
Abstract
Acute myocardial infarction (AMI) is a major contributor to cardiovascular-related mortality, and early diagnosis is crucial for effective treatment and better outcomes. While several biomarkers have been explored for AMI, there remains a need for reliable, non-invasive biomarkers that can accurately differentiate AMI patients from healthy individuals. This study aims to identify potential mRNA biomarkers in peripheral blood that could aid in the diagnosis and monitoring of AMI. We performed transcriptomic analysis of blood samples from 81 individuals, including 16 healthy controls, 58 AMI patients, and 7 post-treated AMI individuals. Through a combination of Sparse Partial Least Squares-Discriminant Analysis (sPLS-DA), random forest (RF), Weighted Gene Co-expression Network Analysis (WGCNA), and LASSO regression, we identified mRNA markers that are significantly correlated with AMI. Specifically, the mRNA expressions of ANKRD52, ART1, NRP2, and PPP1R15A were elevated in AMI patients, whereas BAIAP2L1 and CCNE1 were downregulated. However, while these mRNA biomarkers show potential for distinguishing AMI patients from healthy individuals, further studies are needed to confirm their clinical applicability.
Similar content being viewed by others


Introduction
Acute myocardial infarction (AMI) represents a significant global health challenge, with rising incidence and mortality rates, particularly in regions with low to middle socioeconomic indices1. Early detection of AMI is imperative for the timely initiation of therapeutic interventions to reduce mortality rates. Although troponin and creatine kinase-MB are the conventional markers for AMI diagnosis, their specificity is limited, leading to potential false positives in non-cardiac conditions2. Consequently, the scientific community continues to explore new biomarkers that offer higher sensitivity and specificity for the early detection of AMI.
Previous investigations have explored AMI-specific diagnostic markers through mRNA and single-cell transcriptomics; however, the progression of AMI and cardiac repair is a protracted and dynamic biological process. This complex process spans from the initial transformation of healthy cardiomyocytes under hypoxic conditions, which alters energy metabolism and induces oxidative stress, to the eventual myocardial injury repair. Biomarker levels notably fluctuate throughout these stages, reflecting the dynamic nature of AMI pathophysiology. A study monitoring AMI patients over a 20-week post-event observation period revealed that levels of cardiac-specific miRNA-1 and endothelial-specific miRNA-126 in circulation were significantly elevated compared to healthy control, with a subsequent decline during the observation period3. In ischemic cardiac tissue, HIF-1α protein accumulates persistently, and vascular endothelial growth factor A (VEGFA) is induced in the acute phase of myocardial infarction but not during the chronic phase4. There is an upregulation of insulin-like growth factor 2 (IGF2) and angiopoietin 2 (ANGPT2) in the infarcted region, while expressions of VEGFA and fms-related tyrosine kinase 1 (FLT1, also known as VEGF receptor 1) are diminished5. Single-cell sequencing of peripheral blood has revealed an upregulation of the IL-1 signaling pathway following ST-Elevation Myocardial Infarction (STEMI), indicating an ongoing heart failure process6. Analysis of two GEO high-throughput datasets reveals that post-myocardial infarction, hsa-miR-330-3p exerts regulatory effects on MMP2, leading to macrophage infiltration into atherosclerotic lesions and promoting the secretion of numerous factors such as MMP-2. These factors contribute to endothelial cell damage by cleaving extracellular matrix components, collagen, and elastin, thereby accelerating the formation of intravascular thrombi7. These findings suggest that the potential origin of variability in AMI biomarker studies is attributed to the differing gene expression patterns in myocardial tissues and peripheral blood7. During myocardial infarction, mononuclear cells, macrophages, lymphocytes, and smooth muscle cells accumulates and release of various cytokines and inflammatory biomarkers8. Single-cell transcriptomics provides the necessary resolution to distinguish cell-specific responses, which are often masked in analyses of bulk tissue9. The complexity of myocardial infarction pathophysiology, the dynamic nature of gene expression patterns, and the heterogeneity of patient populations can contribute to the observed variability of identified biomarkers. As myocardial infarction progresses, transcriptional activity undergoes selective regulation, necessitating the identification of more relevant markers through the assessment of myocardial recovery at various post-treatment stages.
The advent of machine learning has markedly altered the stratification of risk and the prediction of mortality in AMI. Chang et al.10 identified a five-feature model including troponin I, HDL cholesterol, HbA1c, anion gap, and albumin as potential biomarkers for the early detection and treatment of AMI. This model utilizes feature selection combined with machine learning techniques, building upon established risk factors for AMI and cardiovascular diseases. A study has corroborated the expression specificity of immune-related genes in AMI, which can facilitate diagnosis in clinical settings through the development of various machine model11. Moreover, the application of machine learning extends beyond biomarkers. Employing algorithms that integrate multimodal data, including electrocardiographic waveforms, demographic information like gender and age, have been shown to enhance the diagnostic accuracy for myocardial ischemia or infarction12. Despite these advances and promising results, characterized by high performance on receiver operating curves, these innovative models necessitate further clinical trials to affirm their applicability and efficacy in real-world medical settings.
In this study, we performed a transcriptomic analysis on peripheral blood samples from AMI patients, individuals seven days post-treatment, and healthy controls. This analysis unveiled AMI-associated transcriptomic signatures, including specific mRNA expression patterns linked to key signaling pathways. Employing a combination of data analytical techniques and machine learning, we executed a multi-step screening process aimed at identifying a set of potential mRNA biomarkers correlated with AMI.
Materials and methods
Study design and sample collection
This study was conducted in accordance with the ethical standards and approval of the Kunming University of Science and Technology (KUST) and the People’s Hospital of Lijiang. All procedures involving human participants were in compliance with the ethical standards of the research committee of KUST and People’s Hospital of Lijiang. This study was conducted in accordance with the principles of the Declaration of Helsinki and informed consent was obtained from all participants involved in this research. For those participants who were unable to provide consent due to their medical condition, consent was obtained from their legal guardians. All subjects were informed about the nature of the study, the procedures involved, potential risks, and their right to withdraw at any time without affecting their medical care.
Patients presenting with AMI were recruited and treated at the People’s Hospital of Lijiang City, Yunnan Province. The inclusion criteria for participation in this study were: (1) a clinical diagnosis of AMI, meeting the standards defined in the “Guidelines for the Diagnosis and Treatment of AMI”; (2) long-term residents living in Lijiang for more than six months; (3) both male and female patients aged 18 years or older; (4) no known hematological diseases or severe liver and kidney dysfunction; (5) no family history of hereditary tumors; (6) no history of radiation therapy or chemotherapy; (7) no occupational exposure to radioactive materials, toxic gases, or other carcinogens; and (8) written informed consent signed by the patient or their legal representative.
Patients were excluded from the study if they met any of the following criteria: (1) a history of diagnosed hematological diseases or severe renal or hepatic dysfunction; (2) recent (within the past six months) coronary intervention treatment (such as stent placement or coronary artery bypass surgery); (3) currently participating in other interventional clinical studies involving AMI; (4) presence of other severe comorbidities, such as active severe infections, advanced malignancies, or immunodeficiency diseases; (5) women who are pregnant or breastfeeding; and (6) any other conditions deemed unsuitable for participation by the researchers, such as poor compliance or significant confounding factors.
Upon confirmation of eligibility, detailed demographic information, chronic disease history, laboratory test results, and diagnostic data were recorded for each patient, and peripheral blood samples were obtained at the time of admission. Peripheral blood was also collected from a subset of seven patients 7–10 days into treatment when their condition had stabilized. The healthy control group was publicly recruited from the local population by our department, consisting of individuals aged 18 or older, excluding those with underlying cardiovascular conditions, pregnant individuals, and patients with cancer. After drawing peripheral blood from participants, each sample was immediately mixed with threefold volume of Trizol (Qiagen, Germany) and stored at -80 °C for subsequent use.
RNA-seq and data analysis
Genomic RNA from all enrolled samples of peripheral blood was extracted using the TRNzol Universal Reagent kit (Tiangen, China). The extracted RNA was quantified and its quality assessed through: (1) Measuring sample concentration with the Qubit 4.0 Fluorometer (Invitrogen, USA) and the Equalbit RNA BR Assay Kit (Vazyme, China). (2) Evaluating RNA integrity using the Qsep1 Bio-fragment Analyzer (Guangding, China). High-quality RNA with a concentration ≥ 80 ng/µl, and a total yield ≥ 1 µg was then used for subsequent library construction. A total of 3 µg of RNA was employed for the library construction process, which included mRNA enrichment with Oligo(dT) magnetic beads (Yesen, China), fragmentation, synthesis of first- and second-strand cDNA, purification with AMPure XP beads (Beckman, USA), end-repair, A-tailing, adaptor ligation, and PCR enrichment. Completed libraries were quantified and quality-assessed using Qubit 4.0, the Agilent 2100 Bioanalyzer (Agilent Technologies, USA), and the Bio-RAD CFX 96 Real-Time System (Biorad, USA), ensuring library concentrations exceeded 10 nM for subsequent sequencing. Sequencing was performed on the NovaSeq 6000 S4 platform (Illumina, USA) using the Illumina NovaSeq 6000 S4 Reagent Kit V1.5, generating PE150 reads. Quality control of all sequencing data was conducted using FastQC software for analysis and filtration. Human sequences were removed by mapping to the human reference genome (GRCh38.p13 https://www.ncbi.nlm.nih.gov/assembly/2334371) using BWA (BurrowsWheeler alignment).
In this study, we conducted various analyses, including RNA-seq analysis, sPLS-DA analysis, WGCNA analysis, and animal analysis. For the detailed methodologies of these analyses, please refer to the Supplementary Methods.
Real‑time quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated using TRIzol (Invitrogen, USA), and 1 µg RNA was used for reverse transcription using a PrimeScript RT Reagent Kit (TaKaRa, JPN) according to the manufacturer’s instructions. RT–qPCR was performed by using TB Green Premix Ex Taq II (TaKaRa, JPN). The samples were processed using a Thermo Q6 Real-Time System. Five genes were selected for validation as potential biomarkers for AMI. Three replicates were run for each sample. The primer sequences are shown in Table.S1.
Statistical analysis
Statistical analyses were performed using R (Version 4.2). Quantitative data were analyzed as appropriate based on their distribution, and categorical variables were assessed accordingly. A p value of ≤ 0.05 was considered statistically significant.
For detailed methodologies related to statistical tests, including the treatment of normally and non-normally distributed data, as well as the handling of categorical variables, please refer to the Supplementary Methods ‘Statistical Analysis’ part.
Results
Subject characteristics
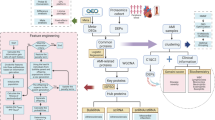
Table 1 presents the demographic and clinical profiles of the study’s participants, comparing healthy controls, AMI patients, and those receiving AMI treatment. The study workflow is illustrated in Fig. 1. A total of 87 participants were recruited, and clinical data - including age, sex, comorbidities, and clinical AMI indicators were collected. Peripheral blood samples were obtained from all subjects, followed by transcriptomic sequencing. To ensure data accuracy, individuals with incomplete clinical information (4 from the control group) and those with poor sequencing quality (2 from the AMI group) were excluded. Ultimately, 58 confirmed AMI patients, 7 post-treatment AMI patients, and 16 healthy controls were included in the analysis. After sequencing, a series of analytical steps were performed, including differential expression analysis, WGCNA, sPLS-DA, and random forest analysis, culminating in LASSO regression for feature selection and validation in a mouse model. This integrated approach aimed to identify key gene expression differences across groups to uncover potential biomarkers and therapeutic targets for AMI. Cardiac biomarkers including Ctn1, CKMB, and MYO were measured between 7 and 10 days post-treatment, while other data were collected at hospital admission. The AMI group (56.21 ± 12.20) was older than the control group (33.94 ± 5.69) (p < 0.001). Given the multi-ethnic composition of the Lijiang region in Yunnan, the proportions of minority groups were catalogued within different cohorts, with the majority being Han, followed by Bai, Naxi, Tibetan, Yi and others ethnicities. Hypertension prevalence was higher in the AMI group (41.38%) compared to the control group (0.00%), while the prevalence of diabetes showed no significant difference across groups (P = 0.136). Smoking history was significantly more common in the AMI group (60.34%), compared to an absence of smoking history in the control group (0.00%) (P < 0.001). The primary diagnosis within the AMI group was Inferior STEMI (55.17%). After treatment, the levels of Ctn1, CKMB, and MYO significantly decreased(P < 0.001), suggesting therapeutic efficacy.

Presented the schematic diagram of the study.
Pairwise differential expression analysis reveals distinct mRNA signatures and treatment effects in AMI
In our RNA-seq data analysis, we compared gene expression across three groups of participants: (1) differences in gene expression between AMI patients and healthy controls, (2) gene expression in patients post-treatment compared to AMI patients, and (3) comparisons of gene expression between post-treatment patients and healthy controls. Initially, Principal component analysis (PCA) was employed to investigate mRNA profiles among individual samples and groups. This unsupervised multivariate approach (Fig. 2A) generates principal component axes capable of elucidating the variability in the data without prior knowledge of the sample groups. Possibly due to sample heterogeneity, the three groups (control, AMI, and post-treatment) could not be distinctly segregated based on their overall transcriptomic landscape. To delve deeper into the data and uncover mRNAs associated with AMI, we subsequently engaged in a supervised analysis, sPLS-DA, a method that identifies the most discriminative mRNAs within the dataset. Through this analysis, we distinguished the groups primarily along component 1, as illustrated in the dot plot of component 1 versus component 2 (Fig. 2B). Although, there was no clear separation into molecular subtypes, our analysis revealed significant differences in gene expression: 1067 genes between AMI patients and controls (Fig. 2C), 477 genes between post-treatment and AMI individuals (Fig. 2D), and 567 genes between post-treatment individuals and controls (Fig. 2E). The threshold for differential gene screening among groups was set as an absolute log2 fold change greater than 2 and p-value < 0.05. The differentially expressed genes (DEGs) identified in pairwise comparisons among the three groups are detailed in Table.S2 (Original). Certain mRNAs were consistently modulated across both comparative analyses (Fig.S1). Certain mRNAs, including HBEGF, G0S2, EREG, FOSB, PLK2, NR4A2, HEY1, SFN, PER1, RNU6-415P, LINC00664, CSAD and CNTNAP3C, were significantly upregulated in the AMI group compared to the controls. However, the mRNAs CSAD, IL32, RPL36, VAMP5, ZNF48, RPS26, UQCC3, RPL13P12, and ZNF593OS showed no significant change compared to the AMI group after treatment (Table.S3). Taking into account the significant age differences between the AMI group and the control group, we included age as a covariate and conducted pairwise differential comparison analyses among the three groups. A total of 3,033 genes were identified between AMI patients and controls (Fig. S2A), 941 genes between post-treatment individuals and AMI patients (Fig. S2B), and 1,342 genes between post-treatment individuals and controls (Fig. S2C). Several differentially expressed genes exhibited consistent expression patterns regardless of age adjustment; for instance, FOSB, G0S2, and CNTNAP3C were significantly upregulated in the AMI group compared to controls and remained significantly upregulated upon re-analysis, with a notable downregulation observed in the treatment group compared to AMI patients. However, another set of genes, such as UTY, ANKRD36BP1, and KDM5D, which were significantly upregulated in the AMI group relative to controls, displayed altered expression patterns when age was considered as a covariate [Table S2 (Age as a covariate)].In the analysis accounting for age as a covariate, we identified a new set of significantly downregulated genes, including PROCR (log2FoldChange = -2.36, p = 0.0488), LINC02987 (log2FoldChange = -2.36, p = 0.0215), and MAD2L2 (log2FoldChange = -2.52, p = 0.0012). Notably, MED18 and ASIC1 were also significantly downregulated in both analyses.

Transcriptomic analysis was conducted on peripheral blood samples from AMI and treatment groups. (A) Depicted the principal component analysis (PCA) plot based on transcriptomics data. (B) The sPLS-DA demonstrated moderate clustering between the three groups. (C) The volcano plots displayed differentially expressed genes (DEGs) between the AMI group, the post-treatment group and the healthy control group. The black line represented the cutoff line with indicated significance criteria. Points with absolute log fold-change ≥ 2 and P < 0.05 were shown in blue, points with absolute log fold-change ≤ -2 and P ≤ 0.05 were in red, and the rest were in green: (C) AMI versus healthy controls, (D) Post-treatment of AMI versus AMI, and (E) Post-treatment of AMI versus healthy controls. AMI Acute Myocardial Infarction, FC fold change, Treat Post-treatment of AMI.
Deciphering molecular pathways and gene functions through enrichment analysis
To elucidate the roles of identified genes, we conducted enrichment analyses using Kyoto Encyclopedia of Genes and Genomes (KEGG) and Gene Ontology (GO) enrichment analyses. GO analysis revealed that, compared to the control group, the upregulated DEG in the AMI group were enriched in 795 terms across Biological Process (BP), Cellular Component (CC), and Molecular Function (MF) categories (p < 0.05). The top 10 enriched terms are depicted in Fig. 3A. Notably, BP was predominantly enriched in positive regulation of calcium ion transport, CC showed enrichment in platelet alpha granule lumen, and MF was enriched for platelet-derived growth factor binding. In the comparison between post-treatment and AMI, upregulated DEGs were associated with an enrichment of 159 terms (p < 0.05), which included cell junction disassembly in BP, blood microparticle in CC and antigen binding in MF. Downregulated DEGs in this comparison were associated with 721 significant terms (p < 0.05), including regulation of epithelial cell proliferation in BB, semaphorin receptor complex in CC, and in transmembrane receptor protein tyrosine kinase activity in MF (Fig. 3B). When comparing treated individuals to controls, the upregulated genes were related to 284 significant terms (p < 0.05), encompassing regulation of epithelial cell proliferation in BP, platelet alpha granule in CC, and in type II transforming growth factor beta receptor binding in MF (Fig. 3C).

GO functional enrichment and pathway enrichment analyses were conducted on DEGs. (A–C) The top 5 GO terms associated with the Molecular Functions (MF), Biological Processes (BP), and Cellular Compartments (CC) in both upregulated and downregulated DEGs were identified. (A) In the comparison between AMI and healthy controls, (B) Post-treatment of AMI versus AMI, and (C) Post-treatment of AMI versus healthy controls. (D–F) KEGG enrichment analyses were performed on the upregulated and downregulated DEGs. The top 10 were shown here. (D) For the comparison between AMI and healthy controls, (E) Post-treatment versus AMI, and (F) Post-treatment of AMI versus healthy controls.
In the pathway enrichment analysis facilitated by KEGG, DEGs between AMI and control groups highlighted enrichment in the top 10 pathways (Fig. 3D). The upregulated DEGs were predominantly enriched in pathways associated with Amoebiasis, Nitrogen metabolism and Glycine, serine, and threonine metabolism. Notably, genes such as CXCL8 (log2FC = 3.38, p. adjust < 0.01) and FOSB (log2FC = 5.86, p < 0.01, p.adjust < 0.01), which were significantly upregulated in AMI group, partook in essential immune-related pathways (Table.S4). Downregulated DEGs were enriched in Cholinergic synapse, Relaxin signaling pathway and Neuroactive ligand-receptor interaction. Comparative pathway enrichment of DEGs between Treatment and AMI groups were depicted in Fig. 3E, with upregulated genes enriched in Inositol phosphate metabolism, Leishmaniasis and Hypertrophic cardiomyopathy, while downregulated genes were enriched in IL-17 signaling pathway, Biosynthesis of amino acids and Leishmaniasis. The top 10 pathways of DEGs enrichment between Treatment and Control comparisons are presented in Fig. 3F, where upregulated differential genes were enriched in TGF-beta signaling pathway, Osteoclast differentiation and non-small cell lung cancer. Downregulated DEGs were enriched in Ribosome, Coronavirus disease COVID-19 and African trypanosomiasis.
Incorporating age as a covariate significantly altered the gene enrichment analysis results for AMI patients. Unlike the age-excluded analysis, both AMI and control groups showed enrichment in the ECM-receptor interaction pathway (Fig. S2C). GO analysis also revealed enrichment in the platelet-related pathway, specifically in the platelet alpha granule lumen (Fig. S2B), highlighting a notable increase in platelet-derived growth factor binding. Additionally, while the positive regulation of calcium ion transport pathway was previously significant, it lost significance under age consideration, shifting to regulation of calcium ion transport. The downregulated genes in the AMI group showed significant enrichment in the Relaxin Signaling Pathway, with RLN2 being significantly downregulated in both analyses. When comparing the treatment group to the AMI group, upregulated genes were enriched in fatty acid metabolism. However, in the age-adjusted treatment group, several immune-related pathways, such as Primary immunodeficiency and Autoimmune thyroid disease, were significantly upregulated—findings not observed in the conventional analysis.
Both analyses revealed enrichment in arginine and proline metabolism for downregulated genes in the treatment group compared to the AMI group. The age-excluded analysis showed downregulation in the IL-17 Signaling Pathway and Cytokine-cytokine receptor interaction, which were upregulated in both AMI and control groups. However, after considering age as a covariate, the enrichment of the Cytokine-cytokine receptor interaction pathway decreased.
Sparse partial least squares regression discriminant analysis (sPLS-DA)
Component 1, encapsulating 38 mRNAs effectively differentiates AMI patients from control subjects. Figure 4A showed the top 25 contributors to this component. Similarly, Component 2, comprising 50 mRNAs detailed in Fig. 4B, distinguishes treatment from controls, emphasizing the leading 25 mRNAs. The efficacy of these two components in classification is underscored by their comprehensive coverage. Data revealed that a very good classification was obtained with two components. Analysis of receiver operating characteristic (ROC) curves conducted for Component 1 revealed that the area under the curve (AUC) for discerning AMI from the other groups was 0.9195 (P < 0.01). The contribution of individual mRNAs to Component 1 was ordered by their importance in mRNA expression, with notable contributors including CC3orf52, CTNNB1, REC114, LINC01736, and POLR3E. The ROC curve for Component 2 demonstrated an AUC of 0.9974 (P < 0.01) for differentiating the AMI group from other groups, and an AUC of 0.9769 (P < 0.01) for the distinction of the control group from others. The top five contributors to Component 2 were ZDHHC3, ZNGB1, ZDHHC6, SPATA7, and SLC31A1. It was noteworthy that certain mRNAs, such as ZFP36 (log2FC = 1.76, p.adjust = 0.012), were significantly upregulated in AMI compared to control and also contributed to Component 2. The standalone contribution of the transcriptomic data to the final sPLS-DA model was demonstrated by score plots of the first two components, indicating the optimal separation capacity of the transcriptomic data (Fig. 4C).

Sparse partial least-squares discriminant analysis. (A, B) Selected features were shown in a pyramid bar plot. The loading plot represented the top 25 mRNAs contributing to group separation (Left). The area under the ROC curve (AUC) values, compared to the grouping level, were shown for the three-group classification (Right). (A) The bar plot showed the contribution value of each mRNA. (B) sPLS-DA contributions to component 2 were depicted. The color in the bar plot represented the highest mRNA expression level in the corresponding group. (C) Displayed the background prediction plot for the sample’s prediction results.
Identification of the most significant module by WGCNA
To identify gene modules of biological significance associated with AMI, we employed WGCNA to construct a gene correlation network. Outlier samples were examined using hierarchical clustering methods, resulting in the exclusion of four outliers (Fig. 5A, h = 15000). To ensure a scale-free co-expression network (Fig. 5B), we selected a soft-thresholding parameter β = 14, achieving a scale-free fit index (R^2) of 0.72. Following the identification and merger of highly similar modules, 34 modules were delineated and color-coded (Fig. 5C). Based on the correlation analysis between modules and traits (Fig. 5D), the tan module, comprising 207 mRNAs, was identified as positively correlated with the clinical trait of chest pain duration (r = 0.73, P < 0.01). The plots of module membership and gene significance further demonstrated significant correlations within the tan module (Fig. 5E).

The results of weighted gene co-expression network analysis (WGCNA). (A) Clustering dendrogram of 81 samples. (B) Analysis of the scale-free index for various soft-threshold powers (left). Analysis of the mean connectivity for various soft-threshold powers(right). (C) WGCNA was performed to identify 28 modules by unsupervised clustering. (D) Heatmap of the correlation between the module eigengenes and clinical traits of AMI, healthy control, and Post-treatment of AMI. The tan module was identified as the positively correlated module with chest pain duration (r = 0.73, P < 0.01). (E) The gene significance and module membership of the genes in the tan module exhibited a positive correlation. (F) KEGG enrichment analyses in tan module.
To elucidate the functional roles of the genes within the tan module, we conducted a KEGG enrichment analysis. The analysis revealed that five mRNAs within the tan module—CD44, FLT3LG, CSF3R, CD36, and HLA-DPB1—were enriched in the Hematopoietic cell lineage pathway (Fig. 5F).
Identification of candidate mRNA biomarkers
Employing a Random Forest (RF) importance score, we prioritized the top 200 mRNAs, illustrating their contribution to classification performance based on disease status (Fig.S3). The top 5 genes with significant contributions under this algorithm are POLR3E, ZNF831, TRIM17, WSB1, and PABPC1(Fig.S3). The out-of-bag (OOB) estimate of error rate was 26.15%, and stability in the ensemble of decision trees was reached at approximately 400 trees (Fig.S4). Subsequently, we integrated the intersections of the top 200 mRNAs from the RF model, the sPLS-DA components 1 and 2, the DEGs of AMI vs. Control, Treatment vs. Control, Treatment vs. AMI, and the mRNAs from the tan module identified in the WGCNA. The differential genes were derived from the analysis that did not consider age as a covariate. The UpSet plot revealed an intersection of 92 mRNAs (Table.S5) across any two significant gene sets (Fig. 6A). Utilizing LASSO regression (Fig. 6B), candidate mRNA biomarkers were filtered from the intersection, yielding six mRNAs with optimal lambda values (Fig. 6C). We validated the expression levels of these mRNAs in peripheral blood samples using quantitative PCR (qPCR) (Fig. 6D). In the AMI group, ANKRD52, ART1, NRP2, and PPP1R15A expressions were elevated compared to the healthy control group, whereas BAIAP2L1 and CCNE1 were reduced. Notably, after treatment, these six mRNAs’ expression levels did not significantly differ from those in the AMI group (Fig. 6E).

Through various data analysis methods, significant gene sets were obtained in AMI, post-treatment of AMI, and healthy control group. (A) The UpSet plot illustrated the intersection and union relationships among DEGs (top 200), sPLS-DA component 1, component 2, tan module of WGCNA, and random forest (RF) top 200 genes. The ribbons between the dots represent the intersections between sets. Genes with an intersection count of two or more groups were selected for further analysis. (B) The extracted features were reduced via the LASSO regression. (C) LASSO coefficients of the variables. (D) The expression level of six mRNA in the AMI, Post-treatment of AMI and healthy controls. (E) Validation of 6 mRNA expression by fluorescence quantitative PCR.
Hub gene expression in the AMI mice model
To further validate the relationship between AMI and the candidate biomarkers, we employed a mouse AMI model. Following immediate ligation of the left anterior descending (LAD) coronary artery, ECG recordings displayed a characteristic ST-segment elevation (Fig.S7A), indicating successful induction of myocardial ischemia. Histological analysis of myocardial sections revealed significant changes in the infarct area when compared to the sham group (Fig.S7B, C). Additionally, elevated serum cTnI levels quantitatively confirmed the occurrence of myocardial damage (Fig.S7 D). M-mode echocardiography was performed to evaluate cardiac structure and function (Fig.S7E). Detailed measurements of interventricular septum (IVS) thickness, left ventricular posterior wall (LVPW) thickness, left ventricular internal diameter (LVID), ejection fraction (EF), and fractional shortening (FS) revealed alterations in cardiac structure and contractility (Fig.S7F-J). Notably, the mRNA levels of ANKRD52 (Fig.S7K) and NRP2 (Fig.S7L) in peripheral blood were significantly elevated in the myocardial infarction model compared to the sham group, consistent with our findings in human data.
Discussion
AMI is a significant contributor to cardiovascular mortality, and early diagnosis is crucial for effective treatment and improved outcomes. Although various biomarkers have been explored for the diagnosis of AMI, there is a continued need for reliable non-invasive biomarkers to accurately differentiate AMI patients from healthy individuals. This study aims to identify potential mRNA biomarkers in peripheral blood to aid in the early diagnosis and monitoring of AMI. In this study, our integrative analysis identified several candidate intersecting genes. ANKRD52, ART1, NRP2, and PPP1R15A were found to be upregulated, while BAIAP2L1 and CCNE1 demonstrated downregulation in the context of AMI. Members of the ANKR family are implicated in a diverse array of functions, including the formation of transcriptional complexes, initiation of immune responses, biogenesis and assembly of cation channels in membranes, and regulation of the cell cycle13,14,15. ANKRD1 is identified as a cardiac-specific ankyrin repeat domain-containing protein principally expressed in the heart and implicated in the morphogenesis and function of cardiomyocytes16. Alterations in both the expression and phosphorylation levels of ANKRD2 protein are known to mediate the balance between muscle physiology and pathological inflammatory responses17. Furthermore, ANKRD26 is associated with thrombopoiesis and the pathogenesis of autoimmune diseases. References 42–44 likely provide further substantiation for these roles18. ANKRD52 has been identified as a regulatory subunit of the protein phosphatase 6 (PP6) holoenzyme, involved in cell cycle regulation and signal transduction19. Data from the Human Protein Atlas (HPA) demonstrate a positive correlation between the expression of ANKRD52 in cardiac muscle and its association with macrophages (Fig.S6, correlation = 0.68). Macrophages serve as pivotal orchestrators within cardiovascular diseases, with the restoration of cardiac function post-myocardial infarction being governed by the cardiac macrophage pool. Following exposure to the post-AMI circulatory environment, macrophages undergo transcriptional and functional repurposing to enhance debris clearance and healing processes20. The upregulation of ANKRD52 in patients with myocardial infarction may activate and modulate the activities and functionalities of macrophages, such as polarization, migration, phagocytosis, or the production of inflammatory factors, thereby influencing cardiac tissue repair and regeneration. However, further experimental investigations are required to substantiate the interactive and functional relationship between them. In skeletal muscle cells, ART1 catalyzes the mono-ADP-ribosylation of integrin α7β1, facilitating its binding with laminins. This interaction triggers the activation of FAK and Rho, leading to the formation of stress fibers and myotendinous contraction21. Similarly, ART1 may exert a regulatory effect on the contractile function of cardiomyocytes, which could be critical for the restoration of myocardial function following myocardial infarction. NRP2 is implicated in cardiovascular development, inflammation, and cardiovascular diseases22,23,24. NRP2’s role encompasses endothelial cell migration, lymphangiogenesis, angiogenesis, monocyte recruitment, and macrophage phagocytic activity. NRP2 may play a multifaceted role in occlusive vascular diseases, such as atherosclerosis22. Furthermore, NRP2 is upregulated in the vascular system in response to injury and inflammation, offering a potential diagnostic tool for identifying diseased arteries. For instance, Chen et al. developed an iodine-131 labeled monoclonal antibody targeting NRP2, which facilitated the imaging of A549 xenograft tumors in mice through single-photon emission computed tomography (SPECT)25. A similar approach might be applicable for visualizing vascular occlusions in patients. Our findings also suggest the potential of elevated peripheral NRP2 as a biomarker for myocardial infarction. Research on BAIAP2L1 has primarily focused on cancer, relating to cellular morphological changes, migration, and adhesion—key processes also in angiogenesis and tissue repair. It may contribute to post-myocardial infarction neovascularization and myocardial repair by influencing the migration and adhesion of cardiomyocytes or endothelial cells26. These processes are critical in angiogenesis and tissue repair, potentially influencing the migration and adhesion of cardiomyocytes or endothelial cells, thereby contributing to neovascularization and myocardial repair following MI. The CCNE1, which encodes cyclin E, is frequently amplified across various histological subtypes and primary disease sites27. In the context of myocardial repair following myocardial infarction, CCNE1 may modulate the G1/S transition of the cell cycle, influencing the proliferation of cardiomyocytes or vascular endothelial cells, thereby affecting the process and outcome of repair. In addition, we observed that the upregulation of ANKRD52 and NRP2 in the peripheral blood of the mouse AMI model is consistent with the corresponding changes in human peripheral blood samples. This may be related to interspecies differences, sample handling, and temporal dynamics. Comprehensive gene expression studies across different models are necessary to better understand the molecular mechanisms of AMI and its potential clinical applications.
Employing a multi-omics and multimarker approach enhances the evaluation of biomarkers for AMI28. Initial assessment of transcriptomic features across three groups was conducted via unsupervised PCA analysis, accompanied by differential analysis based on DEGseq2 statistical testing. Additionally, sPLS-DA facilitated the identification of two sets of candidate mRNA biomarkers, effectively distinguishing between healthy controls, AMI subjects, and those who have undergone treatment. Notably, the protein SLC31A1, marked by substantial contributions in component 2, was observed to have elevated expression in coronary artery sections of a mouse model of AMI. Its elevated expression is linked to significant immunological implications, particularly due to the infiltration of numerous immune cell types, especially monocytes29. Although SLC31A1 (log2FC = 0.66, p = 0.27, p.adjust = 0.59) expression was found to be marginally upregulated in the peripheral blood transcriptome of AMI patients when comparing to healthy controls, the change was not statistically significant.Thus, relying solely on differential analysis may overlook such important genes that exhibit subtle expression regulatory changes in the peripheral blood. It is noteworthy that, while the sPLS-DA method identified AMI-related feature genes and demonstrated high AUC values, indicating strong classification performance on the training data, the model has not been tested on external datasets, which may limit its generalizability. Notably, in the Treat vs. Other(s) group, the AUC value is relatively low (< 0.8), suggesting weaker classification ability for the Treat group, possibly reflecting sample heterogeneity. Future studies should incorporate cross-validation and bootstrap analysis to evaluate AUC stability through repeated sampling and improve model robustness.
Enrichment analyses of DEGs among the three groups revealed that the onset of AMI and its early phase of treatment recovery are specifically associated with genes involved in energy homeostasis, aberrant immune responses, inflammation levels, and amino acid metabolism. The number of upregulated genes in AMI was substantially higher compared to the control, with a greater number of genes downregulated following treatment. The acute phase of AMI triggers an inflammatory response and immune activation, leading to significant upregulation of a multitude of genes associated with immune responses, inflammatory mediators, and cell adhesion factors30. In instances of AMI, there was a notable increase in the expression of CXCL8, which is part of the CXC chemokine family. This factor plays a pivotal role in mediating the invasion of neutrophils during numerous inflammatory processes31. Patients with STEMI who develop heart failure exhibit elevated levels of CXCL8, which correlates with impaired recovery of left ventricular function32. An upsurge in CXCL8 expression may contribute to the onset of heart failure following AMI. Treatment appears to effectively attenuate this expression, as evidenced by a marked reduction in circulating CXCL8 V(log2FC = -3.20, p = 0.002 ,p.adjust = 0.39) levels after seven days, rendering them non-significantly different from levels at admission, suggesting a degree of therapeutic efficacy. During myocardial ischemia, the expression of the pro-inflammatory cytokine IL-17 was elevated, subsequently decreasing after treatment, indicating its critical role in post-infarction inflammation33. Comparative analysis between AMI cases and controls revealed an enrichment of upregulated genes in the glycine, serine, and threonine metabolism pathways. Metabolomic studies have delineated alterations in the metabolic pathways of amino acid biosynthesis, 2-oxocarboxylic acids, tryptophan, aminosugars, and nucleotide sugars in individuals at risk of (AMI) These studies have further identified reduced plasma levels of key metabolites, including pyruvate, alanine, glutamine, tyrosine, and tryptophan34. Our investigation elucidates the significance of amino acid dysregulation in AMI as evidenced by transcriptomic alterations. PPP1R15A was found to be significantly upregulated in the transcriptomic analysis of AMI versus control, and significantly downregulated in treatment versus AMI. Genes that exhibit a sharp increase in AMI and subsequently decrease after treatment may be closely related to the therapeutic response. Identifying candidate genes solely through the intersection of differentially expressed genes may yield numerous similar candidates; however, applying Lasso regression narrows down the selection criteria. Due to significant age differences between the AMI and control groups, we included age as a covariate in our differential analysis. This adjustment notably altered gene expression and enrichment results between the AMI and control groups, as well as the treatment group. Genes such as FOSB, G0S2, and CNTNAP3C were significantly upregulated in the AMI group but downregulated in the treatment group, maintaining this expression pattern after age-related covariance analysis, suggesting their potential roles in the pathogenesis of AMI. FOSB shows sustained activation in both acute and chronic myocardial infarction pig models35. Increased FOS protein levels promote cell survival, inhibit apoptosis, and enhance protection of ischemic cardiomyocytes. While c-Fos, a well-studied member of the FOS protein family, significantly upregulates in rat AMI models, its expression decreases with treatments like metoprolol, correlating with reduced infarct size36.Other upregulated genes, such as UTY, ANKRD36BP1, and KDM5D, displayed altered expression patterns after accounting for age, indicating age-related regulatory effects. KDM5D may activate atherosclerosis by modulating the methylation status of H3K437.Our findings suggest an association between KDM5D activation and age. Both groups exhibited enrichment in ECM-receptor interactions, a pathway commonly activated in ischemic cardiomyopathy38,39. Changes in gene enrichment analysis indicate that previously identified pathways related to immune and inflammatory responses were significantly affected by age, underscoring its critical role in cardiovascular disease research.
The application of WGCNA and machine learning techniques has facilitated the identification of pivotal genes and pathways associated with AMI in various studies40,41,42.Guo et al. utilized WGCNA and Lasso regression to identify SOCS2, FFAR2, and MYO1F as immune-related hub genes43. Concurrently, analysis of GEO datasets with WGCNA and PPI approaches revealed LILRB2, NCF2, and S100A9 as potential biomarkers for AMI44. Further, LASSO and Support Vector Machine-Recursive Feature Elimination (SVM-RFE) identified ACSL1, CH25H, GPCPD1, and PLA2G12A as prospective lipid targets for AMI treatment in cardiac endothelial cells45. Analytical approaches that do not rely on differential gene expression can identify not only genes with substantial expression differences but also those with minor expression changes yet disease relevance. WGCNA, particularly in large sample studies, emphasizes the correlation between gene clusters and disease traits46. SPLS-DA enhances model prediction while reducing variable number through sparsity, facilitating the identification of genes closely associated with disease classification47 Lasso regression employs penalization on coefficients for feature selection and regularization, thereby enhancing the model’s predictive accuracy and interpretability, often used to identify genes strongly associated with response variables, such as disease states, and for the discovery of biomarkers48. Previous research, despite variations in sample types, disease states, techniques, and standardization methods, consistently highlights the significance and specificity of transcriptomic alterations during AMI and implicates multiple genes and pathways in post-infarction cardiac repair and remodeling.
We identified 207 mRNAs significantly positively correlated with the duration of chest pain by WGCNA. In our intersection analysis, we found that both IMPG2 and CA14 are associated with the duration of chest pain and are significantly upregulated in the AMI group compared to controls (Table. S5). The SNP rs17487-G in IMPG2 is significantly related to mean corpuscular volume (MCV)49,potentially influencing systemic inflammatory responses. CA14 decreases in muscle samples from lowlanders at high altitude50, however, its upregulation after AMI may enhance metabolic adaptation of myocardial cells in hypoxic conditions. These mRNAs were enriched in the Hematopoietic cell lineage pathway (genes are enriched in the pathway: CD44, FLT3LG, CSF3R, CD36, HLA-DPB1), which delineates the developmental hierarchy of all blood cells originating from hematopoietic stem cells in the bone marrow and differentiating into red cells, white cells, and platelets. The hypoxic conditions following myocardial infarction precipitate the rapid accumulation of leukocytes in the affected cardiac region51. During the acute phase of myocardial infarction, expression of the granulocyte CSF3R (log2FC = 1.37, p < 0.01 ,p.adjust = 0.03) was significantly upregulated in comparison to the healthy control group, thereby enhancing leukocyte production and function to facilitate the clearance of necrotic cells and initiate the repair process52. After seven days of treatment, CSF3R (log2FC = -0.14, p = 0.83, p.adjust = 0.99) expression levels showed no significant difference from baseline, suggesting that CSF3R continues to play a role in regulating granulocyte generation, neutrophil function, or the mobilization of hematopoietic stem cells, which may be involved in angiogenesis and scar tissue formation. Cardiac repair following AMI is driven by intense sterile inflammation and infiltration of immune cells—the inflammatory phase—responsible for digesting and removing damaged cellular and extracellular matrix components, which spans approximately 3–4 days in mice and is a protracted process in humans53. Interactions mediated by CD44 play a crucial role in resolving post-infarction inflammatory responses and modulating fibroblast function54. During the acute phase of myocardial infarction, CD44 expression was marginally increased in comparison to healthy controls (log2FC = 0.37, p = 0.41, p.adjust = 0.70), and experienced a slight decrease after seven days of treatment (log2FC = 0.16, p = 0.83, p.adjust = 0.99). This may suggest that the cell surface glycoprotein CD44 primarily engages in interactions between cells and the extracellular matrix, with signaling confined to infiltrating leukocytes and infarct fibroblasts. Consequently, the expression differences in peripheral blood are not significant.
Using an RF model, we prioritized the top 200 mRNAs based on their importance scores, highlighting their potential contributions to classification performance in distinguishing disease states. This approach is commonly employed in transcriptomic studies for biomarker selection25,55. likely due to significant sample size differences between groups, necessitating experimental validation of the biomarkers. Among the top-ranked genes, ZNF831, a zinc finger transcription factor, was further analyzed using The Cardiovascular Disease Knowledge Portal. ZNF831 has been associated with myocardial infarction, atrial fibrillation, and related cardiovascular trait56. It shows strong correlations with systolic blood pressure and myocardial infarction (MI), with particularly robust evidence linking it to systolic blood pressure, a major risk factor for AMI (Fig.S5)57. Interestingly, ZNF831 exhibited slightly higher expression in AMI patients compared to controls (log2FC = 0.39, p = 0.50, p.adjust = 0.77), though these differences were not statistically significant. ZNF381 may play different roles in cardiovascular diseases across various physiological or pathological states. While genome-wide association studies (GWAS) have linked ZNF831 to coronary artery disease (CAD) and myocardial infarction, its expression may vary across tissues or cell types, potentially explaining the lack of significant expression changes observed during the acute phase of myocardial infarction. These findings warrant further investigation in diverse pathological conditions and at different time points to better understand the biological role of ZNF831 in cardiovascular diseases.
This study selected representative peripheral blood samples from AMI patients at the onset and post-treatment for transcriptome sequencing and bioinformatics analysis. Despite heterogeneity in some samples, the enrichment results elucidated biological processes associated with AMI. Further research, however, is required to substantiate our findings, including external validation in a more representative population, which remains imperative. Moreover, we have not unraveled the complex mechanisms underlying these results. Potential biomarkers identified in clinical samples and their prospective functions have yet to be confirmed in cellular or animal models. The role of mRNA and associated protein biomarkers in AMI necessitates further elucidation in future research.
Conclusions
In summary, through comprehensive RNA sequencing, our study pinpoint six promising mRNA biomarkers. These biomarkers have shown significant capability in distinguishing AMI patients from healthy individuals. However, to ascertain their true diagnostic potential, further validation in larger and more diverse cohorts is imperative. The amalgamation of these biomarkers may possess the potential to supplement existing non-invasive diagnostic modalities, thereby contributing to the prevention and management of AMI.
Data availability
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation / Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human: HRA007295) that are publicly accessible at https://ngdc.cncb.ac.cn/gsa-human/browse/HRA007295. The Animal data supporting the findings of this study is available from the corresponding author upon reasonable request.
Change history
30 April 2025
The original online version of this Article was revised: In the original version of this Article, an outdated version of the Supplementary Information file 2 was typeset. The correct Supplementary Information file now accompanies the original Article.
References
Global burden of 87 risk factors in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet (London, England). ;396(10258):1223–1249. (2020).
Chen, P. et al. Targeted delivery of extracellular vesicles in heart injury. Theranostics 11 (5), 2263 (2021).
Kazimierczyk, E. et al. Altered MicroRNA dynamics in acute coronary syndrome. Adv. Interventional Cardiology/Postępy W Kardiologii Interwencyjnej. 16 (3), 287–293 (2020).
Wang, T., Xiao, Y., Zhang, J., Jing, F. & Zeng, G. Dynamic regulation of HIF-1 signaling in the rhesus monkey heart after ischemic injury. BMC Cardiovasc. Disord. 22 (1), 407 (2022).
Zhang, W., Zhao, X., Xiao, Y., Han, P. & Kang, J. The association of depressed angiogenic factors with reduced capillary density in Rhesus monkey model of myocardial ischemic infarction. FASEB J. 29, 798796 (2015).
Blokland, I. et al. Single-cell Dissection Immune Response after Acute Myocard. Infarct. (2023).
Hou, R. et al. Screening and analysis of potential critical gene in acute myocardial infarction based on a miRNA-mRNA regulatory network. Int. J. Gen. Med. 15, 2847–2860 (2022).
Lafta, S. A., Issa, A. H., Hazza, M. A. & Abed, A. H. The biomarkers associated with myocardial infarction. Syst. Reviews Pharm. ;12(3). (2021).
Kang, L. et al. Uncovering potential diagnostic biomarkers of acute myocardial infarction based on machine learning and analyzing its relationship with immune cells. BMC Cardiovasc. Disord. 23 (1), 2 (2023).
Mohd Faizal, A. S., Hon, W. Y., Thevarajah, T. M., Khor, S. M. & Chang, S. W. A biomarker discovery of acute myocardial infarction using feature selection and machine learning. Med. Biol. Eng. Comput. 61 (10), 2527–2541 (2023).
Zhu, X. et al. Identification of Immune-Related genes in patients with acute myocardial infarction using machine learning methods. J. Inflamm. Res. 15, 3305–3321 (2022).
Xiao, R. et al. Integrating multimodal information in machine learning for classifying acute myocardial infarction. Physiol. Meas. ;44(4). (2023).
Bork, P. Hundreds of ankyrin-like repeats in functionally diverse proteins: mobile modules that cross phyla horizontally? Proteins 17 (4), 363–374 (1993).
Michaely, P. & Bennett, V. The ANK repeat: a ubiquitous motif involved in macromolecular recognition. Trends Cell Biol. 2 (5), 127–129 (1992).
Li, J., Mahajan, A. & Tsai, M. D. Ankyrin repeat: a unique motif mediating protein-protein interactions. Biochemistry 45 (51), 15168–15178 (2006).
Piroddi, N. et al. Myocardial overexpression of ANKRD1 causes sinus venosus defects and progressive diastolic dysfunction. Cardiovascular. Res. 116 (8), 1458–1472 (2020).
Bean, C. et al. Ankrd2 is a modulator of NF-κB-mediated inflammatory responses during muscle differentiation. Cell Death Dis. 5 (1), e1002–e1002 (2014).
Noris, P. et al. ANKRD26-related thrombocytopenia and myeloid malignancies. Blood 122 (11), 1987–1989 (2013).
Lee, T-F. et al. TAZ negatively regulates the novel tumor suppressor ANKRD52 and promotes PAK1 dephosphorylation in lung adenocarcinomas. Biochim. Et Biophys. Acta (BBA) - Mol. Cell. Res. 1868 (2), 118891 (2021).
Fontaine, M. A. C. et al. Blood Milieu in Acute Myocardial Infarction Reprograms Human Macrophages for Trauma Repair. Advanced science (Weinheim, Baden-Wurttemberg, Germany). ;10(5):e2203053. (2023).
Xu, J. X. et al. Effect of ART1 on the proliferation and migration of mouse colon carcinoma CT26 cells in vivo. Mol. Med. Rep. 15 (3), 1222–1228 (2017).
Harman, J. L., Sayers, J., Chapman, C. & Pellet-Many, C. Emerging Roles for Neuropilin-2 in Cardiovascular Disease. International journal of molecular sciences. ;21(14). (2020).
Islam, R. et al. Role of Neuropilin-2-mediated signaling axis in cancer progression and therapy resistance. Cancer Metastasis Rev. 41 (3), 771–787 (2022).
Alghamdi, A. A. A. et al. NRP2 as an emerging angiogenic player; promoting endothelial cell adhesion and migration by regulating recycling of Α5 integrin. Front. Cell. Dev. Biology ;8. (2020).
Chen, L. et al. 131I-labeled monoclonal antibody targeting neuropilin receptor type-2 for tumor SPECT imaging. Int. J. Oncol. 50 (2), 649–659 (2017).
Chao, A. et al. BAI1-Associated protein 2-Like 1 (BAIAP2L1) is a potential biomarker in ovarian Cancer. PloS One. 10 (7), e0133081 (2015).
Yao, S. et al. Clinical characteristics and outcomes of phase I cancer patients with CCNE1 amplification: MD Anderson experiences. Sci. Rep. 12 (1), 8701 (2022).
Yuan, N., Hu, H-L., Zhai, C-L. & Wang, B. Identification of prognostic and diagnostic signatures for cancer and acute myocardial infarction: multi-omics approaches for Deciphering heterogeneity to enhance patient management. Front. Pharmacol. 14, 1249145 (2023).
Zhou, S. et al. Comprehensive bioinformatics analytics and < italic > In-vivo validation reveal SLC31A1 as an emerging diagnostic biomarker for acute myocardial infarction. Aging.
Chen, L. et al. Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 9 (6), 7204–7218 (2018).
Nymo, S. H. et al. Inflammatory cytokines in chronic heart failure: interleukin-8 is associated with adverse outcome. Results from CORONA. Eur. J. Heart Fail. 16 (1), 68–75 (2014).
Husebye, T. et al. Association of Interleukin 8 and myocardial recovery in patients with ST-elevation myocardial infarction complicated by acute heart failure. PloS One. 9 (11), e112359 (2014).
Chang, S. L. et al. Interleukin-17 enhances cardiac ventricular remodeling via activating MAPK pathway in ischemic heart failure. J. Mol. Cell. Cardiol. 122, 69–79 (2018).
Khan, A. et al. High-resolution metabolomics study revealing l-homocysteine sulfinic acid, cysteic acid, and carnitine as novel biomarkers for high acute myocardial infarction risk. Metab. Clin. Exp. 104, 154051 (2020).
Saddic, L. A. et al. Progression of myocardial ischemia leads to unique changes in immediate-early gene expression in the spinal cord dorsal Horn. Am. J. Physiol. Heart Circ. Physiol. 315 (6), H1592–h1601 (2018).
Zhang, S. et al. The effect of c-fos on acute myocardial infarction and the significance of Metoprolol intervention in a rat model. Cell Biochem. Biophys. 65 (2), 249–255 (2013).
Mokou, M. et al. Proteomics based identification of KDM5 histone demethylases associated with cardiovascular disease. eBioMedicine 41, 91–104 (2019).
Qian, C., Chang, D., Li, H. & Wang, Y. Identification of potentially critical genes in the development of heart failure after ST-segment elevation myocardial infarction (STEMI). J. Cell. Biochem. 120 (5), 7771–7777 (2019).
Wang, L., Zhang, Y., Yu, M. & Yuan, W. Identification of hub genes in the remodeling of non-infarcted myocardium following acute myocardial infarction. J. Cardiovasc. Dev. Disease. 9 (12), 409 (2022).
Li, Y. et al. Combining WGCNA and machine learning to identify mechanisms and biomarkers of ischemic heart failure development after acute myocardial infarction. Heliyon 10 (5), e27165 (2024).
Wu, Y. et al. Construction of novel gene Signature-Based predictive model for the diagnosis of acute myocardial infarction by combining random forest with artificial neural network. Front. Cardiovasc. Med. 9, 876543 (2022).
Sun, Y., Wang, M., Tan, X., Zhang, H. & Yang, S. Identification of oxidative Stress-Related biomarkers in acute myocardial infarction. Int. J. Gen. Med. 16, 4805–4818 (2023).
Liu, J., Chen, L., Zheng, X. & Guo, C. Identification of immune-related genes in acute myocardial infarction based on integrated bioinformatical methods and experimental verification. PeerJ 11, e15058 (2023).
Wang, Y. et al. Identification of potential biomarkers associated with acute myocardial infarction by weighted gene coexpression network analysis. Oxidative Med. Cell. Longev. 2021, 5553811 (2021).
Liu, Z. Y. et al. ACSL1, CH25H, GPCPD1, and PLA2G12A as the potential lipid-related diagnostic biomarkers of acute myocardial infarction. Aging (Albany NY). 15 (5), 1394–1411 (2023).
Langfelder, P. & Horvath, S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Chen, Z. & Boves Harrington, P. Self-Optimizing support vector elastic net. Anal. Chem. 92 (23), 15306–15316 (2020).
Balakrishnan, V., Kherabi, Y., Ramanathan, G., Paul, S. A. & Tiong, C. K. Machine learning approaches in diagnosing tuberculosis through biomarkers - A systematic review. Prog. Biophys. Mol. Biol. 179, 16–25 (2023).
Vuckovic, D. et al. The polygenic and Monogenic basis of blood traits and diseases. Cell 182 (5), 1214–1231e1211 (2020).
Juel, C., Lundby, C., Sander, M., Calbet, J. A. & Hall, G. Human skeletal muscle and erythrocyte proteins involved in acid-base homeostasis: adaptations to chronic hypoxia. J. Physiol. 548 (Pt 2), 639–648 (2003).
Frangogiannis, N. G. The inflammatory response in myocardial injury, repair, and remodelling. Nat. Reviews Cardiol. 11 (5), 255–265 (2014).
Zhang, H. et al. Gain-of-function mutations in granulocyte colony-stimulating factor receptor (CSF3R) reveal distinct mechanisms of CSF3R activation. J. Biol. Chem. 293 (19), 7387–7396 (2018).
Prabhu, S. D. & Frangogiannis, N. G. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circul. Res. 119 (1), 91–112 (2016).
Huebener, P. et al. CD44 is critically involved in infarct healing by regulating the inflammatory and fibrotic response. J. Immunol. 180 (4), 2625–2633 (2008).
Wu, X., Peng, C., Nelson, P. T. & Cheng, Q. Random forest-integrated analysis in AD and LATE brain transcriptome-wide data to identify disease-specific gene expression. PloS One. 16 (9), e0256648 (2021).
Fan, J. et al. Zinc finger protein 831 promotes apoptosis and enhances chemosensitivity in breast cancer by acting as a novel transcriptional repressor targeting the STAT3/Bcl2 signaling pathway. Genes Dis. 11 (1), 430–448 (2024).
Psaty, B. M. et al. Association between blood pressure level and the risk of myocardial infarction, stroke, and total mortality: the cardiovascular health study. Arch. Intern. Med. 161 (9), 1183–1192 (2001).
Funding
This project is supported by Kunming University of Science and Technology & People’s Hospital of Lijiang Joint Special Project on Medical Research (KUST-LJ2022002Z). FH is supported by the Project for Leading Academics and Technologists among Youth and Middle-aged in Lijiang and a recipient of Xingli Talented Youth Program. BS is a recipient of Xingdian Talented Youth Program and supported by National Natural Science Foundation of China (81903611).
Author information
Authors and Affiliations
Contributions
B. S, F. H and ZN. C conceived of the study and carried out its design. R. H, XY. Z, BJ. T, YL.C and JS. G performed experiments and analyzed the data. B. S. wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval
This study was conducted in accordance with the ethical standards and approvals of Kunming University of Science and Technology (KUST) and the People’s Hospital of Lijiang. All procedures involving human participants complied with the ethical standards set by the Ethics Committee of the People’s Hospital of Lijiang and were approved under protocol number 2022002. Additionally, all animal studies were reviewed and approved by the Institutional Animal Care and Use Committees (IACUC) at KUST under Animal Protocol AP-KUST-202412040012. These studies were conducted in line with NIH guidelines and the ARRIVE (Animal Research: Reporting In Vivo Experiments) Guidelines.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Huang, F., Chen, Z., Tan, B. et al. An exploratory study of high-throughput transcriptomic analysis reveals novel mRNA biomarkers for acute myocardial infarction using integrated methods. Sci Rep 15, 8436 (2025). https://doi.org/10.1038/s41598-025-92757-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-92757-4
