
Abstract
Acute myeloid leukemia (AML) is a highly aggressive hematologic malignancy with a poor prognosis for patients. Disulfidptosis response-related long non-coding RNAs (DRLs) have been demonstrated to be closely associated with cancer development. Therefore, this study aims to construct a prognostic DRL signature and investigate the immune microenvironment for AML. RNA-seq and clinical data for AML patients were obtained from The Cancer Genome Atlas (TCGA) database. A total of 344 disulfidptosis-associated lncRNAs were identified, and a prognostic model consisting of seven lncRNAs was constructed and validated. Two risk groups, high-risk and low-risk, were identified. The model demonstrated a robust capacity to predict prognosis, with a worse overall survival for patients in the high-risk group. Additionally, differential expression of the seven lncRNAs were relatively higher in AML samples than in control samples via quantitative polymerase chain reaction(qPCR). The Kyoto Encyclopedia of Genes and Genomes (KEGG) and immune infiltration analysis revealed a substantial infiltration of immune cells and enrichment of immune pathways in the high-risk group. The sensitivity of AML patients to drugs varied according to their risk grade. This study identified a DRL signature, which can effectively predict the prognosis of AML and better understand the mechanism of disulfidptosis in AML. This provides a basis for personalized immunotherapy in AML patients.
Similar content being viewed by others


Introduction
Acute myeloid leukemia (AML) is one of the most lethal hematological malignancies, with an overall survival (OS) rate of only approximately 30% in adults1. AML is a malignant clonal disease originating from myeloid hematopoietic stem/progenitor cells, which is characterized by uncontrolled proliferation and blocked apoptosis of immature myeloid stem/progenitor cells, thereby inhibiting normal hematopoiesis. Although the standard “3 + 7” high-intensity chemotherapy of cytarabine combined with daunorubicin, the application of venetoclax, azacitidine and other targeted drugs, and the promotion of allogeneic hematopoietic stem cell transplantation have improved the prognosis of AML patients, there are still a considerable number of patients with refractory relapse, and the OS rate remains low. Due to the high heterogeneity of AML, different cytogenetic subtypes exhibit distinct gene expression profiles and varying responses to treatment. This article aims to evaluate patients’ prognoses from a molecular level, providing personalized treatment for each AML patient and improving their survival time and quality of life.
Regulatory cell death (RCD) is a precisely regulated mode of cell death that plays an important role in maintaining tissue homeostasis, immune regulation and tumor development2. In RCD, disulfidptosis is a type of death caused by excessive accumulation of disulfide stress, which involves a variety of metabolic pathways of cell death in new ways3. Different from the previously described ferroptosis and cuproptosis, disulfidptosis is characterized by an aberrant accumulation of intracellular disulfide content in tumor cells with high expression of solute carrier family 7 member 11 (SLC7A11) under glucose starvation. This leads to an increase in the content of disulfide bonds in the actin cytoskeleton, which in turn results in excessive contraction of actin filaments. These processes ultimately disrupt cytoskeletal structures and culminate in tumor cell death3. Disulfidptosis represents a promising avenue for the treatment of metabolic cancers. Metabolic therapy using glucose transporter (GLUT) inhibitors has been shown to trigger disulfidptosis and inhibit tumor growth4. In AML, leukemia cells can disrupt systemic glucose metabolism in order to gain a competitive advantage for disease progression5. The most significant regulator of disulfidptosis, SLC7A11, demonstrated a detrimental impact on the viability and colony formation capacity of AML cell lines, with a dependency on cysteine6. This evidence suggests that cystine import may represent a promising drug target for AML. Consequently, the induction of disulfidptosis by glucose starvation has the potential to be a therapeutic strategy for the treatment of AML7. Nevertheless, the mechanism of disulfidptosis and its regulatory network in AML remain unclear. It is imperative to further investigate the regulatory factors associated with disulfidptosis in order to enhance the prognostic evaluation and treatment efficacy of AML.
In the past, the identification of genetic alterations in AML has been primarily focused on protein-coding genes. However, as our understanding of the human genome has improved and more cancer-related genes have been discovered, non-protein-coding parts of our genome are also thought to show different expression profiles in cancer. These breakthroughs in the discovery of functional non-coding RNAs, especially in long non-coding RNAs (lncRNAs), provide researchers and clinicians with new molecular drivers, therapeutic targets, and opportunities for refractory cancers such as AML. LncRNA is a class of non-coding RNA with a length of more than 200 nucleotides and lacks protein-coding function. It primarily regulates gene expression in epigenetics, pre-transcription and post-transcription by interacting with protein, DNA or RNA8. LncRNA plays a pivotal role in AML, and it has been demonstrated that lncRNA expression profile exhibits AML subtype specificity9 and participates in a multitude of biological processes of AML, including driving proliferation, differentiation arrest, treatment resistance, and as a biomarker to predict the prognosis of AML10,11. For instance, HOXA10-AS, a stem cell-specific lncRNA, stimulates proliferation by inducing NF-κB pathway in KMT2A-mutated AML blasts12. LncRNA HOTTIP is one of the lncRNAs that impedes the differentiation of myeloid cells in AML. It enhances the self-renewal ability of hematopoietic cells and causes differentiation block by interacting with hematopoiesis-specific epigenetic regulators and transcription factors, including STAT5a, MYC, RUNX1 and DOT1L13. Moreover, lncRNAs can be employed as biomarkers for cancer diagnosis and risk stratification, with significant therapeutic potential14. For instance, the prediction model of disulfidptosis response-related lncRNAs (DRLs) can accurately predict the prognosis of colon cancer patients15. Recent studies have identified a small number of lncRNAs as diagnostic or prognostic biomarkers in AML16,17,18. A variety of regulatory cell death-related lncRNAs, such as cuproptosis-related lncRNAs, establish AML prognostic models and are potential therapeutic targets19. Nevertheless, the relationship between disulfidptosis-related genes (DRGs) and lncRNAs has not yet been reported in the context of leukemia.
In this study, we employed the Cancer Genome Atlas (TCGA) database to identify DRLs associated with AML prognosis. Furthermore, we developed a prediction model for DRLs. Furthermore, the gene set enrichment analysis (GSEA) and immune infiltration analysis were employed to elucidate the mechanism of action of DRLs in AML. Finally, the expression of lncRNAs in clinical AML samples was validated by quantitative polymerase chain reaction (qPCR). Consequently, our study elucidates the interrelationship between disulfidptosis, AML prognosis and the tumor immune microenvironment, thereby furnishing a robust foundation for the implementation of individualized clinical treatment.
Results
Identification of disulfidptosis-related differentially expressed LncRNAs in AML
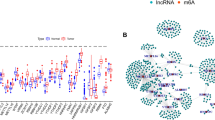
The workflow diagram of this study is shown in Fig. 1. RNA-seq data from 151 samples were downloaded from TCGA. Finally, 128 eligible samples were subjected to analysis. Following normalization, 16,080 lncRNAs and 19,436 miRNAs were identified, respectively. Common platforms for analyzing genetic alterations include Gene4Denovo, VarCards, MethSurv, and cBioPortal. Among these, cBioPortal is distinguished by its significant advantages in multi-dimensional data integration, visualization, survival analysis, and user-friendliness. Specifically, it offers comprehensive access to large-scale cancer genomic data, encompassing mutations, copy number alterations, and expression profiles. Its advanced visualization and analysis tools facilitate effective exploration of genetic alterations in cancer-related genes20,21. In this study, we employed cBioPortal to identify and analyze the mutation status of 28 DRGs in TCGA-AML patients. The study found that the proportion of associated DRGs that were genetically altered in cancer patients was low (Fig. 2A), indicating that their gene sequences were relatively stable. These findings established the necessary research conditions for the subsequent step of screening DRLs based on DRGs. This approach utilized the aforementioned DRLs to construct a disulfidptosis related prognostic model for AML patients, exhibiting genetic and statistical significance. Subsequently, 344 lncRNAs related to 28 DRGs were identified from the TCGA database via Pearson analysis on the basis of R > 0.5 and P < 0.05 (Fig. 2B). The correlation and regulatory relationships between DRGs and these lncRNAs are presented in Table S1.

Workflow diagram of this study.

Screening of disulfidptosis-associated lncRNAs and co-expression analysis with DRGs. (A) The genetic alterations of 28 DRGs in AML patients were evaluated using cBioPortal. The Oncoprint plots indicate that from high to low alterations are MYH10 (1.5%), ACTB (1%), CAPZB (0.5%), and GYS1 (0.5%). (B) A co-expression network was constructed between the 28 DRGs and the 344 lncRNAs identified in AML patients (R > 0.5, P < 0.05). The blue coloration in the figure represents DRGs and the yellow coloration represents lncRNAs.
Construction of the prognostic model based on seven DRLs
A total of 128 patients were divided into a training set (n = 96) and a validation set (n = 32). In order to initially exclude DRLs that may be associated with prognosis, univariable Cox regression analysis identified 26 disulfidptosis-associated lncRNAs that were significantly correlated with OS and an increased risk for AML (Fig. 3A). In order to restrict the scope of prognostic DRLs and prevent the model from becoming overfitted, least absolute shrinkage and selection operator (LASSO) analysis was conducted to extract potential DRL signatures for prognostic prediction in AML patients, resulting in the identification of 12 lncRNAs (Fig. 3B-C). Subsequently, in order to ascertain the dependability of the LASSO Cox regression screening outcomes and to construct the ultimate prognostic model, a seven-lncRNA signature for prognosis was established based on multivariate Cox regression (Fig. 3D). We performed the multicollinearity test with all the variance inflation factor (VIF) < 5 and all covariates in the multivariate Cox regression satisfied the proportional hazard (PH) hypothesis and multicollinearity test (Fig.S1). Regression coefficients were calculated for each enrolled lncRNA to form a prognostic risk score formula to construct a stable prognostic model. The risk score was calculated as follows: risk score = 1.357 × FAM30A expression + 1.423 \(\:\times\:\) MRPL20-AS1 expression + 1.27 × AC022182.2 expression + 1.24 × AL391704.1 expression + 1.481 × LINC01539 expression + 1.381 × AP000439.1 expression + 1.888 × AC113423.1 expression. The regulatory relationships between the DRGs and the seven lncRNAs are illustrated in a Sankey chart (Fig. 3E). Cox regression analysis showed that the seven lncRNAs screened were strongly associated with the prognosis of AML patients, and the stepwise method ensured the reliability and validity of the prognostic models.

Construction of the prognosis of AML patients with DRLs. (A) Forest plot of 26 differential lncRNAs analyzed by Univariate Cox regression. (B, C) Cvfit and lambda curves validate the lncRNA results of LASSO regression analysis. (D) Forest plot of multivariate Cox regression analysis of 7 DRLs on the prognosis of AML patients. (E) Sankey diagram of the correlation between DRGs and DRLs.
Performance of the signature for AML patients in the TCGA database
Based on the previous risk score, 96 AML patients were divided into the high-risk and low-risk groups (48 and 48, respectively). The high-risk group exhibited a lower survival rate, shorter survival time, and worse prognosis in comparison to the low-risk group (Fig. 4A-B). The receiver operating characteristic (ROC) curve was employed to assess the predictive accuracy of the risk score model. The area under the ROC curve (AUC) for the 1-year, 3-year, and 5-year OS rates were 0.82, 0.88, and 0.9, respectively, indicating a high predictive power of this feature in the training set (Fig. 4C).

Validation of the signature in the training set. (A) Distribution of the risk score (top) and survival time of each patient (down). (B) Kaplan-Meier survival analysis of OS in AML patients in the high- and low-risk groups in the training set. (C) ROC curve analysis and AUC projections for 1-, 3- and 5-year OS in the training set.
The same algorithm was employed to calculate the risk scores in both the validation and entire sets. The results of the risk score distribution and scatter diagram analysis were found to be consistent with those of the training set (Fig. 5A-B). Furthermore, the Kaplan-Meier curves of high-risk AML patients demonstrated a worse survival prognosis (Fig. 5C-D). The AUC for the 1-, 3-, and 5-year OS were 0.6, 0.57, and 0.64, respectively, in the validation set and 0.76, 0.8, and 0.89, respectively, in the entire set. This indicates that the risk prediction model has good predictive ability in survival prognosis (Fig. 5E-F).

Validation of the signature in the validation and entire set. (A, B) Distribution of the risk score (top) and survival time of each patient (down) in the validation (A) and entire set (B). (C, D) Kaplan-Meier survival analysis of OS in AML Patients in the high- and low-risk groups in the validation (C) and entire set (D). (E, F) ROC curve analysis and AUC projections for 1-, 3-, and 5-year OS in the validation (E) and entire set (F).
Independent prognostic value of the DRL signature for AML patients
In order to identify independent prognostic factors for AML patients in the TCGA cohort, univariate and multivariate Cox regression analyses were performed. Seven factors were analyzed, including the established lncRNA risk model score, age (≥ 60 years old vs. <60 years old), sex (male vs. female), FAB classification (M4/M5 vs. non-M4/M5 subtype), blast in bone marrow (BM) (≥ 70% vs. < 70%), white blood cell (WBC) counts (≥ 30 vs. < 30 × 109/L) and platelet counts (≥ 50 vs. < 50 × 109/L). The univariate Cox regression analysis demonstrated that the lncRNA risk model and age were significant predictors of OS in AML patients (Fig. 6A). Furthermore, the multivariate Cox regression analysis indicated that the lncRNA risk model and age were independent predictors of OS in AML patients (Fig. 6B). To improve the accuracy of the prognosis of patients with AML, a nomogram was developed which incorporated the aforementioned clinicopathological characteristics as well as the risk score (Fig. 6C). This nomogram was capable of predicting the prognosis of AML patients at 1-, 3-, and 5-years intervals. Furthermore, the calibration curves demonstrated a high degree of congruence between the observed and anticipated survival rates at 1, 3, and 5 years (Fig. 6D).

Analysis of the value of independent prognostic features of the model. (A, B) Univariate (A) and multivariate (B) Cox regression analyses of clinically independent prognostic characteristics. (C) A nomogram that combines risk scores and clinical factors to predict 1-, 3-, and 5-year probability of OS. (D) Calibration curves for 1-, 3-, and 5-years OS.
Relationship between the DRL signature and the clinicopathological characteristics in AML patients
Following the construction of the DRL signature, an investigation was conducted into its correlation with the clinicopathological characteristics of AML patients, including gender, FAB classification, age, blast ratio, WBC counts, platelet counts and molecular biological changes. The high-risk group identified through somatic mutation data analysis exhibited a higher frequency of gene mutations. KIT was the most frequently mutated gene in the high-risk group, while the most common mutation in the low-risk group was observed in NPM1 (Fig. 7A-B). The risk scores for AML patients older than 60 years of age were found to be significantly higher, while no significant differences were observed between risk scores and other clinical variables (Fig. 7C-H).

Differences in Clinical Characteristics and Biological Factors Between AML Patients in the high- and low-risk scoring groups in the TCGA Database. (A, B) Differences in somatic mutation frequency between the high-risk (A) and low-risk(B) scoring groups. (C-H) Differences in risk scores between clinicopathological factors. The interquartile ranges of the values are represented by the upper and lower ends of the boxes. The median values are represented by the lines in the boxes. The statistical P-values are represented by the asterisks (**P < 0.01; ns, no significance).
Validation in a real-world clinical cohort
qPCR was employed to assess the expression levels of the seven DRLs in 40 AML and 10 healthy control samples. The results demonstrated that the expression levels of the seven lncRNAs were relatively higher in AML samples than in control samples. Of the lncRNAs examined, FAM30A, MRPL20-AS1 and AC113423.1 exhibited significantly elevated expression levels in AML patients (Fig. 8A-G). In comparison to the normal group, the risk score obtained from the training set and the significant clinical parameter of age exhibited a notable increase in the AML group (Fig. 8H-I). Furthermore, the relationship between the seven selected lncRNAs and clinical parameters was investigated in 40 AML patient samples. Correlation analysis with clinicopathological factors revealed a positive correlation between the risk score and blast ratio, WBC counts, particularly age (Fig. 8J).

Verification of the correlation between clinicopathological factors and disulfidptosis-related lncRNA prognostic model. (A–G) The expression levels of 7 DRLs in AML samples. The interquartile ranges of the values are represented by the upper and lower ends of the boxes. The median values are represented by the lines in the boxes. The statistical P-values are represented by the asterisks (*P < 0.05; **P < 0.01; ns, no significance). (H,I) Difference analysis of risk score and age in the disulfidptosis prognostic model between normal and AML patient samples. The statistical P-value was represented by the asterisks (*P < 0.05; ***P < 0.001). (J) Correlation analysis between risk scores and clinicopathological factors. The squares were colored from blue to red for increasing correlation and the statistical P-value was represented by the asterisks (*P < 0.05). (K) Expression analysis of SLC7A11 in AML patients in the high-risk and low-risk groups. The statistical P-value was represented by the asterisks (***P < 0.001). (L)The NADP+/NADPH ratio was measured in blood samples from patients in the high-risk and low-risk groups. The statistical P-value was represented by the asterisks (***P < 0.001).
In addition, a series of functional experiments were performed to verify the correlation between the prognostic model and disulfidptosis. A total of 40 AML patients were included in the study to compare the expression levels of the disulfidptosis key gene SLC7A11 and the key enzyme NADPH (nicotinamide adenine dinucleotide phosphate) between the high- and low-risk groups. Our findings revealed that SLC7A11 exhibited elevated expression in the high-risk group (Fig. 8K), and concomitantly, the NADP+/NADPH ratio was found to be significantly elevated in the high-risk group compared to the low-risk group (Fig. 8L). These observations were consistent with the overall changes of the examined lncRNAs, thereby further strengthening the confidence in the efficacy of the constructed lncRNA prognostic model.
KEGG pathway analysis for the high- and low-risk AML patients
In order to gain a deeper insight into the biological processes and pathways associated with the two risk groups identified through the use of the DRL signature, an investigation was conducted using GSEA software to identify enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways22,23. The GSEA analysis revealed that two pathways were enriched in the high-risk group and three pathways were enriched in the low-risk group, respectively (P < 0.05) (Table S2). In the high-risk group, the enriched pathways were predominantly associated with the activation, differentiation, migration, and immune effect of immune cells (Fig. 9). These pathways were closely linked to the core mechanisms of immune response, thereby providing a foundation for immune analysis within this risk model.

GSEA analyses enrichment pathway differences between the high- risk and low-risk groups.
Immune infiltration status and TME of AML patients in different risk groups
The single-sample gene set enrichment analysis (ssGSEA) algorithm was employed to assess the status and disparities in immune cell infiltration and immune-related functions in AML patients between the high-risk and low-risk groups. Tumor-infiltrating immune cells, including B cells, neutrophils, and follicular helper T cells (Tfh cells), were found to be significantly upregulated in the high-risk group. Additionally, MHC class I and T cell co-stimulation were also highly increased. Furthermore, in the high-risk group, tumor lymphocytes, including activated CD8 + T cells, central memory CD4 + T cells, type 2 T helper cells and eosinophils, exhibited significant differences (P < 0.05; Fig. 10A-C). These findings indicate that the infiltration of these immune cells into the tumor microenvironment (TME) may have a significant impact on the prognosis of AML patients.

Immune cell infiltration and TME studies. (A–C) Differences in tumor-infiltrating immune cells, immune-related functions and tumor-infiltrating lymphocytes between AML patients in the high- and low-risk groups. The interquartile range of values was represented by the upper and lower ends of the boxes. The median values are represented by the median lines of the boxes, and the outliers are represented by the black dots. The statistical P-value is represented by the asterisks (*P < 0.05). (D) Differences in immune checkpoint expression between the high-risk and low-risk groups. The statistical differences were tested by the one-way ANOVA between the high- risk and low-risk groups. The statistical P-value was represented by the asterisks (*P < 0.05). (E, F) Comparison of ESTIMATE score and tumor purity in the high- and low- risk group. The statistical differences were tested by the one-way ANOVA between the high- risk and low-risk groups. The statistical P-value was represented by the asterisks (*P < 0.05; ***P < 0.001).
The clinical implications of immunotherapy strategies for AML prompted us to investigate the associations between risk scores and multiple immune checkpoints. Many immunotherapy targets that have proven to be clinically beneficial, such as CD27, PDCD1 (PD-1), CD276, etc., were found to be upregulated in high-risk patients (Fig. 10D). Furthermore, the ESTIMATE score demonstrated that the TME and tumor purity of high-risk patients were significantly higher than those of low-risk patients (Fig. 10E-F).
Prediction of drug sensitivity in AML patients of different risk groups
By predicting the sensitivity of prospective AML medications in different risk groups, it was demonstrated that AML patients exhibited differential drug sensitivity, with patients in the high-risk group demonstrating diminished responsiveness to cytarabine and doxorubicin, coupled with heightened sensitivity to imatinib, cyclopamine, rapamycin, sorafenib, and WZ-1-84, when compared to those in the low-risk group (Fig. 11).

The sensitivity of AML patients in the high- and low-risk groups to various drugs was predicted. The interquartile ranges of the values are represented by the upper and lower ends of the boxes. The median values are represented by the lines in the boxes. The statistical P-values are represented by the asterisks (*P < 0.05; **P < 0.01; ***P < 0.001).
Discussion
Disulfidptosis is a type of cell death caused by the excessive formation of intracellular disulfide bonds. It is characterized by the influence of intracellular redox balance and mitochondrial function3. Its role in the occurrence and development of tumors has attracted increasing attention. Nevertheless, elevated oxidative stress and mitochondrial dysfunction have been documented in AML cells24,25,26, and these abnormalities are pivotal initiators of disulfidptosis, suggesting that this recently identified form of cell death may be a prospective therapeutic target for the treatment of cancers, including AML. It has been demonstrated that a disulfideptosis-based signature can predict the prognosis of a variety of tumor types27,28,29. Furthermore, some studies have indicated a link between DRG and the occurrence and progression of AML, and DRG-based risk score models have been constructed30.
LncRNA is involved in the occurrence and development of tumors and has demonstrated prognostic value in a range of tumors. Nevertheless, no study has yet investigated the prognostic value of lncRNA in disulfidptosis in AML patients. Consequently, the objective was to construct a novel prognostic model based on DRLs in order to more accurately assess the prognosis of AML patients.
In this study, lncRNAs related to DRGs were analyzed in TCGA AML RNA-seq data, and 344 lncRNAs related to disulfidptosis were screened by differential analysis. To construct a disulfidptosis related prognostic model, 128 AML patients from TCGA database were divided into training set and validation set, and 26 lncRNAs with prognostic potential were identified by univariate Cox regression analysis. To avoid overfitting, LASSO regression and multivariate Cox regression were further performed to obtain 7 lncRNA signatures related to disulfidptosis with prognostic value, thereby providing new predictors for the prognosis of AML. By calculating the risk score of each patient, the patients were divided into two groups: a high-risk group and a low-risk group. An OS rate of the high-risk group was inferior to that of the low-risk group. The ROC curve also showed that this predictive feature had good predictive performance and accuracy. With the exception of age, none of the other clinicopathological factors demonstrated as much reliability as lncRNA signatures in terms of prognosis in AML patients.
The model of these seven lncRNAs reflected their powerful predictive power for AML prognosis and their potential as biomarkers for individualized immunotherapy in AML. In order to validate the prognostic value of these lncRNAs in AML and their significant correlation with patient survival through clinical data, the predictive ability of the constructed model was confirmed and a basis for the clinical application of the seven lncRNAs as AML prognostic biomarkers was provided. These seven lncRNAs were validated in a real-world setting, revealing that FAM30A, MRPL20-AS1, and AC113423.1 exhibited notable alterations in AML patients. Among the seven lncRNA signatures related to disulfidptosis, FAM30A has been reported to be a positive risk factor in AML patients, and its high expression indicates a worse prognosis31,32. Furthermore, FAM30A is associated with t [8;21] and RUNX1 mutations. The up-regulation of FAM30A expression in Leukemic stem cells enhances its crosstalk with the bone marrow microenvironment by increasing the expression of intercellular molecules, which may lead to immune escape33. MRPL20-AS1 has been demonstrated to be susceptible to hypoxic damage in endothelial cells34, whereas LINC01539 is highly expressed in thyroid cancer and is associated with a poor prognosis35. Furthermore, other lncRNAs have not been reported in specific studies and their functions require further investigation. It is noteworthy that among the seven screened lncRNAs, such as LINC01539, there appears to be an absence of statistical significance. It is proposed that these lncRNAs function within complex regulatory networks, rather than through direct up-regulation or down-regulation. Consequently, these lncRNAs were identified not on the basis of individual differential expression but rather on the basis of their collective prognostic value as feature lncRNAs. For complex diseases, such as AML, individual molecular markers often do not exhibit significant differences when assessed in isolation. Nevertheless, their integration enhances the prognostic accuracy of prediction models through their combined effects36,37. The enhanced predictive capability of the model is attributed to the integrated patterns of all markers, rather than the individual significance of each component. Accordingly, we determined the prognostic significance of this synergy using the statistical methods of Cox regression and LASSO.
SLC7A11 and NADPH have been shown to play a pivotal role in the processes of disulfidptosis. In the context of glucose starvation, intracellular NADPH is consumed in SLC7A11-overexpressed cells38. Consequently, cystine and other disulfides are abnormally accumulated, inducing disulfidptosis. The notable disparity in SLC7A11 and NADPH, between AML high and low-risk groups in clinical practice aligned with the comprehensive alterations in DRLs, reinforcing the reliability of the prognostic model as a robust tool for forecasting AML prognosis.
To explore the functional mechanism of the constructed DRL signature on AML prognosis, KEGG pathway analysis was performed by GSEA. The results showed that the enriched significant pathways in high-risk patients were closely related to immune-related processes and signaling pathways, suggesting that immune cells, cytokines and their receptors, and allogeneic immune response may play an important role.
A considerable body of research has demonstrated the infiltration and interaction between tumor cells and immune cells in the TME, which plays a pivotal role in the immunotherapy of AML patients with acute myeloid leukemia. Furthermore, the potential value of immune treatment strategies for AML has been substantiated39,40,41,42,43,44. The study revealed that high-risk patients with AML exhibited elevated levels of TME immune activity and immune cell infiltration.
In the high-risk group, Tfh cells, which play a pivotal role in B cell activation and antibody production44, and B cells, were upregulated, reflecting the immune activation status in AML patients. Concurrently, the activation of CD8 + T cells and the central memory CD4 + T cells was observed to be elevated in the high-risk group42. AML cells have the capacity to significantly alter the composition of the bone marrow immune microenvironment. Despite the presence of high levels of immune cell infiltration, this does not appear to impede disease progression, suggesting that the anticancer immune activity of these immune cells may be compromised, potentially leading to unfavorable outcomes. The results demonstrate that the immunogenicity of the tumor itself leads to a more active immune response in high-risk patients with AML. However, this immune response is accompanied by an abnormal immune function and an increase in tumor immune escape. Furthermore, the ESTIMATE algorithm score indicates that the of high-risk patients with AML is significantly more complex and dynamic, creating a favorable environment for tumor growth and potentially interfering with antitumor immune responses, resulting in tumors with higher purity and a poor prognosis.
In addition, in patients with high-risk AML, multiple known immune checkpoint (IC) expressions are significantly elevated, including PD-1, CTLA4, and LAG-3, among others43. The combination of immune checkpoint inhibitors (ICIs) can be achieved by blocking the IC and its ligand, which inhibits tumor immune escape and improves the immune system’s ability to kill tumor cells45. PD-1/PD-L1 inhibitors have been demonstrated to activate T lymphocytes and enhance antibody-dependent cytotoxicity. Some studies have indicated that PD-1/PD-L1 can improve the resistance of AML patients to demethylation drugs46. CTLA4 exerts its regulatory influence on T lymphocyte activation primarily through the modulation of CD4 + T lymphocyte activation and the augmentation of regulatory T cells (Tregs), thereby establishing a tumor immunosuppressive phenotype. CTLA-4 inhibitors, such as ipilimumab, work primarily by blocking or depleting Tregs47. Tregs inhibit the function of antigen-presenting cells by depleting immune-stimulating cytokines, producing immunosuppressive cytokines, and constitutively expressing CTLA-4. LAG-3 is a kind of immune inhibitory checkpoint receptor that is expressed in the activation of T lymphocytes, B lymphocytes, natural killer cells, dendritic cells and tumor-infiltrating T lymphocytes. It competes with CD4 to identify major histocompatibility complex II with high affinity and other ligands. Various phase clinical trials are currently underway, investigating the use of the LAG-3 monoclonal antibody relatlimab, either as a monotherapy or in combination with PD-1/PD-L1 inhibitors, for the treatment of both solid tumors and hematological malignancies48. The developed DRL signature demonstrates the relationship between disulfidptosis and the tumor immune microenvironment, providing a theoretical basis for the implementation of individualized immunotherapy for AML patients.
Finally, we also predicted the drug sensitivity of AML patients in different risk groups and found that the high-risk group was less sensitive to cytarabine and doxorubicin. It was more sensitive to imatinib, cyclopamine, rapamycin, sorafenib and WZ-1-84, including some drugs not used in the clinical treatment of AML. The above findings, combined with the risk model of DRL we have established, may provide more precise and personalized treatment strategies for AML patients.
However, it should be noted that our results are subject to certain limitations. The analysis was conducted using TCGA RNA-seq data, which is relatively limited in terms of the number of samples. Consequently, it is necessary to further expand the test scope of samples and to verify the accuracy of the prediction model through transcriptome data of other independent AML cohorts and real-world clinical follow-up data. Moreover, although the seven lncRNAs used to construct the model demonstrated expression correlation with DRGs, their precise functions and mechanisms in disulfidptosis require further verification and exploration. Similarly, the molecular mechanisms by which these lncRNAs regulate the occurrence and development of AML must be further investigated in the future.
Despite these limitations, this is the first study to construct a DRL signature for AML patients, which demonstrated good predictive power in effectively differentiating AML patients with different prognostic risks and revealing its relationship with the AML immune microenvironment. It provides a new tool for accurate prognosis assessment and individualized treatment in clinical practice. In the future, we will conduct further multi-center large-sample verification to explore the molecular mechanism and evaluate the clinical application value. This will enable us to parse out the relationship between disulfidptosis and AML and facilitate the clinical translation of research results.
Materials and methods
Data collection and processing
The RNA-seq based transcriptome profiling data and clinical data for 151 AML patients were obtained from the TCGA database (https://portal.gdc.cancer.gov). Exclusion criteria included cases without complete clinical data and patients diagnosed with M3 subtype according to the FAB classification methodology, resulting in a final sample size of 128 patients. The clinical characteristics of these AML patients are summarized in Table S3.
Identification of DRLs
To identify the DRLs, 28 DRGs (Table S4) were obtained based on previous studies49,50. Genetic alterations in DRGs of AML patients from the TCGA database were analyzed using the cBioportal network analysis tool (http://www.cbioportal.org/). Pearson correlation analysis was conducted to identify lncRNA co-expressed with DRGs, with a correlation coefficient of > 0.5 and a P-value of < 0.05 set as the screening thresholds. The correlation data were exported and the co-expression networks of DRGs and lncRNAs were mapped using the “igraph R” package.
Construction and validation of the prognostic DRL signature for AML patients
A total of 128 AML samples were randomly divided into two groups using the “caret” R package. One group was used for model construction (training set, n = 96), while the other was used for model validation (validation set, n = 32).
The “survival” R package was employed to define P < 0.05 as screening criteria, after which the disulfidptosis-associated lncRNAs were subjected to univariate cox analysis in order to identify AML patient survival-associated lncRNAs. The optimal prognosis-associated lncRNA subsets were then further screened from the candidate lncRNAs using least absolute shrinkage and selection operator (LASSO) Cox regression analysis. Finally, multivariate cox regression analysis was performed in order to establish the lncRNA prediction model.
The formula for the disulfidptosis-related prognostic risk score was as follows: Risk Score==\(\:\sum\:_{i=1}^{n}\beta\:\times\:lncRNA\) expression. The regulatory Sankey diagrams for DRGs and screened lncRNAs were analyzed and displayed using the “ggalluvial” R package.
Based on the median value of the risk score, patients were divided into the high-risk and low-risk groups, and survival analysis was carried out to evaluate the significance of the prognostic model. The validation set was employed to assess the reliability of this prognostic model, with the entire set of samples undergoing survival analysis.
Assessment of the prognostic model’s independent clinical predictive value
To ascertain the independent prognostic value of the constructed model of lncRNA risk associated with AML, univariate and multivariate Cox regression analyses were conducted in conjunction with clinicopathological parameters, including age, gender, FAB classification, blasts in BM, WBCs and platelets. A nomogram predicting 1-, 3-, and 5-year OS of AML patients was developed based on the aforementioned pathological characteristics using the “rms” R package. The accuracy of the predictions was assessed using calibration curves.
Analysis of mutation
The somatic mutation data was downloaded from the TCGA database (https://portal.gdc.cancer.gov/) and saved in MAF files. The “maftools” R package was employed to examine the differences in mutations between the high-risk and low-risk groups, with oncoplot graphs subsequently generated.
Functional enrichment analysis
In order to investigate the molecular mechanism and biological process involved in the DRL signature, GSEA was performed to identify the significantly enriched pathways between the high-risk and low-risk groups. KEGG dataset c2.cp.kegg.v7.5.symbols.gmt was utilized as a reference. A threshold of P < 0.05 and FDR < 0.25 was employed to identify significant enrichment.
qPCR
The study involving humans was approved by the local institutional review board (number L203154). All research was conducted in accordance with the local regulatory and institutional requirements and “Declaration of Helsinki”. The participants provided their written informed consent to participate in this study. A total of 40 AML patients and 10 healthy volunteers were recruited for the study. Peripheral blood samples were collected from each participant and mononuclear cells were isolated to validate the expression pattern of the identified prognostic lncRNAs and the different expressions of SLC7A11, a key gene for disulfidptosis. Total RNA was extracted from mononuclear cells using the Trizol reagent (15596-026, Invitrogen) and was then reverse transcribed to cDNA using the FastKing RT Kit (KR116, TIANGEN). qPCR was conducted on the CFX Connect Real-Time PCR Detection System (Biorad, USA) using the ChamQ Universal SYBR qPCR Master Mix reagent (Q711, Vazyme). The relative expression of lncRNAs was normalized to GAPDH and calculated by the 2−ΔΔCT method. The primers utilized for qPCR are presented in Table S5.
Verification of the correlation between clinicopathological factors and disulfidptosis-related LncRNA prognostic model
A total of 40 AML patients and 10 healthy volunteers were selected for analysis. The risk score and age of the two groups were compared and analyzed. The correlation between the risk score and other clinicopathological factors in the 40 AML patients was analyzed using the “corrplot” R package. The samples from AML patients were classified into high-risk and low-risk groups based on the median risk score. Subsequently, the expression of SLC7A11 was compared between the two groups.
Furthermore, the NADP+/NADPH ratio was measured in blood samples from patients using the NADP+/NADPH Assay Kit (ab65349, abcam) according to the manufacturer’s instructions. Blood cells were treated and lysed with the provided extraction buffer, followed by centrifugation to obtain the supernatant. The resulting extracts were then divided into two distinct fractions. One fraction was subjected to heating at 60 °C for a duration of 30 min, with the objective of eliminating NADP+. Subsequently, both the heated and unheated samples were added to a 96-well plate, along with G6PDH working solution and the chromogenic agent. The resulting mixture was then subjected to spectrophotometric analysis, with the measurement of light absorption at a wavelength of 450 nanometers being carried out using a dedicated microplate reader. The NADP+/NADPH ratio was subsequently calculated using the following formula: N𝐴𝐷𝑃+/𝑁𝐴𝐷𝑃𝐻 𝑟𝑎𝑡𝑖𝑜 = (𝑁𝐴𝐷𝑃𝑡 ‒ 𝑁𝐴𝐷𝑃𝐻)/ 𝑁𝐴𝐷𝑃H.
Evaluation of TME and immune infiltration status
To investigate the immune infiltration status of AML patients with different risk groups, we performed a ssGSEA analysis of tumor-infiltrating immune cells from AML patients in the high-risk and low-risk groups using the “GSVA” R software package, which uses the 16 tumor-infiltrating immune cells gene set, 13 immune function gene set51 and 28 lymphocytes gene set from TISIDB database (http://cis.hku.hk/TISIDB/). Furthermore, the expression of relevant immune checkpoints was analyzed. Subsequently, the “estimate” R package was utilized to compare TME scores between risk groups, with the objective of assessing the immune and stromal scores of each AML sample.
Assessment of potential drugs sensitivity
The Genomics of Drug Sensitivity in Cancer (GDSC) (https://www.cancerrxgene.org/) database was consulted to obtain the half-maximal inhibitory concentration (IC50) and clinical gene expression data. These were then utilized by the “pRRophetic” R package to predict the sensitivity of AML patients in different risk groups to potential drugs. The IC50 value for each drug was estimated based on the gene expression profiles of AML patients, with the objective of risk stratification. A lower IC50 value is indicative of heightened drug sensitivity. To ensure the reliability of the results, the prediction accuracy was validated using a 10-fold cross-validation approach. Drug sensitivity predictions were performed for several clinically relevant drugs, including conventional chemotherapeutic agents (cytarabine and doxorubicin) and targeted therapies (imatinib, cyclopamine, rapamycin, sorafenib, and WZ-1-84). The statistical significance of differential drug sensitivity between risk groups was evaluated using the Wilcoxon rank-sum test, with a p-value of less than 0.05 considered statistically significant.
Statistical analysis
All statistical analyses were conducted using R software (version 4.4.0). Kaplan-Meier curves and log-rank tests were employed for the purpose of conducting a survival analysis. Univariate and multivariate Cox regression analyses were conducted to assess the prognostic independence and risk model for AML patients. To compare the levels of tumor-infiltrating immune cells, immune checkpoints, and immune function between the two risk groups, the Wilcoxon test was employed. P < 0.05 was considered statistically significant.
Data availability
All data generated or analyzed in the study are included in the article and in the supplementary material. Further inquiries can be directed to the corresponding authors.
References
Kantarjian, H. et al. Acute myeloid leukemia: current progress and future directions. Blood Cancer J. 11, 41 (2021).
Tang, D., Kang, R., Berghe, T. V., Vandenabeele, P. & Kroemer, G. The molecular machinery of regulated cell death. Cell. Res. 29, 347–364 (2019).
Liu, X. et al. Actin cytoskeleton vulnerability to disulfide stress mediates Disulfidptosis. Nat. Cell. Biol. 25, 404–414 (2023).
Zheng, P., Zhou, C., Ding, Y. & Duan, S. Disulfidptosis: a new target for metabolic cancer therapy. J. Exp. Clin. Cancer Res. 42, 103 (2023).
Ye, H. et al. Subversion of systemic glucose metabolism as a mechanism to support the growth of leukemia cells. Cancer Cell. 34, 659–673e6 (2018).
Pardieu, B. et al. Cystine uptake Inhibition potentiates front-line therapies in acute myeloid leukemia. Leukemia 36, 1585–1595 (2022).
Machesky, L. M. Deadly actin collapse by Disulfidptosis. Nat. Cell. Biol. 25, 375–376 (2023).
Huarte, M. The emerging role of LncRNAs in cancer. Nat. Med. 21, 1253–1261 (2015).
Connerty, P. et al. Development of siRNA-Loaded lipid nanoparticles targeting long Non-Coding RNA LINC01257 as a novel and safe therapeutic approach for t(8;21) pediatric acute myeloid leukemia. Pharmaceutics 13, 1681 (2021).
Mishra, S., Liu, J., Chai, L. & Tenen, D. G. Diverse functions of long noncoding RNAs in acute myeloid leukemia: emerging roles in pathophysiology, prognosis, and treatment resistance. Curr. Opin. Hematol. 29, 34–43 (2022).
Zhu, G. et al. HOXBLINC long non-coding RNA activation promotes leukemogenesis in NPM1-mutant acute myeloid leukemia. Nat. Commun. 12, 1956 (2021).
Al-Kershi, S. et al. The stem cell-specific long noncoding RNA HOXA10-AS in the pathogenesis of KMT2A-rearranged leukemia. Blood Adv. 3, 4252–4263 (2019).
Luo, H. et al. HOTTIP LncRNA promotes hematopoietic stem cell Self-Renewal leading to AML-like disease in mice. Cancer Cell. 36, 645–659e8 (2019).
McKiernan, J. et al. A novel urine exosome gene expression assay to predict High-grade prostate cancer at initial biopsy. JAMA Oncol. 2, 882–889 (2016).
Chi, H. et al. Unraveling the role of disulfidptosis-related LncRNAs in colon cancer: a prognostic indicator for immunotherapy response, chemotherapy sensitivity, and insights into cell death mechanisms. Front. Mol. Biosci. 10, 1254232 (2023).
F, Z. et al. m6A-related LncRNAs predict prognosis and indicate immune microenvironment in acute myeloid leukemia. Sci. Rep. 12, (2022).
Mer, A. S. et al. Expression levels of long non-coding RNAs are prognostic for AML outcome. J. Hematol. Oncol. 11, 52 (2018).
Atnaf, A. et al. The role of long noncoding RNAs in the diagnosis, prognosis and therapeutic biomarkers of acute myeloid leukemia. Ann. Hematol. 103, 4931–4942 (2024).
Li, P. et al. A novel cuproptosis-related LncRNA signature: prognostic and therapeutic value for acute myeloid leukemia. Front. Oncol. 12, 966920 (2022).
Cerami, E. et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012).
Gao, J. et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013).
Kanehisa, M. & Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–462 (2016).
Testa, U., Labbaye, C., Castelli, G. & Pelosi, E. Oxidative stress and hypoxia in normal and leukemic stem cells. Exp. Hematol. 44, 540–560 (2016).
Sriskanthadevan, S. et al. AML cells have low spare reserve capacity in their respiratory chain that renders them susceptible to oxidative metabolic stress. Blood 125, 2120–2130 (2015).
Panina, S. B., Pei, J. & Kirienko, N. V. Mitochondrial metabolism as a target for acute myeloid leukemia treatment. Cancer Metab. 9, 17 (2021).
Xiao, L. et al. A disulfidptosis-related LncRNA index predicting prognosis and the tumor microenvironment in colorectal cancer. Sci. Rep. 13, 20135 (2023).
Qu, J., Guan, H., Zheng, Q. & Sun, F. Molecular subtypes of disulfidptosis-regulated genes and prognosis models for predicting prognosis, tumor microenvironment infiltration, and therapeutic response in hepatocellular carcinoma. Int. J. Biol. Macromol. 261, 129584 (2024).
Liang, J. et al. Identification of disulfidptosis-related subtypes, characterization of tumor microenvironment infiltration, and development of a prognosis model in breast cancer. Front. Immunol. 14, 1198826 (2023).
Zhong, F. et al. Development and validation of a disulfidptosis-related scoring system to predict clinical outcome and immunotherapy response in acute myeloid leukemia by integrated analysis of single-cell and bulk RNA-sequencing. Front. Pharmacol. 14, 1272701 (2023).
Zhang, T., Liao, D. & Hu, Y. Cuproptosis-related LncRNAs forecast the prognosis of acute myeloid leukemia. Transl Cancer Res. 12, 1175–1195 (2023).
Wang, F. et al. A three–lncRNA signature for prognosis prediction of acute myeloid leukemia in patients. Mol. Med. Rep. 18, 1473–1484 (2018).
Hornung, R. et al. Mediation analysis reveals common mechanisms of RUNX1 point mutations and RUNX1/RUNX1T1 fusions influencing survival of patients with acute myeloid leukemia. Sci. Rep. 8, 11293 (2018).
Zietzer, A. et al. The LncRNA MRPL20-AS1 is associated with severe OSAS and downregulated upon hypoxic injury of endothelial cells. Int. J. Cardiol. 369, 65–68 (2022).
Ershov, P. V., Yablokov, E. O., Mezentsev, Y. V. & Ivanov, A. S. Long intergenic Non-Coding RNAs of human chromosome 18: focus on cancers. Biomedicines 12, 544 (2024).
Hong, Y. et al. Ferroptosis-Related gene signature for prognosis prediction in acute myeloid leukemia and potential therapeutic options. Int. J. Gen. Med. 17, 3837–3853 (2024).
Li, D. et al. Identification of m6A-Related LncRNAs associated with prognoses and immune responses in acute myeloid leukemia. Front. Cell. Dev. Biol. 9, 770451 (2021).
Liu, X., Zhang, Y., Zhuang, L., Olszewski, K. & Gan, B. NADPH debt drives redox bankruptcy: SLC7A11/xCT-mediated cystine uptake as a double-edged sword in cellular redox regulation. Genes Dis. 8, 731–745 (2021).
Zeng, T. et al. The establishment of a prognostic scoring model based on the new tumor immune microenvironment classification in acute myeloid leukemia. BMC Med. 19, 176 (2021).
Menter, T. & Tzankov, A. Tumor microenvironment in acute myeloid leukemia: adjusting niches. Front. Immunol. 13, 811144 (2022).
Lamble, A. J. & Lind, E. F. Targeting the immune microenvironment in acute myeloid leukemia: A focus on T cell immunity. Front. Oncol. 8, 213 (2018).
Greiner, J. et al. Mutated regions of nucleophosmin 1 elicit both CD4(+) and CD8(+) T-cell responses in patients with acute myeloid leukemia. Blood 120, 1282–1289 (2012).
Bakhtiyari, M. et al. The role of bone marrow microenvironment (BMM) cells in acute myeloid leukemia (AML) progression: immune checkpoints, metabolic checkpoints, and signaling pathways. Cell. Commun. Signal. 21, 252 (2023).
Guo, Z. et al. The association of Circulating T follicular helper cells and regulatory cells with acute myeloid leukemia patients. Acta Haematol. 143, 19–25 (2020).
Singh, S. et al. Immune checkpoint inhibitors: a promising anticancer therapy. Drug Discov Today. 25, 223–229 (2020).
Yang, X., Ma, L., Zhang, X., Huang, L. & Wei, J. Targeting PD-1/PD-L1 pathway in myelodysplastic syndromes and acute myeloid leukemia. Exp. Hematol. Oncol. 11, 11 (2022).
Ramagopal, U. A. et al. Structural basis for cancer immunotherapy by the first-in-class checkpoint inhibitor ipilimumab. Proc. Natl. Acad. Sci. U S A. 114, E4223–E4232 (2017).
Tawbi, H. A. et al. Relatlimab and nivolumab versus nivolumab in untreated advanced melanoma. N Engl. J. Med. 386, 24–34 (2022).
Liu, H. & Tang, T. Pan-cancer genetic analysis of disulfidptosis-related gene set. Cancer Genet. 278–279, 91–103 (2023).
Wang, T. et al. Disulfidptosis classification of hepatocellular carcinoma reveals correlation with clinical prognosis and immune profile. Int. Immunopharmacol. 120, 110368 (2023).
Su, H., Peng, C. & Liu, Y. Regulation of ferroptosis by PI3K/Akt signaling pathway: a promising therapeutic axis in cancer. Front. Cell. Dev. Biol. 12, 1372330 (2024).
Acknowledgements
We would like to acknowledge the contributions of the researchers involved in TCGA.
Funding
This study was supported by the National Natural Science Foundation of China (82170153), Taizhou Clinical Medical College of Nanjing Medical University (TZKY20230302) and Jiangsu Province Traditional Chinese Medicine Technology Development Plan Project (MS2023151).
Author information
Authors and Affiliations
Contributions
J.Z. and S.Q. contributed to conception and design of the study; P.X. and X.S. performed the statistical analysis and wrote the main manuscript text; L.P. obtained clinical samples and conducted the validation experiments. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, P., Sun, X., Pan, L. et al. Disulfidptosis-related LncRNAs forecast the prognosis of acute myeloid leukemia. Sci Rep 15, 13635 (2025). https://doi.org/10.1038/s41598-025-95607-5
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-95607-5
