
Abstract
Early-onset high myopia (eoHM) occurs before school age and is an ideal model for monogenic studies of high myopia due to minimal environmental influence. This study screened for genes and variants associated with eoHM in 47 unrelated Chinese patients with eoHM. Protein–protein interaction (PPI) network analysis was conducted to detect interactions among candidate genes, and protein–protein docking was performed. In 28 patients (28/47, 59.6%), 32 potential pathogenic variants in 22 candidate genes were identified, including 24 novel variants. Among these 28 patients, 64.3% (18/28) carried pathogenic variants in RetNet genes. Of these, 12 patients (42.9%, 12/28) had pathogenic variants in six known genes (TSPAN12, CACNA1F, USH2A, RPGR, COL2A1, and COL11A1), which are responsible for retinal dystrophy and Stickler syndrome associated with eoHM. Additionally, 7 patients (25.0%, 7/28) carried pathogenic variants in seven candidate genes for ocular disease (POLA1, TMEM231, HK1, GSN, COL5A1, CRYBB3, and WDR), which were identified as potentially pathogenic in Chinese eoHM patients for the first time. Phenotype analysis showed that 11 patients presented with only high myopia, 10 patients had inherited retinal diseases (IRDs) with eoHM, and 7 patients had ocular-only Stickler syndrome (Ocular-STL) with eoHM. The initial clinical records of these 17 patients did not show recognizable signs of other primary diseases except for high myopia, and further specific clinical examinations confirmed the diagnosis of IRDs or Stickler syndrome through eoHM. The PPI network analysis identified 87 candidate genes associated with early-onset high myopia (eoHM), grouped into four functional clusters. Thirteen hub genes, including RPGR, COL5A1, CRYAB, and FBN1, were crucial for the pathogenesis of myopia. The network showed strong biological relevance with highly significant enrichment (p-value < 1.0e-16). Our study expands the list of candidate genes associated with eoHM and suggests that eoHM may be the first reason for children to visit an ophthalmology clinic and an important clue for clinicians to detect underlying ocular diseases. These findings highlight the complex interplay of these genes, providing valuable insights into the molecular mechanisms of myopia and potential targets for future therapeutic interventions.
Similar content being viewed by others

Introduction
High myopia (HM) is defined as less than or equal to − 6.00 diopters or an ocular axial length of at least 26 mm. High myopia brings further vision challenges because of increasing the risk of pathologic ocular changes such as cataract, glaucoma, retinal detachment, pathologic myopia choroid neovascularization and myopic macular degeneration, all of which can cause irreversible vision loss, which make HM as major cause of irreversible blindness in the East Asian population1. HM is divided into eoHM and late-onset high myopia (loHM) according to age of onset. eoHM is a refractive error at preschool age (< 7 years)2, while loHM occurs at post-school age. Available evidences suggest that both environmental and genetic factors play important roles in myopia development and progression, although their exact mechanisms are not yet known.The environmental factors include near reading habits3, artificial light exposure4 , heavy academic burden, less outdoor activity5 or diet with high suger intakeS6 , however, preschool children are at less risk of environmental stress. Thus, the etiology of eoHM is driven predominantly by genetic factors with minimal environmental effects.
Many scholars have explored the pathogenesis of high myopia from a genetic perspective, and the available studies have identified 17 pathogenic genes lead to nonsyndromic high myopia, including ZNF644(OMIM614159)7, SCO2(OMIM:608908)8, SLC39A5(OMIM:615946)9, CCDC111(OMIM:615420)10, P4HA2 (OMIM:617238)11, BSG12, CPSF1(OMIM:618827)13, NDUFAF714, TNFRSF21(OMIM:160700)15, XYLT116, DZIP116, LRPAP1(OMIM:160700)17, CTSH(OMIM:615,431)17, LEPREL1(OMIM:614292)18, LOXL3(OMIM:619781)19, ARR3(OMIM:301010)12, and OPN1LW20, but these variants explain only a small part of the pathogenesis of eoHM. Thus, it suggests that eoHM may have a different mechanism from simple high myopia. Recently, several studies have shown that a significant proportion of eoHM is associated with inherited retinal diseases (IRDs) and/or systemic syndromes21,22 and mutations in genes known to be responsible for IRDs were also found in approximately one-fourth of the probands with eoHM23,24. For example, mutations of COL2A1 and COL11A1 genes can cause Stickler syndrome, mutations of RP2, RPGR and EYS genes lead to retinitis pigmentosa (RP), TSPAN12 and FZD4 are common pathogenic genes of familial exudative vitreoretinopathy (FEVR). High myopia is also the most common phenotype of IRDs such as FEVR, RP, and gyrate atrophy of the choroid and retina(GA) or Stickler syndrome.
Although these diseases have a hereditary basis, their genetic mechanisms are intricate and multifaceted. In this study, whole-exome sequencing was used to investigate mutations in a cohort of 47 probands with eoHM.Then we describe the clinical phenotype and genetic analysis of 28 probands with eoHM. To establish the interaction relationship among encoded proteins of genes associated with high myopia and perform functional enrichment analysis on pathogenic genes. And molecular docking is conducted on selected key proteins to validate protein interactions and predict the pathogenesis of pathogenic genes.
Results
Clinical tests
A total of 47 eoHM probands were collected in this study, all of them were diagnosed as “high myopia” at the onset (or met the criteria during follow-up), aged 1–7 years, with a refractive status ≤ − 6.00 DS or axial length ≥ 26 mm. Whole-exome sequencing approach was selectively performed on 47 eoHM probands, and pathogenic variants were detected in 28 probands, including 13 males and 15 females, 5 Hui people, 23 Han people . The average age of onset was 5 years old, mean spherical lens was − 7.00(− 9.00, − 6.00)DS in the right eye and − 8.00(− 9.25, − 6.00)DS in the left eye. The mean axial length was 25.98(25.01,26.41)mm in the right eye and 25.86(24.88,26.66)mm in the left eye. F3, F5, F7, F13, F16, F18, F19, and F23 were high myopia in one eye. The clinical phenotypes are shown in Table 1.
Genetic findings
Thirty-two potential pathogenic variants in 22 candidate genes were identified in 28 probands (28/47, 59.6%) (Table3). Among the 28 probands, 64.3% (18/28) carried pathogenic variants in RetNet genes, of which 12 (42.9%, 12/28) probands had pathogenic variants in 6 known genes (TSPAN12, CACNA1F, USH2A, RPGR, COL2A1 and COL11A1) responsible for retinal dystrophies as well as Stickler syndrome accompanied by eoHM (Fig. 1) , and 7 (32.1%, 9/28) probands had pathogenic variants in 7 candidate genes (POLA1, TMEM231, HK1 , GSN, COL5A1 , CRYBB3 and WDR36 ) (Fig. 1) that were found to be potentially pathogenic in Chinese patients with eoHM for the first time. Of 32 variants, missense variants account for approximately two thirds (71.9%, 23/32) while truncation variants (including, nonsense, frameshift, splicing and intronic change variants) account for 28.1% (74.2%, 9/32) (Fig. 1).

Mutation proportions grouped by the genes reported to cause myopia or not, and mutation types. The variants with identifified in patients with early-onset high myopia, 18 genes that were previously reported to cause phenotypes including myopia,harbored mutations in 9(32%) other genes not previously reported in myopia.
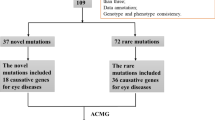
Among 32 variations, 24 variants in 18 candidite genes were not yet included in HGMD, which were novel variants(Table 2). The 24 novel variants were not reported in either the 1000 Genomes Database or the East Asian Population Database (ExAC_EAS), The multiple lines of computational evidence predicted that all of the 24 novel variants would have harmful effects on genes or gene products (Table 2). Further verification were performed in other family members by Sanger sequencing (Fig. 2). Therefore, it was believed that all novel variants were likely pathogenic or pathogenic variants according to ACMG guideline.

The Sanger-sequencing photograph of probands. II1 represents the proband, I1 represents the father of the proband, I2 represents the mother of the proband.In particular, II2 of F9 represents the proband, II1 is the elder brother of the proband; II2 of F11 represents the proband, II1 is the elder sister of the proband; II2 of F13 represents the younger sister of the proband; II2 of F24 represents the younger brother of the proband. The black arrow indicates the site of the mutation. The red arrow in F22 indicates a high frequency mutation, which has been filtered.
In the eoHM cohort of this study, variations in AD genes are the most common in patients with simple eoHM and systemic syndromes (76.5% , 13/17) , while in retinal dystrophies cohort, variations in AR and XL genes are the most common (80.0% , 8/10). The inheritance patterns were autosomal dominant in 21 probands (75.0%, 21/28),and autosomal recessive in 8 probands(23.0%, 7/28), with compound heterozygous or homozygous variants. The normal parents of proband F3 and F16 did not carry the same heterozygous variant as the proband, suggesting that the two variant were de novo and the inheritance patterns were autosomal dominant.
Genotype and phenotype analysis
According to the presence or absence of ocular and systemic abnormalities, eoHM were classified into non-syndromic eoHM and syndromic eoHM . Among the 28 eoHM patients with pathogenic variants,the phenotype analysis showed that 10 probands presented simply high myopia, 10 with inherited retinal disease ( IRDs) with eoHM , 7 Stickler syndrome with eoHM, and 1 with syndromic eoHM. The ocular signs and fundus photograph were showed in Table 1 and Fig. 3.

The Wide-angle fundus photograph of F1,F2,F4,F10,F12,F15,F24,F25. (B,C,D,E,F,G) showed fundus tessellation; (A,F,H) showed fundus tessellation and diffuse retinal atrophy.
Simple high myopia
In this study, 11 probands showed solely high myopia, in which 5 carried the variants in RPGR P4HA2 ,OTX2 and JAG1 genes known to be responsible for high myopia (eoHM), and 6 carried the variants in 6 candidate genes (POLA1, TMEM231, HK1 , COL5A1 , CRYBB3 and GSN) that were found to be potentially pathogenic in Chinese eoHM patients for the first time.
The F7 female proband with RPGR frameshift variant c.2234_2237del(p.Arg745fs) only showed high myopia and her father with the same RPGR hemizygotic variant showed typical RP.
The F4 female proband with RPGR frameshift variant c.2405_2406del(p.Glu802GlyfsTer32) only showed high myopia and her mother with the same RPGR variant also showed sole high myopia.
A heterozygous variant in OTX2 gene c.748G > A(p.Gly250Arg) was detected in F20 proband who was 4 years old at the time of initial diagnosis and had moderate myopia in both eyes and developed to high myopia during the two years of follow-up. No obvious abnormality was found in fundus examination .
Variants in three genes (POLA1 , TMEM231,COL5A1 ) known to be associated with syndromic diseases , such as X-linked reticulate pigmentary25,ciliopathies26 ,different forms of Ehlers-Danlos syndrome (EDS)27 were detected in F12 proband,F19 proband and F10 proband (Table 3), which all showed absence of ocular and systemic abnormalities except high myopia (Table 1).
Variants in three genes (HK1 , CRYBB3, GSN) known to be associated with ocular diseases, such as autosomal dominant retinitis pigmentosa(adRP)28,congenital cataract29 and glaucoma30,31,32, were detected in F22 proband, F27 proband and F21 proband (Table 3), which all presented no ocular abnomolitys cxcept high myopia (Table 1).
Retinal dystrophies accompanied by eoHM
10 probands showed inherited retinal diseases(IRDs) accompanied by eoHM,of which 9 probans carried 11 variants in 9 known genes responsible for (IRDs) , including USH2A, CACNA1F, OAT, PDE6B, RP2, PRPF6, EYS, TSPAN12 and AIPL1. The initial clinical records of the 10 patients did not show recognizable signs of original diseases other than high myopia, and further specific clinical examinations as well as genitic testing determined the diagnosis of IRDs accompanied by eoHM.Among the 10 probands, 5 probands carring variants in USH2A, PDE6B, RP2, PRPF6 and EYS genes showed retinitis pigmentosa (RP) accompanied by eoHM and 4 probands carring variants in CACNA1F, OAT, TSPAN12 and AIPL1 genes showed congenital stationary night blindness(CSNB), gyrate atrophy of the choroid and retina (GA), Familial exudative vitreoretinopathy (FEVR) and Rod-cone dystrophies repectively(Table 3).
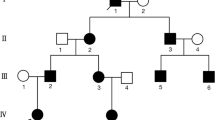
The F23 proband with a missense variant c.452A > T(p.Asn151Ile) in TSPAN12 was diagnosed as high myopia in left eye and severe anisometropia. The fundus flurescein angiography(FFA) revealed the retinal vessels did not fully develop to the periphery fundus and terminated in front of the serrated edge with a clear ridge -like demarcation between the avascular area and the normal vascularized retina in both eyes. The peripheral retinal vascular branches were numerous with changed in brush shape and a small amount of leakage. The proband’s father carring the same TSPAN12 variant had myopia without abnormal findings on fundus photography, but FFA examination suggested that he had FEVR (stage I). (Fig. 4).

The phenotype of F23 pedigree with TSPAN12 mutation. (A) The fundus fluorescein angiography (FFA) of F23 proband: The peripheral blood vessels of both eyes were straight with decreased density, abnormal anastomosis of terminal arteries and veins, and peripheral telangiectasia with a small amount of leakage; (B) The FFA of F23 proband’s father: the vessels in both eyes did not develop to the periphery and terminated in front of the serrated edge, with a clear crest-like demarcation between the avascular area and the normal vascularized retina. The peripheral retinal vascular branches are numerous, with brush-like changes and a small amount of leakage. (C) Pedigree of affected family; (D) Sequence chromatograms of identified mutations; (E) The homology of amino acid sequences between human TSPAN12 and other species. The amino acid at position 151 is highly conserved among species. The mutated residue 151 is boxed and indicated.
The F28 proband with a missense variant c.905G > T(p.Arg302Leu) in AIPL1 was diagnosed as super high myopia in both eye since childhood . Due to the increasing axis and the degree of myopia, she was treated in many hospitals successively and diagnosed as keratoconus and pathological myopia, then she wored rigid gas permeable (RGP) lenses for 10 years. Electroretinogram(ERG) examination showed the moderate decline in scotopic 0.01 ERG and mild decline in Photopic 3.0 ERG.
The F24 proband with a missense variant c.1346C > T(p.Ala449Val) in WDR36 known to be associated with glaucoma and cataract had high myopia (− 6.00DS) when she was 4 years old. Because she could not cooperate to complete the ERG examination, the inicial diagnosis was made as high myopia and amblyopia . When she was 14 years old, full-field electroretinography (ffERG) was performed and found that the a severe decline on both rod and cone cell response in both eyes.The visual field also showed obviously abnormal.
Systemic syndromes accompanied by eoHM
In this study, eoHM had been identified as a symptom of Stickler syndrome caused by known genes COL2A1 and COL11A133. The heterozygous variants in COL2A1 and COL11A1 were identified in 7 probands with eoHM, indluding 4 missense variants , 2 nonsense variant and 1 Intronic variant (Table 3). All of the probands only presented high myopia and ocular changes without other systemic symptom. Of the 7 probands, 5 carring variants in COL2A1 had typical membranous vitreous and 2 carring the variants in COL11A1 had typical beaded vitreous (Table 1).
The heterozygous splicing variants c.1527 + 135G > A in COL2A1 were detected in the F2 proband and her mother who also had high myopia in both eyes and fundus examination showed lattice degeneration of peripheral retina in both eyes with holes in the degeneration area. The heterozygous missense variants c.4384A > G(p.Ile1462Val) in COL2A1 were detected in the F5 proband and her mother who also had high myopia in both eyes. The homozygous missense variant c.1333G > C(p.Val445Leu) in COL11A1 was detected in F13 proband. As validated by Sanger sequencing , the proband’s normal parents carried the same heterozygous variant, Two nonsense variants in COL2A1 (p.Trp1293Ter, p.Arg940Ter) were detected in F3 and F16 probands respectively . Both F3 and F16 probands had typical membranous vitreous without systemic symptoms.The normal parents of proband F3 and F16 did not carry the same heterozygous variant as the probands, suggesting that the two variant were de novo.The phenotype and genetype of F16 proband showed in Fig. 5.

The phenotype of F16 proband with COL2A1 autogenetic mutation. (A) Wide-angle fundus photograph showed fundus tessellation; (B) anterior segment photography showed the vitreous is typically membrane-like; (C) pedigree of affected family; (D) sequence chromatograms of identified mutations; (E) The homology of amino acid sequences between human COL2A1 and other species. The amino acid at position 940 is highly conserved among species. The mutated residue 940 is boxed and indicated.
Protein–protein interaction (PPI)
The PPI network comprised 34 nodes with an average node degree of 3.71; The PPI enrichment p-value was < 1.0e-16, and the local clustering coefficient was 0.612. The Gene Ontology (GO) analysis revealed that all 34 candidate genes divide into 5 clusters as showed by Fig. 6. Cluster I is involved in the transduction of visual signals , retinal feedback machanism, including AIPL1, ARR3, CACNA1F, EYS, GSN, OTX2, PDE6B, RP2, RPGR, TMEM231, USH2A, ZNF644;Cluster II is involved in extracellular matrix (ECM) formation, collagen synthesis and degradation, including ASS1, COL11A1, COL2A1, COL5A1, FBN1, LTBP2, MYOC, OAT, P4HA2, TGFBI, WDR36, ZNF469; Cluster III is involved in Wnt signaling pathway, angiogenesis including FZD4, JAG1, LRP5, TSPAN12;Cluster IV is involved in Energy metabolism, cellular proliferation including GPI, HK1, PCNA, OLA1; Cluster V is involved in lens development and protein stability including CRYAB, CRYBB3.

Cluster I (red node) is involved in the transduction of visual signals , retinal feedback machanism ;Cluster II (yellow node) is involved in extracellular matrix (ECM) formation; Cluster III blue node) is involved in Wnt signaling pathway; Cluster IV (green node) is involved in energy metabolism;Cluster V (purple node is involved in lens development and protein stability.
Protein–protein docking
The docking result between ZNF644 (green) and P4HA2 (blue) shows a favorable interaction, with both proteins aligning in a manner that suggests a stable complex(Fig. 7A). ZNF644 is likely involved in regulatory roles due to its interaction with P4HA2, a key enzyme in collagen biosynthesis.The docking result between ZNF644 (green) and ARR3 (blue) shows a distinct pattern of interaction(Fig. 7B). ZNF644 appears to interact with ARR3, which is a key protein involved in visual signaling. This docking model suggests a potential functional interaction between the two proteins in the visual system.

(A) The green structure represents ZNF644, and the blue structure represents P4HA2. The docking results suggest a stable interaction between these two proteins, where ZNF644 and P4HA2 are positioned in a manner that facilitates their interaction. (B) The docking result between ZNF644 (green) and ARR3 (blue) shows a distinct pattern of interaction. ZNF644 appears to interact with ARR3, which is a key protein involved in visual signaling. This docking model suggests a potential functional interaction between the two proteins in the visual system.
Discussion
EoHM, defifined as a high myopia onset before school age, is considered to be predominantly determined by genetic factors with minimal environmental effects. In this study, whole-exome sequencing was used to investigate mutations in in 47 probands with eoHM. Thirty-two potential pathogenic variants in 22 candidate genes were identified in 28 probands with eoHM (28/47, 59.6%) . Among the 28 probands, pathogenic variants in genes known to be responsible for retinal diseases were found in approximately two-third (64.3% ,18/28) of the probands with eoHM ,including 5 genes (USH2A, PDE6B, RP2, PRPF6 and EYS) responsible for retinitis pigmentosa (RP) and 4 genes (CACNA1F, OAT, TSPAN12 and AIPL1) responsible for congenital stationary night blindness(CSNB), gyrate atrophy of the choroid and retina (GA), Familial exudative vitreoretinopathy (FEVR) and Rod-cone dystrophies repectively.
The high mutation frequency of RetNet genes in eoHM patients have been found in previous studies. RetNet database (https://sph.uth.edu/retnet/) is a website that provides tables of genes and loci causing inherited retinal diseases [in the public domain]) and three other genes, COL9A2, FBN1, and COL18A1, which are responsible for Stickler syndrome, Marfan syndrome, and Knobloch syndrome, respectively.Sun et al.23 performed whole-exome sequencing on 298 eoHM probands and found that pathogenic variants were detected in 34 of 234 genes in 71 probands (23.8%) and variants in genes known to be responsible for retinal diseases were found in approximately one-fourth of the probands with early-onset high myopia. Zhou et al.24 further confirmed this findings on 325 early-onset high myopia probands and identified 76 (23.4%) probands carring RetNet gene variants. Rui et al.34 performed whole-exome sequencing on 20 eoHM probands and found 8 probands were associated with inherited eye diseases carring pathogenic RetNet gene variants. All of these results suggested that variants in RetNet gene is associated with a significant proportion of eoHM and provide clues for genetic screening.
The association of eoHM with ocular and systemic diseases have been revealed in in several previous studies.Marr et al.21 have reported that 112 children with high myopia before the age of 10 years, 92% of them suffered from inherited ocular disease related to high myopia or a syndrome with other systemic abnormalities, among which 14% were inherited retinal dystrophy and 13% were Stickler’s syndrome or Marfan’s syndrome. Logan et al.22 studied 27 probands with eoHM and found that 44% cases had an inherited ocular disease associated with high myopia or syndromes with abnormalities of other systemic systems, of which 25% were inherited retinal diseases manifesting only ocular involvement (simple type) and 19% had syndromes with diseases of other systemic systems, mainly Stickler’s syndrome and Marfan’s syndrome. In this study ,the phenotype analysis showed that 11 probands presented simply high myopia, 10 with inherited retinal disease (IRDs) by eoHM and 7 ocular-only Stickler syndrome (Ocular-STL) by eoHM. For the 10 probands with IRDs by eoHM, the initial clinical records of did not show recognizable signs of original diseases other than high myopia and further specific clinical examinations determined the diagnosis of IRDs ,predominantly RP . The 6 probands, carring variants in USH2A, RPGR, PDE6B, RP2, PRPF6 and EYS, showed RP accompanied by eoHM and 5 probands,carring variants in CACNA1F, OAT, TSPAN12 , WDR36 and AIPL1, showed CSNB, GA, FEVR and rod-cone dystrophies accompanied by eoHM repectively. These findings suggested that eoHM might be a symptom of various forms of retinal dystrophies as well as systemic syndromes caused by a series of known genes. It also implied that eoHM might be the earlier feature than fundus changes as the related gene mutation and the first reason for children visiting ophthalmology clinic , which provide importand clues for genetic screening and further specific clinical examination for definite diagnosis.
Mutations in RPGR are the most common cause of X-linked retinitis pigmentosa (XLRP). In previous study, Sheng et al. have described high myopia as a common symptom of RP in patients with RPGR mutations, especially in carrier females who had high myopia without obvious signs of RP.35 In the present study, mutations in RPGR were identified in 2 of 47 patients with simple eoHM. Both 2 patients were female with heterozygous mutations, and both of the patient’ father carring the same hemizygous mutation showed RP . In Sun et al.study23, mutations in RPGR were identified in 7 of 298 patients with eoHM; 3 were female with heterozygous mutations, and the other 4 were male with hemizygous mutations. All these studys suggest the RPGR as the candidate gene associated with eoHM.
In this study, pathogenic variants in COL2A1 and COL11A1 were detected in 14.9% (7/47) of our cohort with eoHM, which only presented high myopia and ocular changes without other abnormal systemic symptom. COL2A1 and COL11A1 are two common causative genes responsible for Stickler syndrome .Stickler syndrome is an autosomal dominant collagenous connective tissue disease characterized by ocular, facial, articular and auditory lesions,in which high myopia is the most common clinical feature and is the most common cause of inherited retinal detachment. More than 90% of patients with Stickler syndrome have high myopia.36,37
The mutations of the COL2A1 gene usually lead to type 1 Stickler syndrome (STL1) and the mutations of the COL11AL gene cause type 2 Stickler syndrome (STL2). Notably, some mutations in COL2A1 and COL11AL cause solely ocular phenotypes without other sysmetic symptoms36,38,39. The phenotypes of ocular-only Stickler syndrome (ocular-STL) include high myopia and its associated complications (retinal detachment, vitreous degeneration, and cataract). Clinically, it is often difficult to distinguish simple high myopia from Ocular-STL. In the present study, the initial clinical records of 7 probands with variants in COL2A1/COL11AL only show high myopia without the signs of other diseases and the diagnosis as ocular-STL were determined following genitic test and further clinical examinations . Therefore, Whole-exome sequencing were performed for screening candidate genes in 47 probands with eoHM and potential pathogenic variants were identified in 59.6% probands (28 / 47). 31.9% of patients (15/47) with eoHM harbored mutations in 9 genes (COL2A1, COL11A1, TSPAN12, CACNA1F, RPGR , PDE6B, P4HA2 ,OTX2 and JAG1) that were previously reported to cause phenotypes including myopia, and 27.7% of patients (13/47) harbored mutations in 13 other genes not previously reported in high myopia. Of the 34 genes, 11 genes have been reported to cause phenotypes including myopia, Sun et al.23 investigated mutations in 234 genes associated with retinal dystrophies in a cohort of 298 probands with early-onset high myopia using whole exome sequencing and found 14.8% of patients (44/298) with eoHM harbored mutations in 11 genes (COL2A1, COL11A1, PRPH2, FBN1, GNAT1, OPA1, PAX2, GUCY2D, TSPAN12, CACNA1F, and RPGR) that were previously reported to cause phenotypes including myopia, and 9.0% of patients (27/298) harbored mutations in 23 other genes not previously reported in high myopia. Zhou et al.24 investigated mutations in 234 RetNet genes in a cohort of 325 probands with eoHM using whole exome sequencing and the potential pathological variants in 33 genes were detected in 76 of 325 (23.4%) probands with eoHM.Thirty-five of the 76 (46.1%) probands with eoHM had mutations in COL2A1, COL11A1, RPGR, and CACNAIF. Theses researchs suggested that COL2A1, COL11A1, RPGR, CACNA1F genes might be the most common candidate genes of eoHM. Above mententioned previous studies mainly focus on the association between pathogenic variants in RetNet genes and eoHM phenotype, while in this study , to expand our current understanding of the genetic basis of eoHM, we carried out WES study to identify potential causal gene mutations on patients with eoHM and the clinical diagnosis was confirmed by detailed clinical examination and genotype and phenotype analysis. Our results further revealed that eoHM was a symptom of various forms of retinal dystrophies as well as systemic syndromes caused by a series of known genes.
In our study, we used protein–protein interaction (PPI) network analysis to detect interactions between candidate genes and explore possible pathogenic mechanisms.The cluster I analysis show that retinal feedback related gene which is critical in ocular development . Proteins in this cluster are involved in phototransduction—the process by which light signals are converted into electrical signals in the retina. ZNF644 functions as a transcription factor, being retinal feedback mechanisms, which has been identified as a gene associated with high myopia in genetic studies, suggesting it may be involved in the regulation of ocular growth through retinal feedback mechanisms. As protein docking analysis , ZNF644 is bind with P4HA2 which is involved in the structural integrity of the sclera, the remodeling of the collagen matrix in the sclera is essential for maintaining the correct shape and size of the eye. Maybe the transcriptor ZNF644 modulate the P4HA2 expression level, ARR3 is involved in shutting off activated phototransduction proteins, allowing the retina to reset after visual stimuli, critical for light adaptation and retinal feedback regulation. The P4HA2-ZNF644-ARR3 complex may connect retinal feedback with the remodeling of the collagen matrix, ZNF644 play regulate role on sclera response to phototransduction status. Wnt signaling plays a pivotal role in maintaining the vascular integrity of the retina, which is critical for proper retinal function and feedback mechanisms that could indirectly influence ocular elongation by affecting the delivery of visual signals. PCNA and POLA1 are essential for DNA replication and cell cycle regulation, supporting the growth and repair of tissues in the eye. Dysregulation of these proteins could lead to abnormal cell proliferation in the retina or sclera. CRYAB, CRYBB3 was responsible for lens development and protein stability, the dysfunction of these gene may influence the referation status, thereby contributing to myopic development.Through PPI network analysis, we not only identified potential pathogenic genes but also revealed how genes interact with other biological pathways through protein networks, exploring the complexity of the disease from multiple angles and providing a broad mechanistic perspective for eoHM .
As we know, Different ethnic groups vary in genetic backgrounds and other aspects, and these differences may affect the association between genes and diseases. In this study, we recruited 28 families in Ningxia and Gansu regions with a large Hui population. Among the 28 families, 5 are Hui nationality. However, our current study has certain limitations, the sample size of 47 families was insufficient when only targeting Chinese patients with two different ethnic population. To further assess the applicability of these results to other ethnic groups, future research should expand the sample size and include populations from diverse ethnic backgrounds. Another limitation is that the pathogenic mechanism of eoHM caused by caused by a series of known genes remain unclear. Therefore, further functional studies should be conducted to support the pathogenicity and will reveal the pathogenic mechanism of eoHM.
In conclusion, in this study, the potential pathogenic variants in RetNet genes were identified in 59.6% of probands with eoHM and 64.3% eoHM probands with pathogenic variants were confirmed to have inherited retinal diseases.7 pathogenic variants of ocular disease were found to be potentially pathogenic in Chinese eoHM patients for the first time. These findings indicate that the eoHM may be the first reason for children with above mentioned diseases visiting ophthalmology clinic and is often the first diagnosis made by ophthalmologists.Therefore, It is important to screen pathogenic variations in RetNet genes and related genes in eoHM patients that is helpful for the clinicians performing further specific clinical examinations to detect underlying ocular disease or systemic diseases and timely intervention on those diseases.
Materials and methods
Ethical approval
This study was conducted at the Gansu Aier Ophthalmology and Optometry Hospital and People’s Hospital of Ningxia Hui Autonomous Region. The study was approved and reviewed by the Ethics Committee(20190909) on Human Research of above institutions, and it was conducted in accordance with the 1975 Declaration of Helsinki guidelines. Written informed consent was obtained from all included subjects or their legal guardians before participation. Data were collected as previously described in Sheng- XL(https://doi.org/10.3389/fgene.2022.978684).
Probands
The probands who received the initial diagnosis as eoHM and their parents were recruited for both genetic and clinical tests, conducted at Gansu Aier Ophthalmology and Optometry Hospital and Ningxia Eye Hospital, Ningxia Hui Autonomous Region People’s Hospital. In Ningxia Hui Autonomous Region and Gansu Province, in addition to the Han nationality, it is the main settlement of Hui nationality and other ethnic minorities, with a background of multi-cultural communication. The inclusion criteria were as follows: (1) the probands had high myopia (at least one eye with spherical refraction ≤ -6.00D or axial length ≥ 26 mm) at preschool age (< 7 years), (2) absence of congenital diseases, such as congenital cataract, congenital ptosis, and retinopathy of prematurity, (3) absence of ocular traumas or corneal disease that produce secondary high myopia; (4)the probands might have ,or not, other ocular or systemic abnormalities during follow-up.All probands agreed to do whole-exome sequencing.
Clinical evaluations
Routine ophthalmic examinations were performed on all probands and their parents, including slit lamp microscopy, indirect ophthalmoscopy, uncorrected visual acuity (UCVA), best corrected visual acuity (BCVA), visual field examination (750i Carl Zeiss Meditec, USA), color vision examination (5th edition color blindness examination chart, Ziping YU), color fundus photography (TRC-NW300, TOPCON, Japan), optical coherence tomography (OCT) (HD-OCT4000, Carl Zeiss Meditec, USA), fundus fluorescein angiography (FFA), optical coherence tomography angiography (OCTA), and electroretinogram (ERG). The present medical history, previous medical history, personal history, family history and marital history of the probands were recorded and the pedigree chat were drawn.
Whole-exome sequencing
5 ml of peripheral venous blood was collected from each participant, and genomic DNA was extracted using QIAamp DNA Blood Mini Kit (Qiagen, Germany). Whole exonic regions were captured by Agilent SureSelect exome capture kit and sequenced by Illumina at a depth of 100 × (Illumina HiSeq TM2000 paltform, Illumina,America). The raw data were processed by Illumina basecalling Software 1.7 and subsequently compared with the NCBI human genomic DNA reference sequence (GRCh37/hg19). Single nucleotide variation (SNV) information was analyzed using SOAP software (2.x version, http://soap.genomics.org.cn). The information related to inserts and deletions was analyzed by BWA software (0.7.17 version, bio-bwa.sourceforge.net/) to obtain all the variants occurring in the DNA sequences of the samples. Filtered out common variants (usually considered as MAF > 1%) appearing in the database (db135). The variants have no effect on the structure and function of the protein were then filtered out. After stepwise filtering, the unavailable variants with the diseased were eliminated, then filtering to obtain the candidate pathogenic variants. Sanger sequencing was used to exclude false positives for candidate pathogenic variants, while the presence of co-segregation was verified in family members that did not have myopia.40.
In silico analysis
Minimum allele frequency less than 0.005 as the criteria to exclude benign variants by reference to the databases for East Asian populations Allele frequencies available with 1000 Genomes Project (Phase 3, http://browser.1000genomes.org) and Exome Aggregation Consortium (ExAC r1.0, http://exac.broadinstitute.org/). Publicly available servers for bioinformatic prediction tools such as Polyphen2 (2.2.2 version, http://genetics.bwh.harvard.edu/pph2), SIFT (http://sift.jcvi.org), PROVEAN (v1.1.3 version, http://provean.jcvi.org/index.php) and Mutation Taster2 (http://www.mutationtaster.org) were used for to predict the effect of novel mutation sites of the AHCM-associated pathogenic genes on their protein function.
The predicted score of PolyPhen-2 is close to 1, indicating that it is probably damaging (D); otherwise, it is possibly damaging (P) or benign (B). Analyzed by SIFT, amino acids with probabilities < 0.05 are predicted to be deleterious (D). PROVEAN prediction, variants with a score equal to or below − 2.5 are considered deleterious (D), above − 2.5 are considered neutral (N). The scores of Mutation Taster analysis are between 0 and 1, disease probability is higher as the score nears 1.Variants were classified as clinically unclear when at least 1 of 4 predictions had a benign outcome or when there was insufficient evidence of pathogenicity.
Finally ,the pathogenicity of novel variants were classified into five categories “pathogenic,” “likely pathogenic,” “uncertain significance,” “likely benign,” and “benign” according to Standards and Guidelines for Sequence Variants Interpretation issued by American College of Medical Genetics and Genomics (ACMG) in 201533.
Protein–protein interaction (PPI) network analysis
The STRING database(12.5 version, https://string-db.org) (Szklarczyk et al., 2023) applied for detecting the interacting of genes,which integrated both known and predicted PPI. The confidence score greater than 0.4 will be selected for further analysis. Functional enrichment in the network was also showed by STRING under Gene Ontology terms.Visit the String website , to get the myopia-related gene lists under the option “Disease”, followed by a selection of options “Organism” as “Homo sapiens” pair with the term “high myopia” ,then “SEARCH” was clicked and the original data was download. Next, the candidate gene lists were submitted under the option “Multiple proteins”, followed by a selection of options “Organism” as “Homo sapiens”; after that, “SEARCH” was clicked and the original data was download. Clustering of proteins was performed by plugin MCODE(https://apps.cytoscape.org/apps/mcode). The Cytoscape software(3.10.1 version, https://soft.3dmgame.com/down/295981.html ), a bioinformatics software for visualizing and analyzing molecular interaction networks, was used to build a PPI network. The hub genes were detected in Cytoscape with the cytoHubba(https://apps.cytoscape.org/apps/all) ,with a filtering node degree ≥ 2. The criteria for significant gene modules were as follows: degree cutoff = 2, node score cutoff = 0.2, k-core = 2, max depth = 100. In the networks, the nodes correspond to the proteins and the edges represent the interactions.
Protein–protein docking
Predicted three-dimensional structures of ZNF469 (UniProt accession Q9H582), P4HA2 (UniProt accession O15460), and ARR3 (UniProt accession P36575) were generated via AlphaFold online (https://alphafold.ebi.ac.uk/)41 and downloaded in PDB format. These structures were uploaded to the ClusPro Server42 (https://cluspro.org), a protein–protein docking platform that employs rigid-body docking, energy scoring, and clustering to generate optimal interaction models. Default parameters were used to predict binding poses, with the highest-ranked clusters selected based on balanced scoring for electrostatic, hydrophobic, and van der Waals interactions. The resulting docking complexes were downloaded and visualized using PyMOL (version 2.3.2; https://pymol.org)43 to analyze interaction interfaces, render structural images, and validate docking orientations.
Data availability
The datasets generated and analyzed during the current study are available in the [Banklt] repository (BankIt (https://www.ncbi.nlm.nih.gov/WebSub/) ID:2702350, 2702339, 2702333, 2702331, 2702328, 2702313, 2702312, 2702186, 2702178, 2700892, 2704793, 2704969, 2704970).
References
Lipson, M. J., Boland, B. & McAlinden, C. Vision-related quality of life with myopia management: A review. Cont. Lens Anterior Eye 45(3), 101538. https://doi.org/10.1016/j.clae.2021.101538 (2022).
Morgan, I. & Rose, K. How genetic is school myopia?. Prog Retin Eye Res 24(1), 1–38. https://doi.org/10.1016/j.preteyeres.2004.06.004 (2005).
Pärssinen, O. & Kauppinen, M. Risk factors for high myopia: a 22-year follow-up study from childhood to adulthood. Acta Ophthalmol. 97(5), 510–518. https://doi.org/10.1111/aos.13964 (2019).
Czepita, D., Gosławski, W., Mojsa, A. & Muszyńska-Lachota, I. Role of light emitted by incandescent or fluorescent lamps in the development of myopia and astigmatism. Med. Sci. Monit. 10(4), CR68-71 (2004).
Lam, D. S. et al. The effect of parental history of myopia on childrens eye size and growth: Results of a longitudinal study. Invest. Ophthalmol. Vis. Sci. 49(3), 873–876. https://doi.org/10.1167/iovs.06-1097 (2008).
Galvis, V., Tello, A., Camacho, P. A., Parra, M. M. & Merayo-Lloves, J. Bio-environmental factors associated with myopia: An updated review. Arch. Soc. Esp. Oftalmol. 92(7), 307–325. https://doi.org/10.1016/j.oftal.2016.11.016 (2017).
Shi, Y. et al. Exome sequencing identifies ZNF644 mutations in high myopia. PLoS Genet. 7(6), e1002084. https://doi.org/10.1371/journal.pgen.1002084 (2011).
Tran-Viet, K. N. et al. Mutations in SCO2 are associated with autosomal-dominant high-grade myopia. Am. J. Hum. Genet. 92(5), 820–826. https://doi.org/10.1016/j.ajhg.2013.04.005 (2013).
Guo, H. et al. SLC39A5 mutations interfering with the BMP/TGF-β pathway in non-syndromic high myopia. J. Med. Genet. 51(8), 518–525. https://doi.org/10.1136/jmedgenet-2014-102351 (2014).
Zhao, F. et al. Exome sequencing reveals CCDC111 mutation associated with high myopia. Hum. Genet. 132(8), 913–921. https://doi.org/10.1007/s00439-013-1303-6 (2013).
Guo, H. et al. Mutations of P4HA2 encoding prolyl 4-hydroxylase 2 are associated with nonsyndromic high myopia. Genet. Med. 17(4), 300–306. https://doi.org/10.1038/gim.2015.28 (2015).
Xiao, X., Li, S., Jia, X., Guo, X. & Zhang, Q. X-linked heterozygous mutations in ARR3 cause female-limited early onset high myopia. Mol. Vis. 26(22), 1257–1266 (2016).
Ouyang, J. et al. CPSF1 mutations are associated with early-onset high myopia and involved in retinal ganglion cell axon projection. Hum. Mol. Genet. 28(12), 1959–1970. https://doi.org/10.1093/hmg/ddz029 (2019).
Wang, B. et al. A novel potentially causative variant of NDUFAF7 revealed by mutation screening in a chinese family with pathologic myopia. Invest. Ophthalmol. Vis. Sci. 58(10), 4182–4192. https://doi.org/10.1167/iovs.16-20941 (2017).
Pan, H. et al. TNFRSF21 mutations cause high myopia. J. Med. Genet. 56(10), 671–677. https://doi.org/10.1136/jmedgenet-2018-105684 (2019).
Lee, J. K., Kim, H., Park, Y. M., Kim, D. H. & Lim, H. T. Mutations in DZIP1 and XYLT1 are associated with nonsyndromic early onset high myopia in the Korean population. Ophthal. Genet. 38(4), 395–397. https://doi.org/10.1080/13816810.2016.1232415 (2017).
Aldahmesh, M. A. et al. Mutations in LRPAP1 are associated with severe myopia in humans. Am. J. Hum Genet. 93(2), 313–320. https://doi.org/10.1016/j.ajhg.2013.06.002 (2013).
Mordechai, S. et al. High myopia caused by a mutation in LEPREL1, encoding prolyl 3-hydroxylase 2. Am. J. Hum Genet. 89(3), 438–445. https://doi.org/10.1016/j.ajhg.2011.08.003 (2011).
Li, J. et al. Exome sequencing identified null mutations in LOXL3 associated with early-onset high myopia. Mol. Vis. 22, 161–167 (2016).
Li, J. et al. Unique variants in OPN1LW cause both syndromic and nonsyndromic X-linked high myopia mapped to MYP1. Invest. Ophthalmol. Vis. Sci. 56(6), 4150–4155. https://doi.org/10.1167/iovs.14-16356 (2015).
Marr, J. E., Halliwell-Ewen, J., Fisher, B., Soler, L. & Ainsworth, J. R. Associations of high myopia in childhood. Eye (Lond) 15(Pt 1), 70–74. https://doi.org/10.1038/eye.2001.17 (2001).
Logan, N. S., Gilmartin, B., Marr, J. E., Stevenson, M. R. & Ainsworth, J. R. Community-based study of the association of high myopia in children with ocular and systemic disease. Optom. Vis. Sci. 81(1), 11–13. https://doi.org/10.1097/00006324-200401000-00004 (2004).
Sun, W. et al. Exome sequencing on 298 probands with early-onset high myopia: Approximately one-fourth show potential pathogenic mutations in RetNet genes. Invest. Ophthalmol. Vis. Sci. 56(13), 8365–8372. https://doi.org/10.1167/iovs.15-17555 (2015).
Zhou, L., Xiao, X., Li, S., Jia, X. & Zhang, Q. Frequent mutations of RetNet genes in eoHM: Further confirmation in 325 probands and comparison with late-onset high myopia based on exome sequencing. Exp. Eye Res. 171, 76–91. https://doi.org/10.1016/j.exer.2018.02.007 (2018).
Starokadomskyy, P., Escala Perez-Reyes, A. & Burstein, E. Immune dysfunction in Mendelian disorders of POLA1 deficiency. J. Clin. Immunol. 41(2), 285–293 (2021).
Shaheen, R. et al. Mutations in TMEM231 cause Meckel-Gruber syndrome. J.Med. Genet. 50(3), 160–162. https://doi.org/10.1136/jmedgenet-2012-101431 (2013).
Monroe, G. R. et al. Familial Ehlers-Danlos syndrome with lethal arterial events caused by a mutation in COL5A1. Am. J. Med. Genet. 167(6), 1196–1203. https://doi.org/10.1002/ajmg.a.36997 (2015).
Sullivan, L. S. et al. A dominant mutation in hexokinase 1 (HK1) causes retinitis pigmentosa. Invest.Ophthalmol. Vis. Sci. 55(11), 7147–7158. https://doi.org/10.1167/iovs.14-15419 (2014).
Kumar, M. et al. Molecular and structural analysis of genetic variations in congenital cataract. Mol. Vis. 19, 2436–2450 (2013).
Wang, J. et al. Landscape of pathogenic variants in six pre-mRNA processing factor genes for retinitis pigmentosa based on large in-house data sets and database comparisons. Acta Ophthalmol. https://doi.org/10.1111/aos.15104 (2022).
Zhang, R., Shang, F., Li, D., Zhang, Y. & Yuan, L. The first Chinese renal gelsolin amyloidosis with the p.Asp174Asn mutation in the GSN gene: Nephrology picture. J. Nephrol. 34(4), 1257–1259. https://doi.org/10.1007/s40620-020-00873-3 (2021).
Meer, E., Aleman, T. S. & Ross, A. G. WDR36-associated neurodegeneration: A case report highlights possible mechanisms of normal tension glaucoma. Genes (Basel) 12(10), 1624 (2021).
Cheng, W. Y. et al. Identification of genetic variants in five Chinese families with keratoconus: Pathogenicity analysis and characteristics of parental corneal topography. Front. Genet. 6(13), 978684. https://doi.org/10.3389/fgene.2022.978684.PMID:36276932;PMCID:PMC9583916 (2022).
Rui, X. et al. Genotypes and phentypes of hereditary eye diseases associated with early-onset high myopia. Chin J Exp Ohthalmol 41(7), 662–674. https://doi.org/10.3760/cma.j.cn115989-20211216-00695 (2023).
Sheng, X. et al. A novel mutation in retinitis pigmentosa GTPase regulator gene with a distinctive retinitis pigmentosa phenotype in a Chinese family. Mol. Vis. 15(16), 1620–1628 (2010).
Snead, M. P. et al. Stickler syndrome, ocular-only variants and a key diagnostic role for the ophthalmologist. Eye (Lond). 25, 1389–1400 (2011).
Stickler, G. B., Hughes, W. & Houchin, P. Clinical features of hereditary progressive arthro-ophthalmopathy (Stickler syndrome): A survey. Genet. Med. 3, 192–196 (2001).
Liberfarb, R. M. et al. The Stickler syndrome: genotype/phenotype correlation in 10 families with Stickler syndrome resulting from seven mutations in the type II collagen gene locus COL2A1. Genet. Med. 5, 21–27 (2003).
McAlinden, A. et al. Missense and nonsense mutations in the alternatively-spliced exon 2 of COL2A1 cause the ocular variant of Stickler syndrome. Hum. Mutat. 29, 83–90 (2008).
Richards, S. et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 17(5), 405–424. https://doi.org/10.1038/gim.2015.30 (2015).
Jumper, J. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596(7873), 583–589. https://doi.org/10.1038/s41586-021-03819-2 (2021).
Desta, I. T., Porter, K. A., Xia, B., Kozakov, D. & Vajda, S. Performance and its limits in rigid body protein-protein docking. Structure. 28(9), 1071-1081.e3. https://doi.org/10.1016/j.str.2020.06.006 (2020).
Seeliger, D. & de Groot, B. L. Ligand docking and binding site analysis with PyMOL and Autodock/Vina. J. Comput. Aided Mol. Des. 24(5), 417–422. https://doi.org/10.1007/s10822-010-9352-6 (2010).
Acknowledgements
The authors thank all patients and their family members for their participation.
Funding
This work was supported by the National Natural Science Foundation of China (82260206), the National Natural Science Foundation of Ningxia Hui Autonomous Region(2023AAC03485), the key research and development project of Ningxia Hui Autonomous Region(2024BEG02017), the training project of the scientific innovation commanding talented person in Ningxia Hui Autonomous Region (2020GKLRLX13), Major achievement transformation project of Ningxia Hui Autonomous Region(2022CJE09011)
Author information
Authors and Affiliations
Contributions
X.R and W.R wrote the main manuscript, H.L, R.M, S.Y, Y.L, W.C and M.M collected cases data and followed up patients. W.R and X.S polished the article. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval and consent to participate
The Ethics Committee on Human Research at People Hospital in the Ningxia Hui Autonomous Region accepted and examined our work (reference number:20190909), which adhered to the Declaration of Helsinki. Each participant or their legal guardians provided their written informed permission before to taking part.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Rui, X., Li, H., Ma, R. et al. Whole-exome sequencing screening for candidate genes and potential pathogenic variants associated with early-onset high myopia in 47 Chinese families. Sci Rep 15, 11252 (2025). https://doi.org/10.1038/s41598-025-95574-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-95574-x
