
Abstract
Autophagy is a widespread physiological process in the body, which also protects the host by degrading invading pathogens and harmful substances during pathological conditions. Nevertheless, Mycobacterium tuberculosis (MTB), the causative agent of tuberculosis, has evolved strategies to subvert autophagy by modulating microRNA (miRNA) expression, enabling its escape from host defenses. In this study, we established an in vitro model using the human macrophage cell line infected with the highly virulent MTB strain H37Rv. Through RNA sequencing and bioinformatic analysis post H37Rv infection, we screened 14 differentially expressed miRNAs. We predicted and demonstrated that miR-30c-1-3p inhibits autophagy and promotes MTB survival by targeting ATG4B and ATG9B during the infection process. The results showed that miR-30c-1-3p expression was gradually increased before 12 h of H37Rv infection, followed by a decrease. Overexpression of miR-30c-1-3p suppressed autophagic activity. We also identified the targeting of miR-30c-1-3p to ATG4B and ATG9B for the first time, and overexpression of both ATG4B and ATG9B, alone or together, on the basis with upregulation of miR-30c-1-3p reversed the inhibition of autophagy. Autophagy levels were analyzed at different levels by western blot, immunofluorescence, and transmission electron microscopy, all of which showed that upregulation of miR-30c-1-3p inhibited autophagy during H37Rv infection. Additionally, the intervention of miR-30c-1-3p mimics resulted in an increased bacterial load in macrophages, suggesting that MTB achieves immune evasion by upregulating miR-30c-1-3p during infection. In conclusion, our study provides a valuable target for the development of host-directed anti-tuberculosis therapy as well as a new diagnostic marker.
Similar content being viewed by others


Introduction
Tuberculosis (TB) remains a top 10 killer worldwide, posing a substantial threat to human health and an enormous economic burden, particularly in developing countries. The World Health Organization reports that approximately 2 billion people worldwide are infected with Mycobacterium tuberculosis (MTB)1,2. Although only about 10% of infected individuals progress to active TB, the large infected population makes MTB infections the leading cause of human deaths worldwide3. Most infected individuals exhibit latent infection because MTB can evade the host’s immune defenses through multiple pathways, enabling it to survive and multiply in host cells for a long time4. However, these patients with latent TB may develop active or drug-resistant TB when they are immunocompromised, such as when they are co-infected with HIV, or when they do not receive standardized anti-TB treatment5. Elucidating the immune evasion mechanism of MTB and identifying key regulators is significant for the development of new anti-TB drugs.
Autophagy is a physiological process that helps maintain the stability of the internal environment and provides additional energy to cells by breaking down cytoplasmic metabolites, as well as damaged organelles and DNA6. Autophagy can also eliminate foreign pathogens and harmful substances via autophagolysosomes under pathological conditions. Autophagy is a dynamic and continuous process that can be categorized into several important stages, including autophagic precursor formation, autophagosome elongation, autophagosome maturation, and cleavage7. It is important to note that this categorization is subjective and may vary depending on the specific study. During MTB infection, the development of autophagy can be regulated by influencing the expression of microRNAs (miRNAs). For example, ATG4B, the target gene in this study, is an important protein that mediates the extension of autophagosomes. ATG4B processes the LC3 precursor to cytosol-soluble LC3-I, which participates in the expansion of the autophagosome membrane and binds to the newly formed membrane until autophagolysosomes are formed8,9. LC3 is consistently localized on the surface of the autophagosome membrane during autophagy and is a recognized marker of autophagosome formation, reflecting autophagic flux10. Generally, MTB is transmitted through aerosolized particles in the atmosphere and enters the body through the respiratory tract11. Once inhaled, macrophages act as the first line of defense against MTB by recognizing it through various pattern recognition receptors (PRRs) on the surface of their cell membranes, and then phagocytose and directly destroy MTB through autophagy in the lungs. In addition, macrophages can present the bacterial peptide of MTB in an antigenic manner, which activates an adaptive immune response to MTB12. However, MTB evades autophagy through multiple pathways, including downregulation of macrophage PRRs and autophagy-related genes (ATGs), inhibition of autophagy precursor formation and autophagosome elongation, disruption of autophagosome-lysosome fusion, and inhibition of autophagolysosome acidification, which allow MTB to survive and proliferate in the host cell13. Consequently, some researchers consider autophagy induction as a promising host-directed therapeutic strategy against MTB14.
MiRNAs can perform translational repression or cleavage of mRNAs at the post-transcriptional level, and a growing number of researches have revealed that the dysregulated expression of miRNAs is closely related to various human diseases. Early research on miRNAs focused on oncology, and subsequently the critical role of miRNAs in infectious diseases, including viral and bacterial infections, has also received widespread attention15. Autophagy, as a critical part of the immune defense response, significantly affects the fate of MTB after invasion. Dysregulated expression of miRNAs was found to influence the development of autophagy, which helps MTB to survive in the cell16. In this study, we established an MTB-infected macrophage model using human macrophages differentiated from TPH-1 cells and co-cultured with H37Rv. The results showed that the expression of miR-30c-1-3p was gradually increased before 12 h of H37Rv infection, followed by a decrease, and forced upregulation of miR-30c-1-3p led to autophagy inhibition. Furthermore, the intervention of miR-30c-1-3p mimics increased the bacterial load of macrophages, suggesting that MTB achieves immune evasion by upregulating miR-30c-1-3p during infection. Overexpression of both ATG4B and ATG9B alone or together based on upregulation of miR-30c-1-3p reversed the inhibition of autophagy. Autophagy levels were examined at different levels by western blot (WB), immunofluorescence techniques, and transmission electron microscopy. These findings complement the signaling network of MTB regulating the innate immune defense response and identify a new target for the future development of new anti-TB drugs.
Materials and methods
Cells and bacterial culture
THP-1 cells were obtained from the China Center for Type Culture Collection (CCTCC, GDC0100) and cultured in RPMI-1640 medium (Thermo Fisher, USA) containing 10% fetal bovine serum (FBS) (Thermo Fisher, USA) at 37 °C with 5% CO2 for passaging. THP-1 cells were differentiated into macrophages by the addition of phosphatidylinositol amide (PMA) (Sigma Merck, USA) to a final concentration of 5 ng/mL and cultured for passaging with RPMI-1640 medium containing 10% FBS and gentamicin (Sigma Merck, USA) at 37 °C with 5% CO2.
H37Rv was obtained from the China Center for Type Culture Collection (CCTCC, GDC0100). H37Rv was cultured in Middlebrook 7H9 solid medium, which was supplemented with 0.5% glycerol, 0.05% Tween-80, and 10% OADC. Once the growth of colonies was stabilized, individual colonies were selected and transferred to the Mycobacterium complete medium at 37 °C under 5% CO2 until the OD600 of the bacterial solution reached between 0.6 and 0.8.
H37Rv infection of macrophages
After the differentiated macrophage suspensions were added to 6-well plates and cultured for 24 h, they were washed three times with phosphate buffer solution (PBS), and bacterial suspensions at Multiplicity of infection (MOI) = 10 were added dropwise to each well (H37Rv-to-cell ratio of 10:1), and incubated at 37 °C with 5% CO2. Cells were collected after H37Rv infection for 0 h, 6 h, 12 h, 24 h, and 48 h respectively by replacing the complete medium supplemented with gentamicin and continuing the culture for 24 h.
microRNA-sequencing (miRNA-seq) and bioinformatics
Total RNA was extracted from THP-1 cells infected with H37Rv for 2 h and blank control. Starting with total RNA as the initial material, RNA samples were prepared for sequencing libraries utilizing the NEB Next® Multiplex Small RNA Library Prep Set for Illumina® (NEB E7300L). Following reverse transcription and PCR amplification, a cDNA library was obtained. Libraries with insertions between18 and 40 bp were ready for sequencing on Illumina sequencing with SE50. Subsequently, library quality was assessed on the Agilent 5400 system (Agilent, USA) and quantified by qPCR (1.5 nM). The Qualified libraries were pooled and sequenced on Illumina platforms. After cleaning the data, use transcripts per million for normalization expression. The normalized expression level = (read count*105)/libsize. Libsize is the sample miRNA read count. The differentially expressed miRNAs were analyzed by Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment, and further analyzed by TargetScan (https://www.targetscan.org) and ENCORI (https://www.rnasysu.com/encori) databases to predict target genes and screen miRNAs containing autophagy-related target genes. The raw data can be downloaded in the GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE276178).
Real-time quantitative PCR (RT-qPCR)
Total RNA was extracted from THP-1 cells using a total RNA extraction kit (Promega, USA) and converted to cDNA using a reverse transcription kit (Thermo Fisher, USA), followed by the SYBR Green method under the reaction conditions: pre-denaturation 94 °C for 10 min, denaturation 94 °C 15 s, annealing at 60 ℃ for 1 min, and cycling 40 times. The relative expression of the gene to be tested in comparison to the internal reference gene was calculated using the 2−ΔΔCT method, the gene expression levels in the experimental group are relative to the average expression in the control group. The primer sequences and solubility curve can be found in the supplementary file.
Western blot assay
The total protein content of the cells to be tested was extracted by adding a WB-specific cell lysate (Gene-Optimal, China) to the cells. The protein concentration was then quantified using the BCA method (Solarbio, China), and the volume of the upper sample was calculated. Gel electrophoresis was conducted using SDS-PAGE, followed by the transfer of the separated proteins to a nitrocellulose transfer membrane. After closure utilizing 5% skimmed milk, incubation was performed using anti-ATG4B (Abcam, ab223364), anti-ATG9B (Abcam, ab240897), anti-LC3B (Abcam, ab51520) anti-β-actin (Abcam, ab213262) overnight, washed and secondary antibody incubation, and finally detected by exposure using ECL luminescence reagent and analyzed for quantification using Image J software.
Cell transfection and luciferase analysis
miR-30c-1-3p mimics were purchased from Gene-Optimal. The pLVX-NeoR-CLEC4E-WT-P2A-EGFP plasmid was extracted using a plasmid extraction kit (Gene-Optimal Co., Ltd., China). Following the instructions provided with the PEI reagent kit (Thermo Fisher Scientific, USA), 293 T cells were transfected at a PEI:plasmid ratio of 4:1. After overnight incubation, the concentrated lentivirus was collected by centrifugation. THP-1 cells were infected with lentivirus at an MOI of 80. Through 2–4 weeks of antibiotic resistance gene screening, we successfully constructed stable THP-1 cell lines overexpressing ATG4B, ATG9B and ATG4B&ATG9B. To determine luciferase activity, the plasmids pmiRGLO-ATG9B and pmiRGLO-ATG4B were constructed using a plasmid extraction kit. According to the Lipofectamine® 2000 (Thermo Fisher, USA)protocol, the plasmid pEGFP-MN and siRNA were cotransfected into THP-1 cells at a ratio of 1:0.6. Specifically, THP-1 cells were transfected with combinations including pmiRGLO-ATG9B + NC, pmiRGLO-ATG9B + mimics, pmiRGLO-ATG4B + NC, pmiRGLO-ATG4B + mimics, pmiRGLO + NC, and pmiRGLO + mimics. Luciferase activity was measured 24 h post-transfection.
Immunofluorescence staining and confocal microscopy analysis
THP-1 macrophages were collected and fixed using 4% paraformaldehyde, followed by incubation with rabbit anti-LC3 polyclonal antibody (Abcam, ab51520) for primary antibody, Alexa Fluor 488 coupled visualized goat anti-rabbit IgG antibody (Abcam, ab150077) for secondary antibody, DAPI staining of the nuclei, and finally, observation and photographs were taken under a confocal microscope (Olympus, Japan). The number of LC3 puncta was quantified using Image J software, with a minimum of 10 cells counted for each experimental set, repeated three times.
Transmission electron microscopy
THP-1 macrophage or bacterial precipitates were collected by centrifugation, pre-embedded with 2% agarose solution, and then post-fixed with 1% OsO4 for 2 h. Gradient dehydration was performed by adding 30%–50%–70%–80%–95%–100% ethanol sequentially, and the samples were transferred to 818 embedding agents for embedding. The samples were sectioned at a thickness of 60–80 nm using the ultramicrotome (Leica, Leica UC7) and stained with a 2% uranium acetate-saturated alcohol solution to avoid light and 2.6% Lead citrate to avoid CO2. Subsequently, transmission electron microscopy (Hitachi, HT7800) was employed to observe the specimens, and autophagolysosome counting was performed.
Colony-forming unit assay
To assess the effect of miR-30c-1-3p on H37Rv survival, THP-1 macrophages were transfected with miR-30c-1-3p mimics for 24 h. H37Rv infected macrophages at an MOI of 10 for 2 h. Extracellular bacteria were washed away with PBS, and the cell suspension was diluted 100 times. A 1 mL aliquot of the diluted suspension was plated onto agar, cultured for 4–6 weeks, and bacterial colonies were enumerated.
Statistical analysis
The experimental data presented in this paper were analyzed using the SPSS 26.0 statistical package. The experimental results were analyzed using a two-tailed Student’s t-test to identify differences between the two groups and using the Waller-Duncan method for multiple group comparisons. All results were expressed as mean ± standard deviation (means ± SD). Statistically significant differences were considered to be significant when P < 0.05.
Results
Differential expression of miRNAs in H37Rv infected macrophages
To identify differentially expressed miRNAs in MTB-infected macrophages, we used miRNA-seq technology to sequence macrophages infected with H37Rv for 2 h. We found a total of 14 statistically significant differentially expressed miRNAs (Fig. 1A–B). To gain more insight into the biological functions of the differentially expressed miRNAs and target genes, we performed GO and KEGG analyses. The analysis of biological processes, cellular components, and molecular functions revealed that miRNAs and target genes are associated with intracellular plasma membranes and membrane proteins, lysosomes, cellular protein localization, mTOR signaling pathways, and the c lectin-like receptor pathway, which are closely related to autophagy processes (Fig. 2). The regulation of immune functions, such as the differentiation of helper T cells, is also involved (Fig. 2A, C). Partial expression landscapes of genes associated with several signaling pathways can be found in the supplementary files (Supplementary Figure S2–S4). It’s worth noting, however, that since our study exclusively performed miRNA sequencing (miRNA-seq), the expression statuses of target genes depicted in these charts were inferred using bioinformatics methods. The actual expression levels of these targets would necessitate further experimental validation to confirm their accuracy. Targetscan and ENCORI databases were used to predict the target genes of differentially expressed miRNAs. Three miRNAs, namely miR-30c-1-3p, miR-708-5p, and miR-1360b, were identified as potential regulators of the autophagy pathway. Real-time PCR was performed to examine the expression of three candidate miRNAs in macrophages infected with H37Rv for 2 h. The results showed that miR-30c-1-3p was most significantly downregulated (P < 0.001), in addition to the database predicted that miR-30c-1-3p has autophagy-related target genes, ATG4B and ATG9B, which are key genes in the process of autophagy development (Fig. 1C). Therefore, after comprehensive consideration, we selected miR-30c-1-3p for further experimental validation, which has multiple target genes related to autophagy and shows more significant differences in expression.

Analysis of differential miRNAs for H37Rv infection in vitro. (A) Heatmap analysis of differentially expressed miRNAs after H37Rv infection of macrophages for 2 h. (B) differentially expressed miRNAs scatter plot. (C) The expression of LC3B and three miRNAs containing autophagy target genes was quantified by RT-PCR in THP-1 macrophages infected with H37Rv for 2 h. All data above represent the means ± SD of at least three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not statistically different.

GO analysis and KEGG pathway enrichment analysis were conducted on 14 differentially expressed miRNA target genes. (A) GO analysis of upregulated expression of miRNAs. (B) GO analysis of downregulated expression of miRNAs. (C) KEGG analysis of upregulated expression of miRNAs. (D) KEGG analysis of downregulated expression of miRNAs. The autophagy-related biological functions are indicated in red, while the immune defense-related biological functions are indicated in green.
miR-30c-1-3p directly targets ATG4B and ATG9B
To clarify the signaling network of miR-30c-1-3p involved in autophagy regulation, we used Targetscan and ENCORI databases to predict the sequences of miR-30c-1-3p interacting with target genes. The results showed that at a single 7mer site within the 3’-UTR, both ATG4B and ATG9B had matching sequences with miR-30c-1-3p (Fig. 3A). We constructed a pmirGLO dual luciferase reporter vector and examined luciferase activity after transfection of miRNA mimics or mimics negative control (mimics nc). Compared to mimics nc, miRNA mimics significantly inhibited luciferase activity driven by the 3’-UTR structure (WT). In contrast, the intervention of miRNA mimics in the empty plasmid, as well as in mutant 3′-UTR constructs (Mut), did not result in significant differences (Fig. 3B, C). To observe whether miR-30c-1-3p binds to the mRNA 3’UTR of ATG4B and ATG9B, we constructed GFP reporter vectors with putative miR-30c-1-3p binding sites of ATG4B and ATG9B. GFP fluorescence was significantly reduced after co-transfection with miR-30c-1-3p mimics and GFP-containing cells. However, there was no significant alteration when transfected with miR-30c-1-3p mimics nc or binding site mutant cells (Fig. 3F). Additionally, we analyzed the expression of miR-30c-1-3p and its target genes following transfection with miRNA mimics and mimics nc. Following transfection with miR-30c-1-3p mimics, macrophages exhibited elevated levels of miR-30c-1-3p, resulting in a significant reduction in the expression of ATG4B and ATG9B (Fig. 3D, E). In conclusion, these results demonstrate that miR-30c-1-3p directly targets the mRNA 3’TUR of ATG4B and ATG9, inhibiting their expression.

miR-30c-1-3p directly targets ATG4B and ATG9B. (A) Targetscan predicts miR-30c-1-3p interaction sequence with ATG4B and ATG9B. (B, C) miR-30c-1-3p mimics regulation of ATG4B and ATG9B 3’-UTRs reporter. The luciferase reporter assay co-transfected the pmirGLO dual luciferase reporter vector (WT, Mut, and Blank) and miR-30c-1-3p mimics or mimics nc 24 h. (D) The expression of miR-30c-1-3p was observed in THP-1 transfected with miR-30c-1-3p mimics and mimics nc 24 h. (E) Western Blot detection of ATG4B and ATG9B expression in THP-1 transfected with miR-30c-1-3p mimics and mimics nc 24 h. The ATG4B and ATG9B bands were quantified relative to β-actin. (F) Fluorescent microscopic imaging revealed that the expression of GFP in the pEGFP-ATG4B and pEGFP-ATG9B reporters was inhibited by miR-30c-1-3p. THP-1 cells were transfected with GFP reporter vectors (WT and Mut) and fluorescence intensity was subsequently detected by immunofluorescence staining following transfection with miR-30c-1-3p mimics and mimics nc for 24 h. Scale bars: 10 μm. All data above represent the means ± SD of at least three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not statistically different.
H37Rv infection induced altered levels of miR-30c-1-3p, AT4GB, ATG9B and autophagy in macrophages
To investigate the dynamic changes of miR-30c-1-3p and target genes during MTB infection, RT-qPCR was performed on MTB-infected macrophages at different time points. Interestingly, the expression of miR-30c-1-3 was not consistently low as we expected, but gradually increased before 12 h of H37Rv infection and then decreased (Fig. 4A). The expression trend of ATG4B and ATG9B was opposite to that of miR-30c-1-3p, suggesting that their expression is regulated by miR-30c-1-3p (Fig. 4B, C). We hypothesize that H37Rv infection activates macrophage adaptive immune responses, resulting in a decrease of miR-30c-1-3p levels, but the immune escape is achieved by inhibiting autophagy through upregulation of miR-30c-1-3p under the regulation of MTB. Therefore, we decided to use an early infection model infected with H37Rv 2 h in the following experiments, because at this time MTB had not yet upregulated miR-30c-1-3p and external interventions could be more responsive to the regulation of autophagy. We examined the protein levels of LC3 (a marker that responds to autophagosome formation) in macrophages infected with H37Rv for 2 h. The results showed that LC3-II protein levels were elevated in macrophages with H37Rv infection, and the increase was more pronounced with the intervention of bafilomycin A1 (Baf-A1), which exacerbated LC3-II accumulation by blocking autophagolysosome formation (Fig. 4D). These results indicate that H37Rv infection induces a complete autophagic response.

H37Rv infection promotes macrophage autophagy and progressively upregulates miR-30c-1-3p and suppresses ATG4B and ATG9B expression. (A–C) RT-PCR was performed to detect the expression of miR-30c-1-3p and its target genes at different infection time nodes. The expression of miR-30c-1-3p exhibited a gradual increase following infection, reaching a peak at 12 h post-infection, and the expression of ATG4B and ATG9B exhibited an opposite trend. (D) Western Blot demonstrated that H37Rv infection for 2 h promoted macrophage autophagy. H37Rv increased LC3-II protein levels after infection of macrophages for 2 h. The application of Baf-A1 (100 nM) resulted in the accumulation of LC3-II. The LC3 bands were quantified relative to β-actin. All data above represent the means ± SD of at least three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not statistically different. Multiple comparisons between groups were made using the Waller-Duncan method. Different letters represent P < 0.05 and the presence of any same letter represent P > 0.05.
miR-30c-1-3p inhibits macrophage autophagy in H37Rv-infected macrophages by targeting ATG4B and ATG9B
Macrophages were infected with H37Rv for 2 h before being transfected with miR-30c-1-3p mimics and miR-30c-1-3p mimics nc for 24 h to investigate the regulation of macrophage autophagy by miR-30c-1-3p. Western blot showed that miR-30c-1-3p reduced LC3-II protein levels in uninfected and H37Rv-infected macrophages, as well as ATG9B protein levels (Fig. 5A, B). The reduction of ATG4B in macrophages after transfection with miR-30c-1-3p mimics was not statistically different compared to transfection with miR-30c-1-3p mimics nc in the H37Rv-infected group. However, considering the results of the luciferase reporter and WB of the uninfected group, the reduction of ATG4B in macrophages after transfection with miR-30c-1-3p mimics was more pronounced compared to ATG9B. This suggests that ATG4B may be regulated by multiple factors, resulting in multiple signaling pathways upregulating ATG4B expression in macrophages after H37Rv infection, so that a single increase in miR-30c-1-3p is not sufficient to completely inhibit ATG4B expression. Overall, the overexpression of miR-30c-1-3p inhibited macrophage autophagy during H37Rv infection. This finding was verified using confocal microscopy to count LC3 puncta and transmission electron microscopy to directly observe autophagic lysosomes.

miR-30c-1-3p inhibits autophagy by targeting ATG4B and ATG9B during H37Rv infection. (A) Western Blot demonstrated that miR-30c-1-3p mimics intervention reduced H37Rv-induced increase in LC3-II, (B) ATG4B and ATG9B in macrophages. The LC3, ATG4B and ATG9B bands were quantified relative to β-actin. Since A and B are the same experiment with the same batch of cells, the same β-actin blot was used. (C) Confocal microscopy detected LC3 puncta in macrophages infected with H37Rv for 2 h. The number of LC3 puncta was significantly reduced in miR-30c-1-3p mimics transfected macrophages. Scale bars: 2 μm. (D) The inhibition of autophagy by miR-30c-1-3p was confirmed by transmission electron microscopy. In macrophages infected with H37Rv for 2 h, an increase in the number of autophagolysosomes was observed, while a decrease in the number of autophagolysosomes was observed in macrophages transfected with miR-30c-1-3p mimics. Green and red arrows represent suspected autophagosomes and autophagolysosomes respectively, and the number of autophagolysosomes was counted for each cell cross-section. Scale bars: 5 μm. All data above represent the means ± SD of at least three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not statistically different.
Confocal microscopy was used to observe the number of LC3 puncta in macrophages. It was found that miR-30c-1-3p mimics significantly reduced the number of LC3 puncta in macrophages infected or not with H37Rv (Fig. 5C). H37Rv infection also increased the number of LC3 puncta in macrophages, suggesting that H37Rv can induce macrophage autophagy. To demonstrate the inhibitory effect of miR-30c-1-3p on autophagy in MTB-infected macrophages, we observed the number of autophagolysosomes in macrophages transfected with miR-30c-1-3p mimics by transmission electron microscopy, both in infected and non-H37Rv-infected macrophages. The results showed an increase in the number of autophagolysosomes in H37Rv-infected macrophages, but a decrease in the number of autophagolysosomes after transfection with miR-30c-1-3p mimics, more directly demonstrating the inhibitory effect of miR-30c-1-3p on macrophage autophagy during MTB infection (Fig. 5D).
Overexpression of ATG4B and ATG9B reverses autophagy inhibition by miR-30c-1-3p
Previous experiments have clarified the targeting of miR-30c-1-3p to ATG4B and ATG9B as well as the autophagy inhibitory effect of miR-30c-1-3p during MTB infection. To further determine the role of ATG4B and ATG9B on macrophage autophagy, we constructed macrophage-stabilized cell lines overexpressing ATG4B and ATG9B as well as co-overexpressing ATG4B & ATG9B. Plasmid mapping and validation of overexpressing macrophage cell lines can be found in the Supplementary figure S5.
Macrophages, ATG4B-OE, ATG9B-OE, ATG4B&ATG9B-OE were stimulated with rapamycin (50 μg/ml) for 2 h and then transfected with miR-30c-1-3p mimics and mimics nc for 24 h, respectively. WB detection of protein expression in macrophages showed a significant increase in the expression of ATG4B and ATG9B in ATG4B-OE, ATG9B-OE, and ATG4B&ATG9B-OE cells. Following transfection with miR-30c-1-3p mimics, levels of ATG4B and ATG9B were observed to be reduced (Fig. 6A). In comparison to the control group, the level of LC3-II was found to be reduced in cells of ATG9B-OE, with or without transfection of miR-30c-1-3p. An increase in LC3-II levels was observed in ATG4B-OE cells following the intervention with miR-30c-1-3p mimics. Nevertheless, this does not imply that ATG9B exerts an inhibitory effect on autophagy. Because co-overexpression of ATG4B and ATG9B resulted in a more significant elevation of LC3 compared to overexpression of ATG4B alone (Fig. 6B), it suggests that ATG9B plays an important role in macrophage autophagy. Indeed, it has been demonstrated that ATG9B enhances the co-localization of P62 with ubiquitinated proteins, which in turn accelerates the extension of autophagosomal membranes17. The observation of LC3 puncta using confocal microscopy showed an increase in the number of LC3 puncta in ATG4B-OE or ATG9B-OE macrophages, either after transfection with miR-30c-1-3p mimics or mimics nc. More importantly, there was no significant difference in the number of LC3 puncta after transfection with miR-30c-1-3p mimics in ATG4B & ATG9B-OE cells (Fig. 6C), suggesting that co-overexpression of ATG4B and ATG9B reversed the autophagy inhibitory effect of miR-30c-1-3p.

ATG4B and ATG9B overexpression reversed miR-30c-1-3p-mediated autophagy inhibition. (A) The levels of ATG4B and ATG9B proteins were elevated in the corresponding stably transfected cell lines that had been overexpressed, thereby confirming the efficacy of the lentiviral transfection method. ATG4B and ATG9B protein levels were reduced in all cell lines after miR-30c-1-3p mimics intervention. (B) Macrophages were transfected with miR-30c-1-3p mimics for 24 h. Western blot demonstrated that LC3-II protein levels were elevated in ATG4B-OE, ATG9B-OE, and ATG4B & ATG9B-OE macrophages. Furthermore, the inhibition of autophagy mediated by miR-30c-1-3p was completely reversed in ATG4B & ATG9B-OE macrophages. The LC3, ATG4B and ATG9B bands were quantified relative to β-actin. Since A and B are the same experiment with the same batch of cells, the same β-actin blot was used. (C) Confocal microscopy confirmed that ATG4B and ATG9B overexpression reversed miR-30c-1-3p-mediated autophagy inhibition. Overexpression of ATG4B and ATG9B alone or together increased the number of LC3 puncta in macrophages, and there was no significant difference in the number of LC3 puncta in ATG4B & ATG9B-OE macrophages under miR-30c-1-3p mimics intervention. Scale bars: 2 μm. All data above represent the means ± SD of at least three independent experiments. Multiple comparisons between groups were made using the Waller-Duncan method. Different letters represent P < 0.05 and the presence of any same letter represent P > 0.05.
miR-30c-1-3p promotes Mycobacterium tuberculosis survival in macrophages
To clarify the effect of elevated miR-30c-1-3p on the survival of H37Rv in macrophages, we analyzed the change of macrophage bacterial load with transfected miR-30c-1-3p mimics by colony-forming unit (CFU). The results showed that under identical infection conditions, 1 ml of the THP-1 cell suspension treated with miR-30c-1-3p mimics and mimics nc could cultivate 44*102 and 36*102 colonies, respectively (Fig. 7). This indicates that miR-30c-1-3p enhances the bacterial load in H37Rv infected macrophages. Despite the varied colony sizes observed in the media, we speculate that these differences might be attributable to differing bacterial vitality or varying extents of bacterial contact with the medium. This supports our previous hypothesis that miR-30c-1-3p promotes MTB survival in macrophages by inhibiting autophagy.

CFU confirms that miR-30c-1-3p promotes Mycobacterium tuberculosis survival. Following infection at a MOI of 10 for 2 h, cells were lysed and cultured in agar plates for 4–6 weeks before CFU. The results demonstrated that miR-30c-1-3p intervention increased MTB survival. All data above represent the means ± SD of at least three independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001; ns, not statistically different.
Discussion
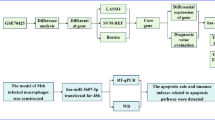
Recently, the research on miRNAs has gradually deepened, showing that miRNAs have an important impact on disease progression and clinical prognosis by regulating autophagy during MTB infection18. Research has shown that miR-33 upregulation following MTB infection inhibits autophagy by targeting several autophagy genes such as ATG5, ATG12, and LC3B19. However, some miRNAs have been identified as differentially expressed and capable of modulating autophagy, their small level of differential expression and targeting only individual ATG suggest that they may not be key genes in promoting MTB immune evasion20. In this study, we identified that miR-30c-1-3p was significantly differentially expressed following H37Rv infection and promoted MTB immune evasion by targeting ATG4B and ATG9B to regulate autophagic processes in human macrophages (Fig. 8). We identified for the first time the targeting of miR-30c-1-3p for ATG4B and ATG9B and also described the significant role of miR-30c-1-3p in the crosstalk between MTB and host innate immune defense responses, which may provide a new target for the development of anti-TB therapies.

Schematic diagram of MTB immune evasion by up-regulation of miR-30c-1-3p. In macrophages, miR-30c-1-3p levels are upregulated after MTB infection, and the exact mechanism is unclear. miR-30c-1-3p inhibited autophagy precursor formation and autophagosome extension by targeting ATG4B and ATG9B. ATG9B is capable of forming the ATG9-ATG2-ATG18 transmembrane transporter protein complex, which connects autophagosomes to the endoplasmic reticulum. ATG4B is responsible for the processing of LC3 precursors and the recycling of LC3-II from the outer membrane of autophagolysosomes.
The expression of miR-30c-1-3p and its target genes in macrophages were examined at different times of H37Rv infection. It was observed that the expression of miR-30c-1-3p increased after infection and remained at a high level, gradually decreasing between 12 and 24 h of infection. Additionally, we have clarified that miR-30c-1-3p inhibits macrophage autophagy and promotes MTB survival during infection. We determined that a portion of the inhibition of autophagy was achieved by targeting ATG4B and ATG9B because overexpression of ATG4B and ATG9B reversed the autophagy inhibition caused by miR-30c-1-3p. Indeed, ATG4B and ATG9B are important proteins in the autophagy process. ATG4B is involved in the process of ubiquitination modification of LC3 and can process LC3 precursors into cytoplasmic soluble LC3-I. LC3 is a key protein that promotes the extension of the autophagosome membrane and is also a key marker for detecting the number of autophagosomes in cells21. It has been shown that miR-193-3p can promote MTB survival by targeting ATG4B to inhibit autophagy22. The role of ATG9B in autophagy is currently unclear. However, studies have shown that ATG9, ATG2, and ATG18 can form a transmembrane transporter protein complex that connects the autophagosome membrane to the endoplasmic reticulum during the early stages of autophagosome formation23,24. Furthermore, ATG9B facilitates the co-localization of P62 with ubiquitinated proteins, thereby accelerating the extension of autophagosome membranes17. This conclusion is supported by the GO enrichment and KEGG enrichment results showing that differentially expressed miRNAs in RNA-seq are associated with the plasma membrane and endomembrane system. Two previous studies have reported the specific down-regulation of miR-30c in the serum of TB patients and in monocytes from individuals with active TB, suggesting its potential diagnostic utility25,26. However, these studies did not further elucidate the regulatory role of downregulated miR-30c in the pathogenesis and progression of TB. We have, for the first time, revealed the autophagy-inhibitory function of miR-30c-1-3p in the context of TB, and elucidated how upregulation of miR-30c-1-3p facilitates MTB immune evasion. It is interesting to note that autophagy promotes tumor proliferation by maintaining intracellular homeostasis and protects the body by removing pathogens in infectious diseases27. The reason for the downregulation of miR-30c-1-3p in tumor cells and its upregulation during H37Rv infection is comprehensible. However, the specific mechanisms that regulate miR-30c-1-3p expression are not yet fully understood. Further studies on miR-30c-1-3p would enhance our comprehension of the intercommunication mechanisms of the innate immune defense response in pathological conditions.
Autophagy represents a crucial component of the innate immune response, with a pivotal role in the direct clearance of MTB. In addition, macrophages can present the bacterial peptide of MTB in an antigenic manner, which activates an adaptive immune response to MTB12. Interestingly, MTB not only evades the immune response by inhibiting autophagy, but also promotes macrophage lipid accumulation by inhibiting the autophagy-mediated transport of lipid to the autophagolysosome and impairing mitochondrial fatty acid oxidation19. The lipid-rich environment provides an optimal environment for MTB to evade innate immune defenses and facilitates its replication28. Lipid-rich MTB exhibit characteristics of dormant mycobacteria, including resistance to anti-TB drugs29. In a separate study, miR-889 expression was observed to be elevated in granuloma tissues during the initial stages of MTB infection. This upregulation of miR-889 was found to facilitate and sustain granuloma organization and inhibiting through the miR-889/TNF-α/TWEAK/AMPK pathway, which, in turn, enhances MTB survival. Elevated miR-889 and TNF-α were associated with maintenance of latent MTB infection. Furthermore, granuloma disruption and reactivation of latent infection were observed after the inhibition of TNF-α expression with adalimumab30. In our previous study, we observed that in osteoclasts from patients with osteoarticular TB, there was an increase in TNF-α expression, an increase in autophagy levels, and a decrease in apoptosis, which ultimately promoted osteoclast survival leading to disruption of bone homeostasis31. In conclusion, the results of these studies indicate that autophagy plays a pivotal role in the pathogenesis of MTB infection and has a significant impact on disease progression and prognosis.
Additionally, miRNAs, which are a large family, exhibit varying expression levels at different stages of MTB infection. Therefore, miRNAs can serve as diagnostic markers for early detection or to distinguish between active and inactive TB, as well as to differentiate drug-resistant MTB32. In comparison to healthy individuals, patients with active TB exhibit overexpression of miR-144 in their sputum and serum, while the expression level of miR-155 is decreased33,34. Despite numerous studies investigating the mechanism of miRNAs in MTB infection, clinical translational therapy still faces significant challenges. As regulatory factors, miRNAs can regulate a variety of gene expression. The application of miRNA mimics systemically will inevitably cause immunotoxicity and other side effects, so how to ensure the safety and efficacy of the drug is also a difficult point in the development of miRNA therapy35. For instance, miR-30c-1-3p can have both anticancer effects and promote MTB survival. Interfering systemically with miR-30c-1-3p to remove MTB may increase the likelihood of cellular carcinogenesis25. However, a study has demonstrated that delivering nanoparticle-packed drugs to treat lung lesions through inhalation can effectively reduce systemic toxicities, making it a safer and more efficient method36,37. In addition, miRNA delivery systems have become a significant research focus to address systemic immunotoxicity. Newly developed exosome/liposome-based nanovesicles are engineered exosome mimics that may be a promising method for drug delivery, reducing dosage, and mitigating systemic immunotoxicity through excellent targeting38,39.
In conclusion, our study reveals a novel pathway for miR-30c-1-3p to inhibit autophagy during MTB infection. In addition to the predicted targets of ATG4B and ATG9B, targetscan also predicts autophagy-associated targets of miR-30c-1-3p, including ATG2 and ATG14. In addition, in another concurrent study, it was also found that miR-30c-1-3p can inhibit autophagy by targeting ATG14 and ULK1, thereby promoting the survival of Helicobacter pylori40. It has also been shown that ATG14 is important for limiting MTB replication in macrophages41. This suggests that miR-30c-1-3p is likely to be a key gene in MTB infection by intervening in the autophagy process through multiple target genes thereby promoting MTB survival. As previously stated, ATG9B can form transmembrane transporter protein complexes with ATG2 and ATG18. Future investigations should explore the impact of miR-30c-1-3p on the plasma membrane system during autophagy. It should also be clarified that the extent of ATG14 inhibition by miR-30c-1-3p in the context of TB and its effect on MTB survival. Animal models should be constructed for in vivo experiments to study the anti-TB effect and systemic response of miR-30c-1-3p interventions in combination with novel drug delivery systems.
Conclusions
In conclusion, our study reveals a novel pathway for miR-30c-1-3p to inhibit autophagy during MTB infection. In addition to the predicted targets of ATG4B and ATG9B, targetscan also predicts autophagy-associated targets of miR-30c-1-3p, including ATG2 and ATG14. This suggests that miR-30c-1-3p can regulate multiple phases of autophagy and therefore be a key regulator. Our study provides valuable targets for the development of host-directed anti-TB therapy as well as new diagnostic markers.
Data availability
The datasets generated and/or analysed during the current study are available in the GEO database (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE276178).
References
Dheda, K. et al. The intersecting pandemics of tuberculosis and COVID-19: Population-level and patient-level impact, clinical presentation, and corrective interventions. Lancet Respir Med 10(6), 603–622 (2022).
Bañuls, A. L. et al. Bacterium tuberculosis: Ecology and evolution of a human bacterium. J Med Microbiol 64(11), 1261–1269 (2015).
Hmama, Z. et al. Immunoevasion and immunosuppression of the macrophage by mycobacterium tuberculosis. Immunol Rev 264(1), 220–232 (2015).
Liu, C. H., Liu, H. & Ge, B. Innate immunity in tuberculosis: Host defense vs pathogen evasion. Cell Mol Immunol 14(12), 963–975 (2017).
Bell, L. C. K. & Noursadeghi, M. Pathogenesis of HIV-1 and Mycobacterium tuberculosis co-infection. Nat Rev Microbiol. 16(2), 80–90 (2018).
Mizushima, N. & Komatsu, M. Autophagy: Renovation of cells and tissues. Cell. 147(4), 728–741 (2011).
Shariq, M. et al. The exploitation of host autophagy and ubiquitin machinery by Mycobacterium tuberculosis in shaping immune responses and host defense during infection. Autophagy 19(1), 3–23 (2023).
Ho, P. T. B., Clark, I. M. & Le, L. T. T. MicroRNA-based diagnosis and therapy. Int J Mol Sci 23(13), 7167 (2022).
Park, N. Y., Jo, D. S. & Cho, D. H. Post-translational modifications of ATG4B in the regulation of autophagy. Cells. 11(8), 1330 (2022).
Fan, S. et al. Inhibition of autophagy by a small molecule through covalent modification of the LC3 protein. Angew Chem Int Ed Engl. 60(50), 26105–26114 (2021).
Bussi, C. & Gutierrez, M. G. Mycobacterium tuberculosis infection of host cells in space and time. FEMS Microbiol Rev 43(4), 341–361 (2019).
Bo, H. et al. Mycobacterium tuberculosis-macrophage interaction: Molecular updates. Front Cell Infect Microbiol 13, 1062963 (2023).
Zhai, W., Wu, F., Zhang, Y., Fu, Y. & Liu, Z. The immune escape mechanisms of Mycobacterium Tuberculosis. Int J Mol Sci. 20(2), 340 (2019).
Adikesavalu, H. et al. Autophagy induction as a host-directed therapeutic strategy against mycobacterium tuberculosis infection. Medicina (Kaunas). 57(6), 522 (2021).
Yang, H. et al. MiRNA-based therapies for lung cancer: Opportunities and challenges?. Biomolecules. 13(6), 877 (2023).
Peng Xianglin, Pu. et al. Research progress of non-coding RNA in autophagy of macrophages during Mycobacterium tuberculosis infection. Acta Med. Univ. Sci. et Technol. Huazhong 53(1), 117–122 (2024).
Wang, S. et al. Upregulation of ATG9b by propranolol promotes autophagic cell death of hepatic stellate cells to improve liver fibrosis. J Cell Mol Med. 28(2), e18047 (2024).
Sampath, P., Periyasamy, K. M., Ranganathan, U. D. & Bethunaickan, R. Monocyte and macrophage miRNA: Potent biomarker and target for host-directed therapy for tuberculosis. Front Immunol. 12, 667206 (2021).
Ouimet, M. et al. Mycobacterium tuberculosis induces the miR-33 locus to reprogram autophagy and host lipid metabolism. Nat Immunol. 17(6), 677–686 (2016).
Sinigaglia, A. et al. Tuberculosis-associated MicroRNAs: From pathogenesis to disease biomarkers. Cells 9(10), 2160 (2020).
Agrotis, A., Pengo, N., Burden, J. J. & Ketteler, R. Redundancy of human ATG4 protease isoforms in autophagy and LC3/GABARAP processing revealed in cells. Autophagy. 15(6), 976–997 (2019).
Qu, Y. et al. MiR-129-3p favors intracellular BCG survival in RAW264.7 cells by inhibiting autophagy via Atg4b. Cell Immunol. 337, 22–32 (2019).
Gómez-Sánchez, R. et al. Atg9 establishes Atg2-dependent contact sites between the endoplasmic reticulum and phagophores. J Cell Biol. 217(8), 2743–2763 (2018).
Sawa-Makarska, J. et al. Reconstitution of autophagosome nucleation defines Atg9 vesicles as seeds for membrane formation. Science. 369(6508), eaaz7714 (2020).
Gao, S. H. et al. Integrating serum microRNAs and electronic health records improved the diagnosis of tuberculosis. J Clin Lab Anal. 35(8), e23871 (2021).
Spinelli, S. V. et al. miR-30c is specifically repressed in patients with active pulmonary tuberculosis. Tuberculosis (Edinb). 105, 73–79 (2017).
Peng, X. et al. Nrf2: A key regulator in chemoradiotherapy resistance of osteosarcoma. Genes Dis. https://doi.org/10.1016/j.gendis.2024.101335 (2024).
Pandey, A. K. & Sassetti, C. M. Mycobacterial persistence requires the utilization of host cholesterol. Proc Natl Acad Sci USA. 105(11), 4376–4380 (2008).
Rodríguez, J. G. et al. Global adaptation to a lipid environment triggers the dormancy-related phenotype of Mycobacterium tuberculosis. mBio. 5(3), e01125-e1214 (2014).
Chen, D. Y. et al. MicroRNA-889 inhibits autophagy to maintain mycobacterial survival in patients with latent tuberculosis infection by targeting TWEAK. mBio. 11(1), e03045-e3119 (2020).
Liu, W. et al. Mycobacterium tuberculosis infection increases the number of osteoclasts and inhibits osteoclast apoptosis by regulating TNF-α-mediated osteoclast autophagy. Exp Ther Med. 20(3), 1889–1898 (2020).
Agrawal, P., Upadhyay, A. & Kumar, A. microRNA as biomarkers in tuberculosis: A new emerging molecular diagnostic solution. Diagn Microbiol Infect Dis 108(1), 116082 (2024).
Lv, Y. et al. Sputum and serum microRNA-144 levels in patients with tuberculosis before and after treatment. Int J Infect Dis 43, 68–73 (2016).
Wagh, V., Urhekar, A. & Modi, D. Levels of microRNA miR-16 and miR-155 are altered in serum of patients with tuberculosis and associate with responses to therapy. Tuberculosis (Edinburg) 102, 24–30 (2017).
Kara, G., Calin, G. A. & Ozpolat, B. RNAi-based therapeutics and tumor targeted delivery in cancer. Adv Drug Deliv Rev 182, 114113 (2022).
Man, D. K. et al. Potential and development of inhaled RNAi therapeutics for the treatment of pulmonary tuberculosis. Adv Drug Deliv Rev 102, 21–32 (2016).
Liu, J. et al. ROS-responsive liposomes as an inhaled drug delivery nanoplatform for idiopathic pulmonary fibrosis treatment via Nrf2 signaling. J Nanobiotechnol. 20(1), 213 (2022).
Yan, Y. et al. Inhibiting collagen I production and tumor cell colonization in the lung via miR-29a-3p loading of exosome-/liposome-based nanovesicles. Acta Pharm Sin B 12(2), 939–951 (2022).
Duan, L. et al. Exosome-mediated delivery of gene vectors for gene therapy. Nanoscale 13(3), 1387–1397 (2021).
Deng, Q., Xu, Y., Zhong, Y., et al. miR-30c Increases the intracellular survival of Helicobacter pylori by inhibiting autophagy. Cell. Microbiol., 2022, 4536450.
Aylan, B. et al. ATG7 and ATG14 restrict cytosolic and phagosomal Mycobacterium tuberculosis replication in human macrophages. Nat Microbiol. 8(5), 803–818 (2023).
Funding
This study was funded by the Medical Research Project of Wuhan Municipal Health Commission (grant number WX21M02).
Author information
Authors and Affiliations
Contributions
P.X. and J.F. designed the study. X.P. and F.P. conducted the experiments, analyzed the data and wrote the first draft of the manuscript. F.Z. and F.X. helped collect the data. X.D. and J.W. provided technical and methodological support. all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Peng, X., Pu, F., Zhou, F. et al. Has-miR-30c-1-3p inhibits macrophage autophagy and promotes Mycobacterium tuberculosis survival by targeting ATG4B and ATG9B. Sci Rep 15, 10240 (2025). https://doi.org/10.1038/s41598-025-94452-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-94452-w
