney, we performed nutritional intervention in mice. After 19 weeks of HFD, only a slight, albeit significant, gain in body weight was observed in mice on HFD compared to the chow diet (CD) group (Fig. 1a). These results confirmed previous studies reporting female mice being rather resistant to diet-induced obesity32. Fasting blood glucose level (Fig. 1b) and urinary excretion of albumin (Fig. 1c) were significantly higher in the mice fed a HFD compared to the CD group. Notably, cystatin C (Fig. 1d) and serum creatinine (Fig. 1e) values, thus the estimated glomerular filtration rate (eGFR) (Fig. 1f) were comparable between the two groups. Taken together, the data indicate that mice fed a HFD were characterized by defective protein reabsorption by the proximal tubule.

Metabolic and renal function parameters. (a) Body weight over time. Data are presented as mean ± SD (n = 4). (b) Fasting blood glucose level. (c-e) Urinary Albumin-to-Creatinine (Alb/Cr) (C), serum Cystatin C (D), and serum creatinine (E) level. (f) Estimated glomerular filtration rate (eGFR) calculated as follows: CKD-EPI creatinine-cystatin Eq. (2021): eGFRcr-cys = 135 × min(Scr/κ, 1)α × max(Scr/κ, 1)−0.544 × min(Scys/0.8, 1)−0.323 × max(Scys/0.8, 1)−0.778 × 0.9961Age × 0.963 [if female]; Scr = standardized serum creatinine in mg/dL, κ = 0.7 (females) or 0.9 (males), α = − 0.219 (female) or − 0.144 (male), min(Scr/κ, 1) is the minimum of Scr/κ or 1.0, max(Scr/κ, 1) is the maximum of Scr/κ or 1.0, Scys = standardized serum cystatin C in mg/L, Age (years). Comparisons of the means was performed by unpaired Student’s t-test. In the scatter plots, each dot represents one individual sample.
Kidney of mice fed a HFD are hypo-acylated
To ascertain changes in levels of S-acylation and/or acylated renal proteins following dietary intervention, we performed MS analysis of the total and acyl resin-assisted captured (RAC) proteins from the crude membrane fractions isolated from the kidney of HFD and CD fed mice (Supplementary Fig. 1 and supplementary Fig. 2). A total of 3246 S-acylated proteins were identified, of which 335 were uniquely S-acylated in the CD or in the HFD groups. These data suggest an ‘on/off switch’ mechanism for regulation of S-acylation in the mice kidney under these conditions (Fig. 2a). Comparative analysis of the S-acylated protein pool, normalized to the total protein levels in the input fraction (Integrated S-acylated proteome), revealed another 211 proteins found S-acylated in both groups but in significantly different abundance (Fig. 2b). Overall, the data indicate that the kidney proteins from the mice fed a HFD were hypo-acylated in comparison to those from the CD group. To map the S-acylation changes along the nephron, the proteomic data were integrated with the publicly available anatomy-guided single-cell RNA sequencing database of mouse kidney33. S-acylation changes were evenly distributed among the glomerulus, the proximal tubule, the loop of Henle, and the collecting system, with only a minority of the differentially S-acylated proteins being expressed in the distal tubule (Fig. 2c and Supplementary Fig. 3).

S-acylated proteomic profile in CD and HFD groups. (a) Matrix bubble plot of the proteins found S-acylated exclusively either in the CD or in the HFD group (S-acylated proteins, dark blue and red), and their relative abundance in the input fraction (total protein, light blue and yellow). (b) Volcano plot comparing abundance of the S-acylated form normalized for the abundance in the input fraction (total protein) of S-acylated proteins found in the kidney of both CD and HFD mice (Integrated S-acylated proteome). The relative abundance (log2 FC) is reported on the x-axis, the significance (-log10 FDR) on the y-axis. The vertical and horizontal lines show the cut-off of FC = ± 1.0, and of FDR = 0.1, respectively. (c) Anatomy-guided single-cell transcriptomic-derived pie-chart of the localization of the differentially S-acylated proteins throughout the nephron and collecting system, generated from the Kidney Cell Explorer open access software (https://github.com/qinzhu/kidneycellexplorer).
HFD represses Acsf2 expression and lipid metabolism
To further investigate the molecular mechanism underlying the protein hypo-acylation in kidneys of HFD fed mice, we inspected the total expression levels of the enzymes involved in S-acylation, including fatty acid-CoA condensation and hydrolysis, as well as of the enzymes involved in S-acylation/deacylation cycle. The two most abundant (zDHHC) motif-containing S-acyltransferases, Zdhhc9 and Zdhhc17, were significantly upregulated in the kidney of mice fed a HFD, whereas the two main acyl protein thioesterases, Apt1 and Apt2 were significantly underrepresented in the HFD group (Fig. 3, Supplementary Fig. 4). Moreover, acyl-CoA Synthetase Family Member 2 (Acsf2), one of the highly expressed acyl-CoA synthetases in renal tubular cells34 was significantly repressed in the HFD group (Fig. 3, Supplementary Fig. 4). These results suggest that the hypo-acylated proteome could be attributable to a reduced acyl-CoA synthesis. CoA-conjugation and activation of fatty acids is necessary for their incorporation into the de novo synthesized lipids. We probed whether lipid levels could be used as a readout for lower acyl-CoA availability in the kidneys of HFD fed mice. Mammalian lipidomes represent fatty acids of varying chain lengths, unsaturation levels and modifications. Palmitic and stearic acid, the most abundant identified protein acylation derivatives29, are also abundant fatty acids in membrane lipids35. We reasoned, if lower Acsf2 activity is linked directly to palmitoyl-CoA or stearoyl-CoA generation, we might observe changes in lipid species that directly involve these acyl-CoA’s. Indeed we observed a stark difference in lipid levels from membrane lipidomes of kidneys, involving abundant phospholipids and sphingolipids. Most identified lipids species were observed to decrease (Fig. 4). A comparison of lipid species between mice that are fed a chow or HFD by a volcano plot showed significant decrease in saturated and monounsaturated ceramides (Cer) and sphingomyelin (SM) species that carry conjugated palmitic acid (Fig. 4b, c). Similarly, changes were observed in C16:0 lyso phospholipids (LPs) (Fig. 4b, c). Unlike other phospholipids that carry multiple fatty acids, these species are unambiguously detected in our LC–MS workflow.

Total expression level of S-acylation-related enzymes. (a, b) Heatmap cluster analysis and scatter dot plots of the relative total abundance of the indicated proteins in the input fraction (total protein) from crude membranes isolated from the kidney of CD and HFD mice. In all scatter plots, each dot represents one individual sample. Comparisons of the means was performed by unpaired Student’s t-test.

Diet-induced renal lipid signature shift. (a) Heatmap cluster analysis of the top 70 lipid species in the crude membrane fraction isolated from the kidney of CD and HFD groups. Absolute levels of each species were measured relative to an internal lipid standard. (b) Volcano plot comparing the 382 lipid species identified by LC–MS/MS between CD and HFD groups. The fold-change of concentration (log2 FC) is reported on the x-axis, the significance (-log10 FDR) on the y-axis. The vertical and horizontal dotted lines show the cut-off of fold-change = ± 0.5, and of FDR = 0.1, respectively. (c) Scatter dot plots of the total relative abundance of the indicated lipids in crude membranes isolated from the kidney of CD and HFD mice. In all scatter dot plots, absolute levels of each lipid species relative to an internal lipid standard are shown. Each dot represents one individual sample. Comparisons of the means was performed by unpaired Student’s t-test.
S-acylation influences megalin protein expression
In disease enrichment analysis (disgenet2r)36, diabetes and obesity were among the most represented kidney-related diseases (Fig. 5a, b). GO enrichment analysis of the on/off S-acylated proteome (Fig. 5c) and the Integrated S-acylated proteome (Fig. 5d) indicated that intracellular sorting machinery and vesicle-mediated transport (e.g., endocytosis) are main targets of the S-acylation machinery. Protein/polypeptide reabsorption from the proximal tubular fluid require endocytosis primarily mediated by the multiligand receptor low-density lipoprotein receptor protein 2 (LRP2), called megalin37,38,39,40,41. The kidney of the mice fed a HFD, which are characterized by elevated albuminuria (Fig. 1c), displayed a marked decrease in the total and S-acylated megalin level (Fig. 6a). Interestingly, the swisspalm database predicted several S-acylation sites within the murine and the human megalin protein sequence (Fig. 6b). Moreover, human megalin was previously found S-acylated in human prostate cancer LNCaP cells42. To assess the link between S-acylation and megalin protein expression, we evaluated the effect of the S-acylation inhibitor 2-bromopalmitate (2-BP) on megalin protein levels in vitro. HEK293 cells were previously shown to express megalin43. 2-BP, at 100 μM supplemented concentration, was well tolerated by HEK293 cells and significantly inhibited the S-acylation of caveolin-1 and flotillin-1, two S-acylated proteins highly expressed in HEK293 cells (Supplementary Fig. 5). Under these conditions, megalin protein levels were markedly reduced (Fig. 6c and Supplementary Fig. 6). To further confirm that inhibition of S-acylation caused a reduction in megalin surface expression, we assessed the ability of HEK293 cells to bind [3H]gentamicin, a substrate of megalin44,45. Pre-incubation with 2-BP significantly decreased the [3H]gentamicin bound fraction (Fig. 6d).

Pathway and disease enrichment analysis. (a, b) DisGeNET (disgenet2r package) kidney-related disease enrichment analysis with respect to the main site of expression of the differentially S-acylated proteins. The significance (FDR) of enrichment gradually increases from light to dark color, and the size of the dots indicates the number of genes contained in the corresponding pathway. (c, d) Circle plot of GO enrichment analysis. Each spot in the circle represents an S-acylated protein, and the outer circle refers to significantly enriched biological pathways. The inner circle shows the Z-score, the color intensity corresponds to the value of the Z-score. Top-ten significantly enriched GO pathways are listed.

S-acylated and total protein level of megalin. (a) Megalin position in Volcano plots. The relative abundance (log2 FC) is reported on the x-axis, the significance (-log10 FDR) on the y-axis. The vertical and horizontal lines show the cut-off of fold-change = ± 1.0, and of FDR = 0.1, respectively. (b) Computational prediction of S-acylated cysteine residues of megalin protein sequence using the SwissPalm open access database (https://swisspalm.org). (c) Representative Western blotting of megalin and Na+/K+-ATPase expression in HEK293 cells incubated for 48 h with 100 μM 2-BP. Densitometric quantification of the megalin band normalized by that of the Na+/K+-ATPase. (d) Gentamicin surface bound fraction in HEK293 cells incubated for 48 h with 2-BP. In scatter plots, each data point represents one independent experiment. The indicated p-values were calculated from the unpaired Student’s t-test.
Discussion
Our study in mice with HFD-induced albuminuria showed that the protein levels of renal Zdhhcs and Apts are, respectively, higher and lower in the injured than in the normal kidney. It has been recently shown that kidney biopsies from patients with CKD as well as the kidneys of unilateral ureter obstruction (UUO) or ischemia/reperfusion (IR) mice were characterized by a concomitant downregulation of Zdhhc9 and upregulation of Apt130. Moreover, overexpression of Zdhhc9 or pharmacological inhibition of Apt1 protected the animals against unilateral ureter obstruction (UUO) or ischemia/reperfusion (IR)-induced renal fibrosis through regulating β-catenin S-acylation30. Taken together, the data suggest that the induction of the S-acylation machinery might represent an attempt at protection during the early stage of the kidney injury, when the kidney function (eGFR) is still within the normal range. However, in the presence of a more advanced damage with reduced kidney function and fibrosis, the S-acylation is also repressed, perhaps contributing to the progressive and irreversible loss of kidney function. It is well known that the transforming growth factor-β (TGF-β) signalling pathway is a key mediator of the fibrotic process46 and that the intracellular signaling by TGF-β is inhibited by inhibitory Smad proteins in a feedback control mechanism. Notably, S-acylation of inhibitory Smads is required to inhibit TGF-β-driven fibrosis47,48.
Lipidomic analysis suggested decrease in levels of lipid molecular species containing long-chain fatty acids, such as conjugated palmitic acid. The differences were the most striking for Cer or their downstream SM metabolites (Fig. 4). Mammalian Cer are composed of a long chain base (LCB) of 18 carbons, and fatty acids of variable (C16 to C26) length. LCBs themselves are derived from palmitoyl CoA and L-serine, in a reaction catalysed by the the serine palmitoyl transferases49. Thus relative changes in activated fatty acids such as palmitoyl-CoA are expected to be reflected strongest by C16-fatty acids containing sphingolipid species (Fig. 4b, c). Arguably, the repression of renal Acsf2 induced by HFD represents a protective mechanism against the ongoing renal injury. Indeed, ACSF2 is involved in ferroptosis and the silencing thereof is able to rescue from erastin-induced ferroptosis50. Ferroptotic death is characterized by the overwhelming accumulation of oxidized lipids. It has been proposed that ACSF2 catalyzes the synthesis of lipid precursors that are necessary for the execution of the cell death50. In the kidney, ferroptosis mediates renal tubule necrosis51. The expression of ACSF2 was found increased in the kidneys of db/db mice and individuals with diabetic nephropathy52, while ACSF2 knockdown increases renal tubular cell resistance to ferroptosis, and confers renoprotection in IR-induced acute kidney injury34. However, it is likely that Acsf2 downregulation and the overall decrease in levels of lipid molecular species containing long-chain fatty acids, resulted in renal protein hypoacylation in mice fed a HFD, despite the induction and repression of Zdhhc and Apt protein levels, respectively.
Protein S-acylation acts as a facilitator for preparing and sorting anterograde cargo to cholesterol-sphingolipid-rich “raft-like” membranes53,54. In line with the proposed role, the shortage of acyl-CoA, required for protein S-acylation, has a major impact on endosomal and vesicular transport. Notably, endocytosis plays a key role in the reabsorption of albumin and low-molecular-weight plasma proteins that are filtered through the glomerular basement membrane, avoiding the loss of proteins and of the ligands thereof55. Megalin is a single transmembrane domain protein with a complex binding pocket conferring polyspecificity for a wide range of proteins, including albumin38,39,56, insulin37, haemoglobin40, vitamin D-binding protein55, and retinol-binding protein41. Upon binding, megalin internalizes into coated vesicles, which are subsequently delivered to endosomes and further to lysosomes for ligand processing and receptor degradation or recycling. Patients with Donnai-Barrow syndrome (MIM #222,448) carry recessive mutations in the LRP2 gene encoding megalin and display low-molecular weight proteinuria57. Inducible megalin-deficient mice were characterized by a 16-fold higher urinary excretion of albumin than wild-type mice58. Mice fed a HFD were characterized by normal eGFR but albuminuria, a phenotype compatible with a less efficient tubular reabsorption of proteins (Fig. 1). Our in vitro data indicate that inhibition of S-acylation caused a reduction in megalin total protein level. Consistently, the level of both total and S-acylated megalin in the kidneys of mice fed a HFD is markedly lower than that in the kidneys of mice fed a CD. Notably, we have previously shown that female C57/BJ mice fed a CD (11 kcal% Fat) or a HFD (58 kcal% Fat) for 16 weeks, showed a comparable LRP2 mRNA level59. Premature degradation of S-acylated-deficient mutants has been observed for a variety of membrane proteins60,61,62. Moreover, it has been previously shown that S-acylation affects the ubiquitination and the degradation pathway of LRP6, another member of the LDL receptor-related protein (LRP) family63. Based on our findings, we speculate that impaired S-acylation may potentially affect the degradation of megalin (LRP2) (Fig. 7), though further investigations specifically targeting megalin protein are necessary to confirm this hypothesis.

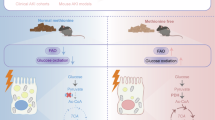
Summary. In mice fed high fat diet (HFD) with elevated albuminuria, the acyl-CoA Synthetase Family Member 2 (ACSF2) renal protein expression level is lower than in mice fed a chow diet (CD), resulting in lower level of acyl-CoA and protein S-acylation. Megalin hypo-acylation causes a reduction in megalin total protein expression level, which may contribute to the elevated albuminuria. Created in BioRender. Visentin, M. (2025) https://BioRender.com/e18k722.
Proteinuria is strongly associated with the risk of CKD progression64,65,66. The extent of short-term proteinuria reduction upon angiotensin converting enzyme (ACE) inhibitor therapy significantly correlated with the reduction in the rate of eGFR decline, progression to end-stage renal disease, and reduction in cardiovascular and heart failure risk, indicating that proteinuria plays an actual pathogenic role in renal disease progression and in cardiovascular co-morbidities risk67,68,69,70. Notably, polymorphisms in the LRP2 gene (encoding megalin) are associated with an increased risk of developing CKD71. Our results suggest that re-activation of the S-acylation machinery might present as an intriguing druggable target to reduce proteinuria. We speculate that supplementation with longer chain fatty acids, which bind with stronger affinity to the long-chain acyl-CoA synthetases (ACSLs) than to ACSF272, might increase acyl-CoA availability and megalin S-acylation, thereby reducing protein loss.
Materials and methods
Animal husbandry and treatment
Animal experiments and protocols conformed to the Guide for the Care and Use of Laboratory Animals (US National Institutes of Health), conformed to the Swiss animal protection laws and were approved by the Cantonal Veterinary Office (ZH145/17). All experiments were performed in accordance with relevant guidelines and regulations. Mice were housed in individually ventilated cages with access to food and water ad libitum. Female C57/BJ mice (Charles River Laboratories, Wilmington, MA, USA), 12 weeks of age, were randomly assigned to a diet with 11 kcal% Fat (chow, D12329; ResearchDiets, NJ, USA) or to a 58 kcal% Fat (high-fat diet, D12331; ResearchDiets, NJ, USA) for 19 weeks. Thereafter 16 h of fasting, spontaneous urination samples were collected during handling prior to euthanization via CO2 inhalation. After euthanization, whole blood was collected and the serum stored at − 80 °C. Kidneys were excised, snap-frozen and stored at − 80 °C.
In vitro S-acylation inhibition
Human embryonic kidney 293 cells (HEK293) (#CRL-1573, American Type Culture Collection, Rockville, MD, USA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, #22,320–022) at 37 °C in a humidified atmosphere of 5% CO2. The medium was supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin (penicillin/streptomycin, #15,140–122), and 10% (v/v) fetal bovine serum (FBS, Biowest, Nuaillé, France, #S181C). Ninety-percent confluent HEK293 cells were incubated for 48 h with 2-bromopalmitate (2-BP) at the extracellular concentration of 100 μM (#238,422, Sigma-Aldrich, St. Louis, MO, USA), and then used in surface binding assay or western blotting.
[3H]Gentamicin surface binding assay
HEK293 cells grown in 12-well plates were placed on ice and washed twice with 2 mL of ice-cold binding buffer (20 mM HEPES, 5.5 mM D-glucose, 116.4 mM NaCl, 5.3 mM KCl, 0.8 mM MgSO4, 2 mM NaH2PO4, with pH adjusted to 7.4 with KOH), and once with acidic buffer (10 mM sodium acetate, 116.4 mM NaCl, adjusted to pH 3.5 with acetic acid), each for 5 min. Cells were then incubated with 100 μM non-labelled gentamicin sulfate (#G3632, Sigma-Aldrich, St. Louis, MO), spiked with 1.6 μCi/mL of [3H]gentamicin (Gentamicin sulfate [3H(G)], Specific Activity: 200 mCi/g, #ART0690, American Radiolabeled Chemicals, St. Louis, MO), in ice-cold binding buffer (pH 7.4) for 20 min. After three washes with ice-cold binding buffer (pH 7.4, 2 mL, 5 min), surface-bound [3H]gentamicin was released by incubation with 0.5 mL of ice-cold acidic buffer (pH 3.5) for 5 min. The acidic buffer was mixed with scintillation liquid Ultima Gold™ fluid (#6,013,329, Perkin Elmer, Waltham, MA, USA) and radioactivity measured with a liquid scintillation analyzer (Tri-Carb 2250 CA, Canberra Packard, Schwadorf, Austria). For normalization, cells were lysed in 0.5 mL of 1% (w/v) Triton X-100 and the bicinchoninic acid protein assay was performed (#UP95424A and #UP95425A, Interchim, Montluçon Cedex, France).
Determination of serum and urine parameters
Serum creatinine was measured using a colorimetric/fluorometric kit (#K625, Biovision, Milpitas, CA, USA). Blood glucose was assessed using a glucometer (#7,101,520, mylife Unio; Ypsomed Distribution AG, Burgdorf, Switzerland). Urinary albumin (#ab108792; Abcam, Cambridge, UK) and serum cystatin C (#ab201280, Abcam, Cambridge, UK) were measured by enzyme-linked immunosorbent assay (ELISA).
Crude membrane preparation from mouse kidney
The isolation of total membrane fractions was performed as described previously73, with some modifications. Briefly, the kidney was resuspended in 5 mM sucrose supplemented with protease inhibitors (#11,836,153,001, Roche Diagnostics GmbH, Mannheim, Germany). After homogenization of the cells with a motor-operated tight-fitting glass-teflon potter, the homogenates were centrifuged for 10 min at 900gav. The supernatant was collected and centrifuged for 20 min at 8,500gav. The supernatant was transferred and centrifuged for 1 h at 100,000gav. The pellet containing the crude membranes was resuspended in 250 mM sucrose with a 25G needle and the protein concentration was determined by bicinchoninic acid protein assay. Samples were stored at − 80℃ until use.
Pull-down of the S-acylated protein fraction
S-acylated proteins were isolated by resin-assisted capture (RAC) using the CAPTUREome™ S-Palmitoylated Protein Kit (Badrilla, Leeds, UK). Briefly, kidney crude membranes (equivalent to 1.5 mg of proteins) were subjected to simultaneous lysis and blocking of free thiols for 4 h at 40 °C. Then, proteins were precipitated with ice-cold acetone and pelleted at 16,000gav for 5 min. The pellet was washed five times with 70% (v/v) ice-cold acetone, and then resuspended in binding buffer. An aliquot (Input fraction) was collected and kept on ice. The remainder was subjected to thioester bond cleavage and resin capture. Then, the samples were washed with binding buffer and then with 50 mM triethylammonium bicarbonate (TEAB), and prepared for proteomic analysis on the same day.
Proteomics sample preparation and data acquisition
For total proteome analysis, samples were reduced and alkylated with Tris(2-carboxyethyl)phosphine and 2-Chloroacetamide to a final concentration of 5 mM and 15 mM, respectively. The samples were shaken at 700 rpm for 30 min at 30 °C. Then samples were diluted with pure ethanol to reach a final concentration of 60% EtOH (v/v), mixed with carboxylated magnetic beads (hydrophobic and hydrophilic) using the KingFisher Flex System (ThermoFisher Scientific, Waltham, MA, USA). After 30-min incubation at room temperature, samples were washed 3 times with 80% EtOH (v/v). For the enzymatic digestion the beads were added to trypsin in 50 mM TEAB. Samples were digested overnight at 37 °C. The supernatant was removed, and the remaining peptides were extracted from beads with H2O. The two elutions were combined and concentrated. The peptide concentration was estimated using the Lunatic UV/Vis absorbance spectrometer (Unchained Lab, Pleasanton, CA, USA). The resin containing the S-acylated protein fraction and the Input fraction were digested overnight at 37 °C with 20 ng/μl of trypsin. The supernatants were collected, dried, and desalted using the Phoenix kit (PreOmics, Planegg, Germany). The peptides were re-dissolved in 3% (v/v) acetonitrile / 0.1% (v/v) formic acid, and the peptide concentration was estimated with the Lunatic UV/Vis absorbance spectrometer (Unchained Lab, Pleasanton, CA, USA). Peptides were separated on an M-class UPLC (Waters, Milford, MA, USA) and analyzed on an Orbitrap Fusion Lumos Tribrid mass spectrometer (ThermoFisher Scientific, Waltham, MA, USA). Acquisition and analysis of mass spectrometric data are described in the Supplementary Materials and Methods.
Lipidomics sample preparation and data acquisition
Total lipids were extracted from crude membranes (equivalent to 200 μg of proteins) with 1 mL Methanol/MTBE (methyltert-butyl ether)/Chloroform (MMC) [4:3:3; (v/v/v)] at 37 °C (250gav, 60 min) as described previously74. Internal lipid standards include D7SA (d18:0), D7SO (d18:1), dhCer (d18:0/12:0), ceramide (d18:1/12:0), glucosylceramide (d18:1/8:0), and SM (d18:1/18:1(D9)). The single-phase supernatant was collected, dried under N2 and dissolved in 70 μL methanol. Untargeted lipid analysis was performed on a high-resolution Q Exactive MS analyzer (ThermoFisher Scientific, Waltham, MA, USA) as described earlier74.
SDS gel electrophoresis and Western blotting
Proteins were resolved onto a gradient 3–8% NuPAGE Tris–Acetate gel (#EA0375PK2, ThermoFisher Scientific, Waltham, MA, USA). The gel was cut and the upper part was electroblotted for 2.5 h in a transfer buffer containing 10% methanol. The bottom part was electroblotted for 70–75 min in a transfer buffer containing 20% methanol. After the transfer, the PVDF membranes (#10,600,023, Cytiva, Marlborough, MA, USA) were blocked in 5% (w/v) skimmed milk dissolved in PBS supplemented with 0.1% (w/v) Tween-20 (PBS-T). Membranes were then incubated overnight at 4 °C with the respective primary antibodies. The rabbit anti-megalin (#ab313741, Abcam, Cambridge, UK) and the mouse anti-Na+/K+ ATPase (#ab7671, Abcam, Cambridge, UK) were diluted 1:1,000. The next day, membranes were washed three times, each of 5 min, at room temperature with PBS-T and then incubated for 1 h at room temperature with an ECL™ Anti-Mouse IgG horseradish peroxidase linked whole antibody (1:3,000, #NA931, Sigma-Aldrich, St. Louis, MO, USA) or an ECL™ Anti-Rabbit IgG horseradish peroxidase-linked whole antibody (1:30,000, #G21234, ThermoFisher Scientific, Waltham, MA, USA). After three washing steps with PBS-T, each of 10 min, membranes were exposed to the SuperSignal West Femto Maximum Sensitivity Substrate (34,096, ThermoFisher Scientific, Waltham, MA, USA), and the signal acquired in a Fusion FX7 instrument (Vilber Lourmat, Eberhardzell, Germany).
Data analysis
All plots and statistical analyses were performed with GraphPad Prism Version 9 or the open-access language programming software R Version 4.3.1 (R Project for Statistical Computing). The figure legends provide details on the statistical analysis. Proteomics data analysis is described in the Supplementary Materials and Methods. The mass spectrometry proteomics data were managed using the local laboratory information management system b-fabric (LIMS)75. The acquired DIA MS data were processed for identification and quantification using DIANN76. The R package prolfqua77 was used to analyze the differential expression for all quantifiable proteins.
Data availability statement
We uploaded the raw data and the DIANN identification and quantification results to the PRoteomics IDEntification Database (PRIDE) Projekt number PXD052634. The files 2371711.zip and 2371714.zip contain the DIA-NN identification and quantification results for the total proteome and enriched palmitoylated samples, respectively. The files DiaNN_total_proteome.zip and DiaNN_palmitoylated_proteins.zip contain the results of the differential expression analysis using the R prolfqua package, respectively. Finally, the file IntegrateProteomeAndEnriched.zip includes the analysis results to compare the differences in the differential expression levels of the enriched and whole proteome samples.
References
Levey, A. S. et al. Nomenclature for kidney function and disease: Report of a Kidney Disease: Improving Global Outcomes (KDIGO) Consensus Conference. Kidney Int. 97, 1117–1129. https://doi.org/10.1016/j.kint.2020.02.010 (2020).
Thomas, M. C. et al. Diabetic kidney disease. Nat. Rev. Dis Primers 1, 15018. https://doi.org/10.1038/nrdp.2015.18 (2015).
Jha, V. et al. Chronic kidney disease: Global dimension and perspectives. Lancet 382, 260–272. https://doi.org/10.1016/S0140-6736(13)60687-X (2013).
Adler, A. I. et al. Development and progression of nephropathy in type 2 diabetes: The United Kingdom Prospective Diabetes Study (UKPDS 64). Kidney Int. 63, 225–232. https://doi.org/10.1046/j.1523-1755.2003.00712.x (2003).
Wong, T. Y., Shankar, A., Klein, R. & Klein, B. E. Retinal vessel diameters and the incidence of gross proteinuria and renal insufficiency in people with type 1 diabetes. Diabetes 53, 179–184. https://doi.org/10.2337/diabetes.53.1.179 (2004).
Valmadrid, C. T., Klein, R., Moss, S. E. & Klein, B. E. The risk of cardiovascular disease mortality associated with microalbuminuria and gross proteinuria in persons with older-onset diabetes mellitus. Arch. Internal Med. 160, 1093–1100. https://doi.org/10.1001/archinte.160.8.1093 (2000).
Klein, S., Gastaldelli, A., Yki-Jarvinen, H. & Scherer, P. E. Why does obesity cause diabetes?. Cell Metab. 34, 11–20. https://doi.org/10.1016/j.cmet.2021.12.012 (2022).
Guasch-Ferre, M. et al. Total and subtypes of dietary fat intake and risk of type 2 diabetes mellitus in the Prevencion con Dieta Mediterranea (PREDIMED) study. Am. J. Clin. Nutrit. 105, 723–735. https://doi.org/10.3945/ajcn.116.142034 (2017).
Odegaard, J. I. & Chawla, A. Pleiotropic actions of insulin resistance and inflammation in metabolic homeostasis. Science 339, 172–177. https://doi.org/10.1126/science.1230721 (2013).
Gai, Z. et al. Lipid accumulation and chronic kidney disease. Nutrients https://doi.org/10.3390/nu11040722 (2019).
Mortality, G. B. D. & Causes of Death, C. Global, regional, and national age-sex specific all-cause and cause-specific mortality for 240 causes of death, 1990–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 385, 117–171, https://doi.org/10.1016/S0140-6736(14)61682-2 (2015).
Yamagata, K. et al. Risk factors for chronic kidney disease in a community-based population: A 10-year follow-up study. Kidney Int. 71, 159–166. https://doi.org/10.1038/sj.ki.5002017 (2007).
Lim, A. Diabetic nephropathy - complications and treatment. Int. J. Nephrol. Renovasc. Dis. 7, 361–381. https://doi.org/10.2147/IJNRD.S40172 (2014).
Lewis, E. J., Hunsicker, L. G., Bain, R. P. & Rohde, R. D. The effect of angiotensin-converting-enzyme inhibition on diabetic nephropathy. The Collaborative Study Group. New Engl. J. Med. 329, 1456–1462. https://doi.org/10.1056/NEJM199311113292004 (1993).
Brenner, B. M. et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. New Engl. J. Med. 345, 861–869. https://doi.org/10.1056/NEJMoa011161 (2001).
Holman, R. R., Paul, S. K., Bethel, M. A., Matthews, D. R. & Neil, H. A. 10-year follow-up of intensive glucose control in type 2 diabetes. New Engl. J. Med. 359, 1577–1589. https://doi.org/10.1056/NEJMoa0806470 (2008).
Zoungas, S. et al. Follow-up of blood-pressure lowering and glucose control in type 2 diabetes. New Engl. J. Med. 371, 1392–1406. https://doi.org/10.1056/NEJMoa1407963 (2014).
Yen, C. L. et al. Fibrate and the risk of cardiovascular disease among moderate chronic kidney disease patients with primary hypertriglyceridemia. Front. Endocrinol. Lausanne 15, 1333553. https://doi.org/10.3389/fendo.2024.1333553 (2024).
Perkovic, V. et al. Effects of semaglutide on chronic kidney disease in patients with Type 2 diabetes. New Engl. J. Med. https://doi.org/10.1056/NEJMoa2403347 (2024).
Kang, H. M. et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 21, 37–46. https://doi.org/10.1038/nm.3762 (2015).
Zadoorian, A., Du, X. & Yang, H. Lipid droplet biogenesis and functions in health and disease. Nat. Rev. Endocrinol. 19, 443–459. https://doi.org/10.1038/s41574-023-00845-0 (2023).
van der Rijt, S., Leemans, J. C., Florquin, S., Houtkooper, R. H. & Tammaro, A. Immunometabolic rewiring of tubular epithelial cells in kidney disease. Nat. Rev. Nephrol. 18, 588–603. https://doi.org/10.1038/s41581-022-00592-x (2022).
Herman-Edelstein, M., Scherzer, P., Tobar, A., Levi, M. & Gafter, U. Altered renal lipid metabolism and renal lipid accumulation in human diabetic nephropathy. J. Lipid Res. 55, 561–572. https://doi.org/10.1194/jlr.P040501 (2014).
Nuskova, H. et al. Competition for cysteine acylation by C16:0 and C18:0 derived lipids is a global phenomenon in the proteome. J. Biol. Chem. 299, 105088. https://doi.org/10.1016/j.jbc.2023.105088 (2023).
Thorne, R. F. et al. Palmitoylation of CD36/FAT regulates the rate of its post-transcriptional processing in the endoplasmic reticulum. Biochimica et biophysica acta 1298–1307, 2010. https://doi.org/10.1016/j.bbamcr.2010.07.002 (1803).
Ren, W., Sun, Y. & Du, K. Glut4 palmitoylation at Cys223 plays a critical role in Glut4 membrane trafficking. Biochem. Biophys. Res. Commun. 460, 709–714. https://doi.org/10.1016/j.bbrc.2015.03.094 (2015).
Pouliot, J. F. & Beliveau, R. Palmitoylation of the glucose transporter in blood-brain barrier capillaries. Biochimica et biophysica acta 1234, 191–196. https://doi.org/10.1016/0005-2736(94)00272-q (1995).
Zhao, Y., McCabe, J. B., Vance, J. & Berthiaume, L. G. Palmitoylation of apolipoprotein B is required for proper intracellular sorting and transport of cholesteroyl esters and triglycerides. Mol. Biol. Cell 11, 721–734. https://doi.org/10.1091/mbc.11.2.721 (2000).
Chamberlain, L. H. & Shipston, M. J. The physiology of protein S-acylation. Physiol. Rev. 95, 341–376. https://doi.org/10.1152/physrev.00032.2014 (2015).
Gu, M. et al. Palmitoyltransferase DHHC9 and acyl protein thioesterase APT1 modulate renal fibrosis through regulating beta-catenin palmitoylation. Nat. Commun. 14, 6682. https://doi.org/10.1038/s41467-023-42476-z (2023).
Schunk, S. J., Floege, J., Fliser, D. & Speer, T. WNT-beta-catenin signalling - a versatile player in kidney injury and repair. Nat. Rev. Nephrol. 17, 172–184. https://doi.org/10.1038/s41581-020-00343-w (2021).
Yang, Y., Smith, D. L. Jr., Keating, K. D., Allison, D. B. & Nagy, T. R. Variations in body weight, food intake and body composition after long-term high-fat diet feeding in C57BL/6J mice. Obesity 22, 2147–2155. https://doi.org/10.1002/oby.20811 (2014).
Ransick, A. et al. Single-cell profiling reveals sex, lineage, and regional diversity in the mouse kidney. Dev. Cell 51, 399–413. https://doi.org/10.1016/j.devcel.2019.10.005 (2019).
Shi, H. et al. Inhibition of ACSF2 protects against renal ischemia/reperfusion injury via mediating mitophagy in proximal tubular cells. Free Radic. Biol. Med. 198, 68–82. https://doi.org/10.1016/j.freeradbiomed.2023.02.003 (2023).
Cockcroft, S. Mammalian lipids: Structure, synthesis and function. Essays Biochem 65, 813–845. https://doi.org/10.1042/EBC20200067 (2021).
Pinero, J. et al. The DisGeNET knowledge platform for disease genomics: 2019 update. Nucleic Acids Res. 48, D845–D855. https://doi.org/10.1093/nar/gkz1021 (2020).
Orlando, R. A. et al. Megalin is an endocytic receptor for insulin. J. Am. Soc. Nephrol. JASN 9, 1759–1766. https://doi.org/10.1681/ASN.V9101759 (1998).
Zhai, X. Y. et al. Cubilin- and megalin-mediated uptake of albumin in cultured proximal tubule cells of opossum kidney. Kidney Int. 58, 1523–1533. https://doi.org/10.1046/j.1523-1755.2000.00314.x (2000).
Cui, S., Verroust, P. J., Moestrup, S. K. & Christensen, E. I. Megalin/gp330 mediates uptake of albumin in renal proximal tubule. Am. J. Physiol. 271, F900-907 (1996).
Gburek, J. et al. Megalin and cubilin are endocytic receptors involved in renal clearance of hemoglobin. J. Am. Soc. Nephrol. JASN 13, 423–430. https://doi.org/10.1681/ASN.V132423 (2002).
Christensen, E. I. et al. Evidence for an essential role of megalin in transepithelial transport of retinol. J. Am. Soc. Nephrol. JASN 10, 685–695. https://doi.org/10.1681/ASN.V104685 (1999).
Zhou, B. et al. Low-background acyl-biotinyl exchange largely eliminates the coisolation of non-S-acylated proteins and enables deep S-acylproteomic analysis. Anal. Chem. 91, 9858–9866. https://doi.org/10.1021/acs.analchem.9b01520 (2019).
Gao, Y., Zhou, S., Luu, S. & Glowacki, J. Megalin mediates 25-hydroxyvitamin D(3) actions in human mesenchymal stem cells. FASEB J. 33, 7684–7693. https://doi.org/10.1096/fj.201802578R (2019).
Dagil, R., O’Shea, C., Nykjaer, A., Bonvin, A. M. & Kragelund, B. B. Gentamicin binds to the megalin receptor as a competitive inhibitor using the common ligand binding motif of complement type repeats: insight from the nmr structure of the 10th complement type repeat domain alone and in complex with gentamicin. J. Biol. Chem. 288, 4424–4435. https://doi.org/10.1074/jbc.M112.434159 (2013).
Nagai, J., Tanaka, H., Nakanishi, N., Murakami, T. & Takano, M. Role of megalin in renal handling of aminoglycosides. Am. J. Physiol. Renal Physiol. 281, F337-344. https://doi.org/10.1152/ajprenal.2001.281.2.F337 (2001).
Akhmetshina, A. et al. Activation of canonical Wnt signalling is required for TGF-beta-mediated fibrosis. Nat. Commun. 3, 735. https://doi.org/10.1038/ncomms1734 (2012).
Voytyuk, O., Ohata, Y., Moustakas, A., Ten Dijke, P. & Heldin, C. H. Smad7 palmitoylation by the S-acyltransferase zDHHC17 enhances its inhibitory effect on TGF-beta/Smad signaling. J. Biol. Chem. 300, 107462. https://doi.org/10.1016/j.jbc.2024.107462 (2024).
Li, W. et al. Membrane targeting of inhibitory Smads through palmitoylation controls TGF-beta/BMP signaling. Proc. Natl. Acad. Sci. USA 114, 13206–13211. https://doi.org/10.1073/pnas.1710540114 (2017).
Lone, M. A. et al. Subunit composition of the mammalian serine-palmitoyltransferase defines the spectrum of straight and methyl-branched long-chain bases. Proc. Natl. Acad. Sci. USA 117, 15591–15598. https://doi.org/10.1073/pnas.2002391117 (2020).
Dixon, S. J. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. https://doi.org/10.1016/j.cell.2012.03.042 (2012).
Linkermann, A. et al. Regulated cell death in AKI. J. Am. Soc. Nephrol. JASN 25, 2689–2701. https://doi.org/10.1681/ASN.2014030262 (2014).
Chen, J. et al. ACSF2 and lysine lactylation contribute to renal tubule injury in diabetes. Diabetologia https://doi.org/10.1007/s00125-024-06156-x (2024).
Ernst, A. M., Toomre, D. & Bogan, J. S. Acylation - a new means to control traffic through the golgi. Front. Cell Dev. Biol. 7, 109. https://doi.org/10.3389/fcell.2019.00109 (2019).
Ohno, Y., Kihara, A., Sano, T. & Igarashi, Y. Intracellular localization and tissue-specific distribution of human and yeast DHHC cysteine-rich domain-containing proteins. Biochimica et biophysica acta 1761, 474–483. https://doi.org/10.1016/j.bbalip.2006.03.010 (2006).
Nykjaer, A. et al. An endocytic pathway essential for renal uptake and activation of the steroid 25-(OH) vitamin D3. Cell 96, 507–515. https://doi.org/10.1016/s0092-8674(00)80655-8 (1999).
Ren, Q. et al. Distinct functions of megalin and cubilin receptors in recovery of normal and nephrotic levels of filtered albumin. Am. J. Physiol. Renal Physiol. 318, F1284–F1294. https://doi.org/10.1152/ajprenal.00030.2020 (2020).
van der Wijst, J., Belge, H., Bindels, R. J. M. & Devuyst, O. Learning physiology from inherited kidney disorders. Physiol. Rev. 99, 1575–1653. https://doi.org/10.1152/physrev.00008.2018 (2019).
Mori, K. P. et al. Increase of total nephron albumin filtration and reabsorption in diabetic nephropathy. J. Am. Soc. Nephrol. JASN 28, 278–289. https://doi.org/10.1681/ASN.2015101168 (2017).
Gai, Z. et al. Organic Cation Transporter 2 (OCT2-SLC22A2) overexpression may confer an increased risk of gentamicin-induced nephrotoxicity. Antimicrob. Agents Chemotherapy https://doi.org/10.1128/AAC.00907-16 (2016).
Percherancier, Y. et al. Palmitoylation-dependent control of degradation, life span, and membrane expression of the CCR5 receptor. J. Biol. Chem. 276, 31936–31944. https://doi.org/10.1074/jbc.M104013200 (2001).
Maeda, A. et al. Palmitoylation stabilizes unliganded rod opsin. Proc. Natl. Acad. Sci. USA 107, 8428–8433. https://doi.org/10.1073/pnas.1000640107 (2010).
Abrami, L., Leppla, S. H. & van der Goot, F. G. Receptor palmitoylation and ubiquitination regulate anthrax toxin endocytosis. J. Cell Biol. 172, 309–320. https://doi.org/10.1083/jcb.200507067 (2006).
Abrami, L., Kunz, B., Iacovache, I. & van der Goot, F. G. Palmitoylation and ubiquitination regulate exit of the Wnt signaling protein LRP6 from the endoplasmic reticulum. Proc. Natl. Acad. Sci. USA 105, 5384–5389. https://doi.org/10.1073/pnas.0710389105 (2008).
Iseki, K., Iseki, C., Ikemiya, Y. & Fukiyama, K. Risk of developing end-stage renal disease in a cohort of mass screening. Kidney Int. 49, 800–805. https://doi.org/10.1038/ki.1996.111 (1996).
Ruggenenti, P. et al. Proteinuria predicts end-stage renal failure in non-diabetic chronic nephropathies. The "Gruppo Italiano di Studi Epidemiologici in Nefrologia" (GISEN). Kidney international. Supplement 63, S54–57 (1997).
Peterson, J. C. et al. Blood pressure control, proteinuria, and the progression of renal disease. The Modification of Diet in Renal Disease Study. Ann. Internal Med. 123, 754–762. https://doi.org/10.7326/0003-4819-123-10-199511150-00003 (1995).
Ruggenenti, P., Perna, A., Remuzzi, G. & Investigators, G. G. Retarding progression of chronic renal disease: The neglected issue of residual proteinuria. Kidney Int. 63, 2254–2261. https://doi.org/10.1046/j.1523-1755.2003.00033.x (2003).
Bjorck, S., Mulec, H., Johnsen, S. A., Norden, G. & Aurell, M. Renal protective effect of enalapril in diabetic nephropathy. Bmj 304, 339–343. https://doi.org/10.1136/bmj.304.6823.339 (1992).
de Zeeuw, D. et al. Albuminuria, a therapeutic target for cardiovascular protection in type 2 diabetic patients with nephropathy. Circulation 110, 921–927. https://doi.org/10.1161/01.CIR.0000139860.33974.28 (2004).
Ibsen, H. et al. Reduction in albuminuria translates to reduction in cardiovascular events in hypertensive patients: losartan intervention for endpoint reduction in hypertension study. Hypertension 45, 198–202. https://doi.org/10.1161/01.HYP.0000154082.72286.2a (2005).
Wuttke, M. et al. A catalog of genetic loci associated with kidney function from analyses of a million individuals. Nat. Genet. 51, 957–972. https://doi.org/10.1038/s41588-019-0407-x (2019).
Kuwata, H. & Hara, S. Role of acyl-CoA synthetase ACSL4 in arachidonic acid metabolism. Prostaglandins Other Lipid Mediators 144, 106363. https://doi.org/10.1016/j.prostaglandins.2019.106363 (2019).
Visentin, M. et al. Fluorocholine Transport Mediated by the Organic Cation Transporter 2 (OCT2, SLC22A2): Implication for Imaging of Kidney Tumors. Drug Metab. Disposit. 46, 1129–1136. https://doi.org/10.1124/dmd.118.081091 (2018).
Lone, M. A. et al. SPTLC1 variants associated with ALS produce distinct sphingolipid signatures through impaired interaction with ORMDL proteins. J. Clin. Investig. https://doi.org/10.1172/JCI161908 (2022).
Panse, C., Trachsel, C. & Turker, C. Bridging data management platforms and visualization tools to enable ad-hoc and smart analytics in life sciences. J. Integr. Bioinform. https://doi.org/10.1515/jib-2022-0031 (2022).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods 17, 41–44. https://doi.org/10.1038/s41592-019-0638-x (2020).
Wolski, W. E. et al. prolfqua: A comprehensive R-package for proteomics differential expression analysis. J. Proteome Res. 22, 1092–1104. https://doi.org/10.1021/acs.jproteome.2c00441 (2023).
Funding
This work was supported by the University Hospital of Zurich. F.X. is the recipient of a scholarship from the Chinese Scholarship Council (#202108370215). M.A.L. is supported by the Swiss Foundation for Research on Muscle Disease (FSRMM) the EMPIRIS foundation, Zurich.
Author information
Authors and Affiliations
Contributions
F.X. and M.V. conceptualized the project; F.X., Z.G., P.G., W.E.W., M.A.L. performed experiments, and data analysis and interpretation; F.X., P.G., W.E.W., M.A.L., M.V. wrote the initial draft of the manuscript; All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
Experiments involving animals are described in accordance with ARRIVE guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xiu, F., Gai, Z., Gehrig, P. et al. The landscape of renal protein S-acylation in mice with lipid-induced nephrotoxicity. Sci Rep 15, 7689 (2025). https://doi.org/10.1038/s41598-025-92530-7
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-92530-7
