
Abstract
Given the severe side effects of prolonged morphine use, the search for safer alternatives is a global priority. This study investigates marine bacteria from sediments along the Bejaia coast, Algeria, to identify opioid-like bioactive compounds with potential analgesic properties. A total of 45 bacterial strains were isolated using six different culture media, with 18 strains exhibiting antimicrobial activity. Molecular identification based on 16S rRNA gene sequencing classified these strains into six genera: Streptomyces, Nocardiopsis, Alloalcanivorax, Pseudonocardia, Sinomicrobium, and Lysinibacillus. One strain, S5T2H1, was identified as a new Streptomyces species through a polyphasic approach. LC-HRESIMS analysis of secondary metabolites revealed that strain S56T3J31 produced A58365A, antimycin A, and three potentially novel compounds. However, strain S5T2H1 synthesized cyclo(phenylalanyl-prolyl) and niphimycin Ia, along with three unidentified metabolites, while strain S7T2H1 secreted a single compound identified as berberifuranol. Molecular docking and molecular dynamics simulations demonstrated that A58365A exhibited strong interactions with the μ-opioid receptor (5C1M), showing a stable binding affinity comparable to morphine. These findings highlight marine-derived bacterial compounds as promising candidates for opioid drug development.
Similar content being viewed by others
Introduction
The opioid drugs, such as morphine, heroin, and fentanyl, are widely used as potent analgesics for managing moderate to severe pain1. In addition to their primary role in pain relief, these drugs are also utilized in other medical applications, including the treatment of drug dependence2. Morphine, the prototypical opioid, is a naturally occurring alkaloid derived from the opium poppy (Papaver somniferum)3. It exerts its analgesic effects by binding activating the μ-opioid receptor, which is primarily located in the nervous system. This interaction triggers a cascade of physiological responses, ultimately leading to pain relief4. However, prolonged use of morphine is associated with severe side effects, including respiratory depression, sedation, constipation, and nausea5. As a result, morphine-related deaths have risen dramatically in recent years.
Given these challenges, the search for alternative analgesics that retain the therapeutic efficacy of morphine while minimizing its adverse effects has become a global priority. In this context, marine environments have emerged as a valuable source of bioactive secondary metabolites with diverse pharmacological activities6. Notably, several marine-derived compounds have demonstrated opioid-like activity, presenting promising leads for the development of safer analgesics7.
Among these marine ecosystems, the Algerian coast, which forms part of the larger Mediterranean coastline, spans approximately 1600 km from the western border of Tunisia to the eastern border of Morocco8. This region harbors a wealth of untapped habitats that support unique microbial communities, many of which remain largely unexplored9. Due to the specific and diverse microclimatic conditions of these coastal environments, some microbial species found in these regions may be endemic10. Therefore, exploring marine microorganisms isolated from Algerian coastal ecosystems could lead to the discovery of novel, safer alternatives to conventional opioid drugs, offering a potential solution to the severe side effects associated with morphine. In this context, this study focuses on culturable bacteria from marine sediments along the Bejaia coastline (Algeria) to identify bioactive compounds with pharmaceutical relevance as potential alternatives to morphine. The main goals of this study were to: (i) Investigate the biodiversity of culturable bacteria isolated from marine sediments collected from the Bejaia coastline (Algeria). (ii) Assess their antimicrobial potential against various target microorganisms, in order to select the potent strains. (iii) Identify and annotate secondary metabolites from the selected strains using High-Performance Liquid Chromatography coupled with High-Resolution Mass Spectrometry (HPLC-ESI-HRMS), and. (iv) Exploring the identified marine-derived compounds secreted by selected marine bacterial strains as potential alternatives to morphine, through in silico approaches, including docking studies, molecular dynamics simulations and MM/GBSA binding free energy calculations.
Materials and methods
Sampling of marine sediments
The isolation of marine bacterial strains was carried out from marine sediments samples collected from four different sites, located on the west coast of Bejaia, Algeria: Tazboujth (36°48′13 0.96″N and 5°00′52.17″E), Cap Sigli (36°53′52 0.65″N and 4°45′31.00″E), fishing port (36°49′31.68″N and 4°56′ 49.97″E), Saket (36°49′50 0.44″N and 4°56′10.35″E). The samples were collected at a depth of 2 to 3 m, packed in sterile bags, and stored at 4 °C for further analysis.
Determination of physicochemical properties of sediments
The physicochemical parameters of the sediments, including pH, salinity (g/L), and organic matter (%), were analyzed for each sediment sample collected from Tazeboujth, Port de Pêche, Saket, and Cap Sigli in Bejaia, Algeria. pH was measured using a pH meter (Hanna HI5120), and salinity was determined using an electrical conductivity meter (Hanna HI5120). However, organic matter content was determined by drying the samples at 100 °C for 24 h, followed by ashing at 550 °C, with calculations based on weight loss. Peat humification was assessed using the method of Schumacher11.
Isolation of marine bacterial strains
The marine sediment samples were air-dried at room temperature to reduce moisture content and eliminate fast-growing, non-spore-forming bacteria. One gram of each dried sediment sample was diluted in 3 mL of sterile seawater (dilution 1/3) to resuspend microbial cells while maintaining osmotic balance. This was followed by moderate heat treatment at 55 °C for 6 min to selectively enrich heat-resistant and spore-forming bacteria, such as actinobacteria and certain Bacillaceae members12. An aliquot of 100 µL was taken from this suspension and spread in duplicate on agar plates containing six different media: Casein Starch Agar (SCA)13, Marine 2 Medium (M2)14, NaST21Cx Agar15, Nutrient-Poor Sediment Medium (NPS), Nutrient-Rich Sediment Medium (NRS), and Selective Mannitol Peptone Medium (SMP)12, each designed to support the growth of diverse marine bacterial taxa with potential bioactive metabolite production.
Screening for antimicrobial activity of marine bacterial strains
Preparation of bacterial suspension
The antibacterial activity of the marine bacterial strains was evaluated against nine pathogenic bacterial species. Among them, five Gram-positive bacteria, including Staphylococcus aureus ATCC 25923, methicillin-resistant Staphylococcus aureus (MRSA) ATCC 43300, Listeria innocua CLIP 74915, Bacillus subtilis and Enterococcus faecalis, were tested. Similarly, four Gram-negative bacteria were also assessed: Escherichia coli ATCC 25922, Klebsiella pneumoniae ATCC 700603, Pseudomonas aeruginosa ATCC 27853, and Proteus sp. It is worth mentioning that two of the tested strains, Enterococcus faecalis and Proteus sp., were obtained from the University Hospital Center Khellil AMRANE in Bejaia, having been isolated from clinical samples collected from patients. Furthermore, the antifungal activity was tested against nine microscopic fungi, including seven filamentous fungi, such as Aspergillus niger 939N, Aspergillus carbonarius A731C, Aspergillus flavus NRRL, Aspergillus ochraceus NRRL3174, Aspergillus parasiticus CB5, Mucor rammanianus NRRL 1829, Fusarium polyferatum, Botrytis cinerea, and one yeast, Candida albicans.
To prepare microbial suspensions, the tested bacterial strains were inoculated into nutrient broth and incubated at 37 °C for 18 h. However, the yeast Candida albicans was seeded in MYG medium (1.0% phytone peptone, 1.0% glucose, 50 mM HEPES [11.9 g/L], pH 7.0) and incubated at 30 °C for 48 h. In addition, the tested microscopic filamentous fungi were grown in Malt Extract Agar (MEA) at 25 °C for 5 days.
For each tested bacterial and yeast suspension, the turbidity was adjusted to 0.5 McFarland and 0.1 McFarland, respectively, measured at 600 nm using a Shimadzu UV spectrophotometer 1240. However, for the tested microscopic fungi, spore suspensions were adjusted to 107 spores/mL in physiological saline using a hemocytometer16. This standardized suspension was then utilized in subsequent antimicrobial activity assays.
Antimicrobial activity of marine bacterial strains
The marine bacteria isolates were inoculated on 5294 plates medium (Glucose: 15.0 g/l; Soymeal 15.0 g/l; Corn steep 5.0 g/l; CaCO3 2.0 g/l; NaCl 5.0 g /l; Eau distillée: 1000 ml. pH = 7.2), and incubated at 30 °C for 14 days. The antibacterial activity of marine bacteria strains was evaluated by agar cylinder method. 6 mm cylinders were cut from the well-developed culture of bacteria strains and then placed on the surface of Muller Hinton medium for bacteria17, MYG for yeast and MEA medium for microscopic filamentous fungi18, already seeded with test microorganisms. The plates were maintained at 4 °C for 4 h to ensure good diffusion of bioactive compounds secreted by bacteria strains, followed by incubation to the suitable temperature. The inhibition areas were measured after 24 h (bacteria and yeasts) and 48 h (microscopic fungi)19.
Molecular identification of active marine bacterial strains
The genomic DNA of bacteria strains has been extracted using Invisorb Spin Plant Mini Kit (Invitek. GmbH, Berlin, Germany), following the manufacturer’s protocol. The quality of the achieved DNA was checked via an 0.8% agarose gel electrophoresis. The 16S rRNA gene region was amplified through PCR with 2 universal primers on the positions 27F (AGA GTT TGA TCC TGG CTC AG) and 1525R (AAG GAG GTG WTC CAR CC). The PCR reaction was conducted using a special master mix containing primers, PCR water and “Jump Start Ready Mix (JSRM)” including, 99% pure deoxynucleotides, JumpStart Taq DNA polymerase, and buffer in an optimized reaction concentrate20.
The PCR reaction was performed in 200 µL microtubes containing: 22 µl PCR water, 25 µl JSRM, 1 µl reverse and forward primer dilution as well as 1 µl of the template DNA. One tube remains without DNA and therefore serves as control. The PCR reaction was conducted in a Mastercycler Gradient (Eppendorf, Hambourg, Germany) under the program: Initial denaturation at 95 °C for 5 min, 35 cycles of denaturation at 94 °C for 30 s, annealing at 52 °C for 30 s, elongation at 72 °C for 120 s, and a final extension at 72 °C for 10 min. Before 16S rRNA sequencing, the PCR products have been purified from the reaction residues using NucleoSpin® Gel and PCR Clean-up kit (MachereyNagel, Düren, Germany). The success of the PCR was checked by electrophoresis. 16S rRNA genes of bacteria strains were sequenced using two primers such as 27F and R5188.
The obtained sequences were checked for quality and assembled using the program ‘‘Bioedit alignment, v 7.0.5.3”. The obtained sequences were compared to an online databank using the “Basic Local Alignment Search Tool” (BLAST) of the National Center for Biotechnology Information (NCBI)20. Selected strains, which exhibited low percentages of similarity with the closest type species, were subjected to morphological, physiological, and biochemical characterization. The results were compared to the closest species described in the Compendium of Actinobacteria: https://www.dsmz.de/collection/catalogue/microorganisms/special-groups-of-organisms/compendium-of-actinobacteria.
Cultural characteristics were determined according to the methods described by Shirling and Gottlieb21. Physiological characterization, including growth at different temperatures and NaCl concentrations, was assessed using GYM medium as the basal medium after two weeks of incubation. The ability of the selected strains to utilize ten different carbon sources (glucose, arabinose, sucrose, xylose, inositol, mannose, fructose, rhamnose, raffinose, cellulose) was tested at 30 °C, following the methods outlined by Shirling and Gottlieb21. Enzymatic activities, including nitrate reductase, urease, gelatin liquefaction, and H₂S production, were determined using API® CORYNE, API® CAMPY, and API® Zym test strips (bioMérieux, Marcy-l’Étoile, France).
HPLC–UV-HRMS analysis of crude extracts from selected marine bacterial strains
Bioactive compounds in crude extracts from selected marine bacterial strains were annotated and identified through analysis using HPLC [Agilent 1260 series RP-HPLC system: column 50 × 2.1 mm Acquity UPLC BEH C18 (Waters); solvent A: 0.1% formic acid in H2O, B: 0.1% formic acid in acetonitrile; gradient system: 5% B for 0.5 min, in 19.5 min to 100% B and maintained for 5 min at 100% B; flow rate 0.6 mL/min; 40 °C; DAD-UV detection at 200–600 nm], coupled to mass spectrometry, High resolution electrospray ionization mass spectrometry (HRESIMS), data were recorded on a MaXis ESI-TOF-mass spectrometer (Bruckers Daltonics). Molecular formulas were calculated including the isotopic pattern with the Smart Formula algorithm (Bruckers). Comparison of results was performed by Dictionary of Natural Product. Compounds annotation were only be concluded if the respective mass shown distinctive molecular ion cluster ([M+H]+, [M+Na]+, [2M+H]+, or [2M+Na]+.
In silico study
Molecular docking
The Glide program, part of the Schrödinger suite and enhanced with Extra Precision (XP) capabilities, was used to attach ligands into the crystal structure of the mu-upload receptor in association with the BU72 agonist protein. XP docking, which is more thorough and selective than the standard precision (SP) technique, takes longer due to its complex methodology. It is designed for ligands that have already obtained high scores in SP docking, and it uses a comprehensive scoring methodology that requires stronger compatibility between ligand and receptor geometries. This meticulous process aims to eliminate the false positives often encountered with SP, by imposing penalties on ligands that poorly match the chosen receptor conformation22. To perform the molecular docking both the receptor (Crystal structure of active mu-opioid receptor bound to the agonist BU72 with 5C1M PDB) and ligands, were prepared through addition of hydrogen, removal of water molecules and energy minimization. The active site of the receptor was identified via existing binding affinity of 4VO ((2S,3S,3aR,5aR,6R,11bR,11cS)-3a-methoxy-3,14-dimethyl-2-phenyl-2,3,3a,6,7,11c-hexahydro-1H-6,11b-(epiminoethano)-3,5a-m ethanonaphtho[2,1-g]indol-10-ol). As a result, docking against numerous receptor conformations is advised in order to find ligands with appropriate binding energies for molecular dynamic simulation analysis.
Molecular dynamics simulations
The Desmond software, version 2.0 (Schrödinger Release 2024-4) https://www.schrodinger.com/platform/products/desmond/, was used to evaluate the stability of receptor-ligand complexes generated by molecular docking23,24,25,26,27. The simulation setup included the TIP3P water model within a cubic periodic box measuring 10 Å on each side, utilizing the Simple Point Charge (SPC) technique, combined with the OPLS all-atom force field from 200528. To achieve charge neutrality, sodium ions were introduced. A number of regulated processes were used to reduce the energy of the complexes and prepare them for equilibrium. Molecular dynamics (MD) simulations were run using periodic boundary conditions, using the OPLS 2005 force field parameters, a 1 ps relaxation duration, and a constant temperature of 300 K and volume in the NPT ensemble29. To achieve charge neutrality, sodium ions were introduced. A number of regulated processes were used to reduce the energy of the complexes and prepare them for equilibrium. Molecular dynamics (MD) simulations were run using periodic boundary conditions, using the OPLS 2005 force field parameters, a 1 ps relaxation duration, and a constant temperature of 300 K and volume in the NPT ensemble.
MM-GBSA (molecular mechanics generalized born surface area) binding energies
The MM-GBSA and MM-PBSA approaches were used to calculate the binding free energies of protein–ligand complexes. The PRIME module in Maestro 11.4, together with the OPLS-2005 force field, was utilized to calculate the binding energies of the ideally docked ligand-receptor couples. The binding energy is calculated using a particular formula:
where ∆EMM denotes the energy difference after minimization of the protein–ligand complex, ∆GSolv is the discrepancy in GBSA solvation energy between the complex and the individual protein and ligand energies, and ∆GSA refers to the change in surface area energies within the protein–ligand complex compared to the sum of their separate surface energies. The minimization of the protein–ligand complexes was achieved through the local optimization feature in PRIME30.
Results and discussion
Physicochemical analysis of marine sediment
The physicochemical analysis, presented in Supplementary Table S1, includes pH, salinity, and organic matter, was conducted for four marine sediment samples collected from different regions of Bejaia, Algeria. These parameters are key environmental factors influencing microbial diversity31. Results indicated that all sites exhibited alkaline conditions (pH 7.78–8.13), likely due to carbonate-rich coastal sediments, which can support alkaliphilic bacteria. However, Tazeboujth had the lowest salinity (0.468 g/L) and organic matter content (1.07%), which may limit microbial diversity due to nutrient scarcity. In contrast, Saket (8.248 g/L) and Cap Sigli (7.216 g/L) exhibited high salinity and organic matter content, potentially favoring halotolerant microbial communities32. These findings suggest that each site harbors a distinct microbial ecosystem shaped by variations in acidity/alkalinity, salinity, and nutrient availability. The interplay between these factors likely influences the composition and adaptability of microbial communities in each marine sediment32,33.
Isolation of marine bacterial strains
The investigation of microbial diversity in marine sediment collected from four different sites in the Bejaia region, Algeria, using different culture media led to the isolation of 45 distinct marine strains, selected based on their diverse morphological characteristics (Supplementary Fig. S1). Exploring the microbial diversity of the underexplored Algerian coast presents a unique opportunity to identify potent marine-derived compounds targeting the μ-opioid receptor, potentially providing innovative scaffolds for safer and more effective analgesics4,6. To date, few studies have investigated bacterial diversity in this region, making our findings valuable for expanding the current understanding of marine microbial communities in Algeria8,9.
The comparison between the efficiency of the culture media used indicates that Casein Starch Agar (SCA)13,34 was the most effective medium for isolating bacteria from marine sediments collected in the Bejaia region, Algeria (Fig. 1). A total of 24 isolates were obtained using this medium, while 14 isolates were recovered from M214, 5 from NaST21Cx15, and 2 from NRS6. No isolates were obtained using NPS or SMP media12. This difference could be explained by the presence of starch and casein in SCA medium, which stimulate the growth of several bacteria, including actinomycetes19. Several studies have confirmed that this medium is effective for the selective isolation of bacteria from various ecosystems35,36,37,38,39.

Distribution of the number of bacteria isolates according to the culture medium.
Media with a low concentration of organic nutrients, such as NPS and NRS, delay the growth of fast-growing bacteria for a sufficient period, enabling slow-growing bacteria to emerge40. However, in our case, we were only able to obtain two isolates using NRS. Different pretreatment procedures and selective isolation media are recommended for the isolation of marine bacteria, such as Actinobacteria. There is no universal isolation technique, but it is always necessary to vary the methods and media in the same screening41.
Screening for antimicrobial activity
Antibacterial activity
The antibacterial activity of the 45 marine bacterial strains was assessed against four Gram-negative and five Gram-positive bacteria, with the results presented in Supplementary Table S2. Among the 45 tested isolates, 11 strains exhibited antimicrobial activity against at least one tested bacterium. Among them, three strains demonstrated activity exclusively against Gram-positive bacteria, while two strains showed activity only against Gram-negative bacteria. Additionally, six strains displayed activity against both Gram-positive and Gram-negative bacteria. Among the marine bacterial isolates, strain S5T2H1 was the most active, exhibiting significant inhibition zones, particularly against Gram-positive bacteria. It showed an inhibition zone of 35 mm against Staphylococcus aureus, 30 mm against both MRSA and Enterococcus faecalis, and 28 mm against Listeria innocua. Additionally, this strain demonstrated activity against the Gram-negative bacterium Pseudomonas aeruginosa, with an inhibition zone of 23 mm (Supplementary Fig. S2).
Regarding the resistance of the tested bacteria, both K. pneumoniae and Proteus sp. exhibited the highest resistance, as none of the marine bacterial strains showed activity against them. In contrast, Bacillus cereus was the most susceptible. The susceptibility of Gram-positive bacteria to the bioactive compounds secreted by marine bacterial strains, compared to Gram-negative bacteria, may be attributed to differences in their cell wall structure. The presence of lipopolysaccharides in the outer membrane of Gram-negative bacteria reduces permeability to lipophilic compounds, making them more resistant42.
The stronger the antimicrobial activity exhibited by the marine bacterial strains, the more it suggests that these isolates secrete a diverse array of bioactive compounds, potentially extending beyond antibacterial properties to include metabolites with pharmacological potential43,44,45. Therefore, given that many secondary metabolites from marine microorganisms interact with human receptors, the marine strains isolated from the Algerian coast could serve as a valuable source of novel compounds for targeting the μ-opioid receptor, providing promising scaffolds for the development of safer and more effective analgesics46,47.
Antifungal activity
The antifungal activity of the 45 marine bacterial strains was evaluated against eight filamentous fungi and one yeast. The results are presented in Supplementary Table S3. The results indicate that eleven strains exhibited antifungal activity against at least one of the eight tested filamentous fungi (Supplementary Fig. S3). Among these, S5T2H1 was the most active strain, exhibiting potent antifungal activity against five tested filamentous fungi, along with the yeast Candida albicans, with inhibition diameters ranging from 13 to 35 mm (Supplementary Fig. S3). Additionally, Candida albicans exhibited greater sensitivity to the bioactive compounds secreted by marine actinomycete strains compared to the tested filamentous fungi, as indicated by the inhibition zones ranging from 15 to 28 mm (Supplementary Fig. S3).
Marine bacteria are a valuable resource for drug discovery against various diseases48,49. Therefore, the antifungal activity of the secondary metabolites secreted by marine bacteria isolated from the Algerian littoral suggests their ability to interact with eukaryotic cells, such as filamentous fungi and Candida albicans. This indicates that these marine strains could be promising candidates for producing compounds that may interact with the human μ-opioid receptor, offering potential scaffolds for the development of new analgesics50,51.
Molecular identification and phylogenetic study of selected actives strains
The eighteen marine bacterial strains, selected from the 45 obtained isolates for their significant antimicrobial activity, were subjected to molecular identification based on 16S rRNA gene sequencing, as they could be promising candidates for producing compounds that may interact with the human μ-opioid receptor. The obtained sequences were compared to the closest homologs registered in the GenBank database using the BLAST tool, with the results presented in Supplementary Table S2. The results revealed that the identified isolates belong to six different genera, including seven Streptomyces strains, six Nocardiopsis strains, and three strains each affiliated with Alloalcanivorax and Pseudonocardia. Additionally, a single strain was identified as Sinomicrobium, and another as Lysinibacillus.
Among the identified strains, four Nocardiopsis isolates were assigned to N. terra (S29T3J31, S57T3J31; 99.65–99.70% similarity) and N. umidischolae/N. alborubida (S3T3I1, S7T2H1; 99.40% similarity) (Supplementary Table S4). Three strains (S5T3I1, S1T2Kmr, S6T3I1) showed high similarity to Pseudonocardia species (99.80–100%), supported by strong phylogenetic bootstrap values (Fig. 2). Additionally, two strains, S1PHmr, S1PKmr, were related to the species Streptomyces antimycoticus and Streptomyces aculeolatus, respectively, with 100% similarity. Meanwhile, strains S56T3J31, S2T2H1, and S6T2H1 were identified as S. humidus, S. albogriseolus, and S. chitinivorans, respectively, with 99.30–99.75% similarity.

Molecular phylogenetic analysis by maximum likelihood method, based on almost-complete 16S rRNA gene sequences showing the position and phylogenetic relationship between the some selected strains and the type strains of the closest species. Numbers at the nodes are bootstrap values; expressed as a percentage of 1000 re-samplings (only values 50% are shown).
Phylogenetic analysis confirmed the molecular identification of S37T3J31 and S28T3J31, clustering them with Sinomicrobium oceani (99.90% similarity) and Lysinibacillus fusiformis (99.80%) (Fig. 2, Supplementary Table S4). Additionally, Alloalcanivorax strains S1T3J31, S4T3I1, and S2T3I1 formed a stable phylogenetic clade with A. dieselolei, A. balearicus, and A. xenomutans (Fig. 2). The Actinobacteria genera Streptomyces and Nocardiopsis have been previously reported in Algerian marine environments. However, this study marks the first isolation of Pseudonocardia, Sinomicrobium, Alloalcanivorax, and Lysinibacillus from Algerian marine sediments8,52. The latter three genera have been previously found in deep-sea sediments53,54, methane cold seep sites55,56, and tidal flats57,58.
One strain, S5T2H1, shares 98.5% 16S rRNA gene sequence similarity with Streptomyces coelicoflavus, which is below the threshold for differentiating between bacterial species. According to Kim et al.59, this threshold is set at 98.65%, meaning that strains with lower similarity are likely considered different species. Fourthermore, the phylogenetic analysis (Fig. 2) demonstrated that this strain forms a stable and distinct lineage separate from neighboring species within the tree (bootstrap value > 80%). Therefore, it may represent a new species within the genus Streptomyces. However, to better differentiate strain S5T2H1 from its closest phylogenetic neighbors, Streptomyces antimycoticus and Streptomyces aculeolatus, additional characterization was conducted, with the main differences highlighted in Supplementary Table S5.
Several macromorphological differences were observed between strain S5T2H1 and its closest species, Streptomyces coelicoflavus and Streptomyces fragilis (Supplementary Table S5). The aerial mycelium of strain S5T2H1 appeared pebble grey on ISP2, white on ISP3, ISP4, and ISP5, and was absent on ISP6 and ISP7. In contrast, Streptomyces coelicoflavus formed aerial mycelium only on ISP3 and ISP4, with a window grey color. Meanwhile, the aerial mycelium of Streptomyces fragilis ranged from grey on ISP2, ISP3, and ISP6, to white on ISP4 and ISP5. The differences between strain S5T2H1 and its two closest species extend to biochemical characteristics. Strain S5T2H1 was able to utilize all the tested carbon sources except lactose. In contrast, Streptomyces coelicoflavus could not utilize xylose and succinate, while Streptomyces fragilis exhibited a limited metabolic profile, utilizing only three carbon sources: D-galactose, l-arabinose, and sucrose. Additionally, Streptomyces fragilis was the only species that exhibited gelatin liquefaction (Supplementary Table S5).
Based on the results of molecular identification, phylogenetic studies, and the observed morphological and biochemical differences from Streptomyces coelicoflavus and Streptomyces fragilis, strain S5T2H1 is a strong candidate to be a new species within the genus Streptomyces. However, further investigation, including whole-genome sequencing and digital DNA-DNA hybridization (dDDH) with these species, is necessary to confirm its classification.These findings highlight the diversity of bacterial genera in the Algerian marine environment, including previously unreported taxa. Therefore, these marine bacteria may serve as a valuable source of potent bioactive metabolites targeting the μ-opioid receptor.
Metabolic profile of bioactive compounds secreted by selected strains
The three Actinobacteria strains, S5T2H1, S7T2H1, and S56T3J31, were selected for their potent inhibitory effects against the test microorganisms and their distinct taxonomic positions. Their crude extracts were analyzed using high-performance liquid chromatography coupled with high-resolution mass spectrometry (HPLC-HRESIMS) to annotate and identify their secreted secondary metabolites.
Compound annotation of the ethyl acetate extract prepared from strain S56T3J31 revealed nine compounds, including two known compounds, three unknown compounds, and five originating from the culture medium itself (Supplementary Fig. S4, Table S6). The medium-derived compounds were detected at retention times of 4.47, 5.20, 6.30, 7.15, and 7.72 min, corresponding to the known isoflavones genistein, genistin, 6″-O-acetyldaidzin, 6″-O-acetylgenistin, and glycitein, respectively. These findings were established based on LC-HRESIMS analysis of the crude extract prepared from the negative control—5294 medium without strain S56T3J31(Supplementary Fig. S9, Table S8). This analysis enabled the detection of the same four compounds at identical retention times, with similar UV–Vis spectra, molecular weights, and molecular formulas. The major natural source of isoflavones is soybean, which was used as an ingredient in the 5294 medium; therefore, this ingredient is the source of the three isoflavone compounds detected in the crude extract prepared from strain S56T3J31.
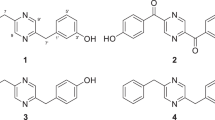
The two known peaks, 8 and 77, detected at retention times of 3.82 and 13.11 min, exhibited molecular ion clusters of [M+H] + at m/z 268.0811 and [M+H] + at m/z 507.2336, corresponding to molecular formulas C12H13NO6 and C25H34N2O9, respectively. A search for similar molecules with these characteristics in natural product databases, such as Antibase and the Dictionary of Natural Products, revealed that compounds 8 and 77 correlate with A58365A and antimycin A, respectively (Fig. 3, Supplementary Fig. S4, Table S6).

Structures of compounds identified from crude extract prepared from selected strains: (1): A58365A; (2): Antimycin A4; (3): Cyclo(phenylalanyl-prolyl); (4): Niphimycin Ia; (5): Berberifuranol.
The compound A58365A is a fused tetrahydro-indolizine with carboxyl (position 3), hydroxyl (position 8), and ketone (position 5) functional groups, along with a propanoic acid side chain. It was isolated from Streptomyces chromofuscus and is primarily known as an angiotensin-converting enzyme (ACE) inhibitor60. Antimycin A, on the other hand, belongs to the macrodiolide (bis-lactone) family, characterized by a nine-membered lactone ring. It is produced by Streptomyces spp. and functions as a mitochondrial respiratory inhibitor. As the active ingredient in Fintrol®, Antimycin A is a restricted-use piscicide employed in fisheries management to eliminate invasive species and protect native fish populations61.
The molecular formulas of additional peaks found at retention times of 5.25, 6.42, and 12.06 min were established using HPLC–UV-HRESIMS as follows: C30H17NO2, C21H22O7, and C18H20N2O7 (Supplementary Fig. S4, Table S6). A search across chemical databases revealed no correlated compounds related to actinobacteria or any other microorganisms with similar properties. Therefore, these compounds can presumably be considered unknown.
The LC-HR-ESIMS analysis of the crude extract from strain S5T2H1 revealed several distinct peaks, indicating a complex mixture of metabolites. The compounds identified include both known and potentially novel metabolites, along with two originating from the culture medium at retention times of 5.53 and 7.61 min, corresponding to genistin and glycitein, respectively (Supplementary Fig. S5, Table S7).
Two known compounds, detected at retention times of 4.43 and 11.00 min, were identified as cyclo(phenylalanylprolyl) and the non-ribosomal peptide niphimycin Ia, respectively (Fig. 3, Supplementary Table S7, Fig. S5). Compound Cyclo(phenylalanyl-prolyl), Fig. 3, is a cyclic lipopeptid belonging to the class of diketopiperazines62. It is isolated from various microorganisms, including Actinobacetria63,64, Batrachochytrium dendrobatidis65, and Lactobacillus plantarum66. This compound exhibits a broad spectrum of biological activities, including antimicrobial64, anti-wax moth larvae (Galleria mellonella)65, and antifungal activities66. However, niphimycin Ia is synthesized by several Streptomyces species, such as Streptomyces noursei. This guanidylpolyol macrolide antibiotic is characterized by its macrolide ring structure, which incorporates guanidyl and polyol groups that contribute to its bioactivity67.
Furthermore, compounds detected at retention times of 8.36, 8.97, and 11.61 min exhibited molecular ion clusters of [M+H]+ at m/z 356.2908, 381.3036, and 1047.5862, with molecular formulas determined by HPLC–UV-HRESIMS as C19H37N3O3, C21H39N3O3, and C53H78N10O12, respectively. A search in chemical databases revealed no compounds with similar properties, suggesting that these three compounds may represent novel entities (Supplementary Table S7). These findings align with taxonomic characterization results indicating that strain S5T2H1 may represent a new species within the genus Streptomyces, potentially harboring a unique biosynthetic gene cluster responsible for synthesizing these metabolites. This discovery opens the possibility of identifying novel bioactive compounds from this strain68. However, confirming the novelty of these metabolites requires purification and structural elucidation using NMR analysis.
LC-HRESIMS analysis of the crude extract secreted by strain S7T2H1 (Supplementary Fig. S6, Table S8), revealed a single active peak with a retention time of approximately 9.17 min. This peak was identified as berberifuranol (Fig. 3) (Supplementary Table S8), exhibiting a molecular ion cluster [M+H]+ at m/z 245.1282, along with UV maxima at 200 nm, 256 nm, and 358 nm (Supplementary Fig. S6). Berberifuranol is a heterocyclic molecule derived from plants, notably isolated from the roots of Berberis lycium69. This study marks the first reported instance of microbial strain involvement in the biosynthesis of berberifuranol.
In silico study
Conventional opioids act as agonists by interacting with the μ-opioid receptor, providing effective pain relief but often causing severe side effects. Consequently, there is an urgent need to identify new agonists that can deliver analgesic effects without the adverse side effects associated with traditional opioids70. A promising approach involves exploring secondary metabolites secreted by marine bacteria as potential sources of novel μ-opioid receptor agonists. In this context, the binding affinity and stability of interactions between the μ-opioid receptor and five distinct marine bacteria-derived compounds—A58365A, Antimycin A4, Cyclo(phenylalanyl-prolyl), Niphimycin Ia, and Berberifuranol—were compared to each other and to the reference molecule, morphine, using a combination of molecular docking, molecular dynamics simulations and MM/GBSA calculations.
Molecular docking
Molecular docking was performed using the Schrödinger Glide software with XP mode to estimate the binding affinity of five secondary metabolites secreted by marine bacterial strains against the active μ-opioid receptor (5C1M). The results, expressed in kcal/mol, are presented in Table 1:
Based on the docking results (Table 1), the compound A58365A exhibited the strongest binding affinity among the five ligands, with a docking score of − 5.84 kcal/mol, followed closely by Antimycin A4 with a score of − 5.6 kcal/mol. Both ligands displayed slightly weaker affinities compared to the reference molecule, morphine, which exhibited the lowest docking score of − 7.04 kcal/mol, indicating the strongest binding interaction. Lower docking scores reflect stronger predicted interactions between a ligand and the target protein, as less energy is required for the ligand to bind effectively71. Therefore, among the tested ligands, compound A58365A is considered to have the highest binding affinity, suggesting it is more likely to form stable interactions through the formation of a stable complex within the receptor’s active site of the target protein 5C1M. In contrast, Cyclo(phenylalanyl-prolyl) and Berberifuranol exhibited weaker interactions, with docking scores of − 3.73 and − 3.95 kcal/mol, respectively. However, Niphimycin Ia did not dock successfully, possibly due to its high molecular weight (1142 g/mol) and lack of compatibility with the receptor’s binding site (5C1M).
The 3D visualizations were generated to examine the molecular architecture of the binding interactions between the five secondary metabolites and the key residues within the binding cavities of the μ-opioid receptor’s active site (5C1M), as illustrated in Fig. 4.

Docked poses of: 1. A58365A, 2. Antimycin A4; 3. Cyclo(phenylalanyl-prolyl). 5. Berberifuranol and 6. Morphine program (Maestro version 14.2.118, Schrödinger Release 2024-3).
The results in Fig. 4 highlight the critical shared residues, such as Asp147, Met151, Lys233, His297, Val300, and Tyr326, involved in the interactions of all studied ligands within the binding pocket of the target protein 5C1M. These results align with previously published studies, underscoring the importance of these same key residues in target-ligand interactions72. Understanding these interactions provides insights into the potential effects of these compounds on the receptor’s function. Among the tested compounds, A58365A and Antimycin A4 demonstrated strong interactions with key amino acids within the binding pocket of the μ-opioid receptor (5C1M), as evidenced by their interaction profiles (Fig. 4.1 and 4.2). A58365A primarily engaged through its indolizine ring, forming water-mediated hydrogen bonds with Tyr-148 and Asp-147, hydrophobic contacts with Ile-296, and hydrogen bonding via its propionic acid chain with Lys-269, complemented by a hydrophobic interaction with Val-236 (Fig. 4.1). Similarly, Antimycin A4 exhibited robust binding, primarily through hydrophobic contacts with Leu-291, Asp-147, Val-300, Lys-233, and Ile-296, alongside hydrogen bonds with Cys-217 and Thr-218, and an ionic interaction with Lys-233 (Fig. 4.2). These findings, depicted in Fig. 4, align with the detailed interaction profiles shown in Figs. 5.

Protein–ligand interaction profile in complexes: (A) 5C1M-A58365A, (B) 5C1M -Berberifuranol; (C) 5C1M-Cyclo(phenylalanyl-prolyl). (D) 5C1M-Antimycin A4 and (E) 5C1M-Morphine. Different bar colors represent different types of connections: hydrogen bonds (green), hydrophobic contacts (purple), ionic bonds and water bridges in blue.
Compound A58365A, Fig. 5A, displayed a high interaction strength due to its diverse and dominant water bridges, hydrogen bonds, and hydrophobic contacts, while Antimycin A4’s strong binding affinity arose from a combination of substantial water bridges, hydrophobic contacts, and hydrogen bonds (Fig. 5D). In comparison with the reference molecule, (Fig. 5E), both A58365A and Antimycin A4 exhibited interactions comparable to those of morphine, sharing several key amino acids within the active site of 5C1M. These interactions were characterized by a balanced array of hydrogen bonds with Lys-233, hydrophobic interactions with Trp-293, Ile-296, and Tyr-326, and an ionic bond with Asp-147 (Fig. 5E). Similarly, Dumitrascuta et al.70 reported interactions with key amino acids of 5C1M for structurally related compounds of morphine, such as N-methylmorphinans-6-ones, which engaged hydrophobic residues V236, I296, and V300. Thereby, these findings suggest a potential similarity in the mechanisms of action and binding properties of A58365A and Antimycin A4 with Morphine73.
Berberifuranol, Fig. 5B, and cyclo(phenylalanyl-prolyl), Fig. 5C, exhibited moderate to low interaction profiles, primarily characterized by water bridges and hydrophobic contacts. Berberifuranol formed fewer hydrogen bonds involving residues such as Val-236 and Tyr-326, slightly reducing its binding strength compared to A58365A. Similarly, cyclo(phenylalanyl-prolyl) demonstrated limited hydrogen bonding with Asp-147 and Val-296, and the absence of ionic bonds further weakened its overall interaction with the receptor.
Molecular dynamic simulation analysis
To investigate the dynamic properties of the docked complexes, Root-Mean-Square Deviation (RMSD) analyses were performed using the Schrödinger Desmond program to evaluate the stability of the μ-opioid receptor (5C1M) in complex with six different ligands, including the reference molecule, over a 100 ns simulation period71. Figure 6 presents the RMSD graphs of these complexes during the 100 ns timeframe.

RMSD plot of μ-opioid receptor target and five marine bacteria-derived compounds as a function of simulation time. (A) 5C1M-A58365A, (B) 5C1M -Berberifuranol; (C) 5C1M-Cyclo(phenylalanyl-prolyl). (D) 5C1M-Antimycin A4 and (E) 5C1M-Morphine.
The RMSD plot of 5C1M-A58365A (Fig. 6A) shows an initial fluctuation between 20 and 30 ns before reaching a relatively stable conformation. After the equilibration phase, the RMSD remains stable, fluctuating modestly between 3.8 and 4.0 Å. A lower and more stable RMSD indicates minimal fluctuation of the complex. Therefore, the 5C1M-A58365A complex demonstrates greater stability and structural integrity throughout the interaction74,75. In contrast, the reference molecule morphine (Fig. 6E) exhibits higher RMSD fluctuations, ranging from 2.5 to 4.7 Å, compared to A58365A. This suggests that the 5C1M-morphine complex undergoes weaker dynamic binding interactions.
Furthermore, both complexes, 5C1M-Cyclo(phenylalanyl-prolyl), Fig. 6C, and 5C1M-Antimycin A4, Fig. 6D, show noticeable fluctuations during the early stages of the simulation, with partial stabilization occurring after ~ 40 ns, eventually exhibiting moderate stability. In contrast, the RMSD values of 5C1M-Berberifuranol complex, Fig. 6B, remain relatively high with significant fluctuations, indicating a less stable complex76. The docked complex was used to analyze the Root Mean Square Fluctuation (RMSF) behavior, and a plot was generated incorporating the RMSF, B factor, and interactions to assess the flexibility of individual residues in the ligand–protein complexes. The detailed results are depicted in Fig. 7.

MD simulation protein–ligand interaction root-mean-square fluctuation (RMSF) profile. (A) 5C1M-A58365A, (B) 5C1M -Berberifuranol; (C) 5C1M-Cyclo(phenylalanyl-prolyl). (D) 5C1M-Antimycin A4 and (E) 5C1M-Morphine.
The Root Mean Square Fluctuation (RMSF) analysis evaluates the average positional deviation of residues in the μ-opioid receptor (5C1M) relative to their reference positions throughout the molecular dynamics simulation77. For the 5C1M-A58365A complex, RMSF values range from 0.8 to 4.0 Å (Fig. 7A), with the highest fluctuation observed at residue 210, which is located in a non-critical region of the receptor, neither contributing to the active site nor forming direct interactions with A58365A.
In contrast, the RMSF profile for the 5C1M-morphine complex, Fig. 7E, and the four other marine-derived compounds exhibits higher fluctuations, ranging from 0.9 to 6.0 Å. Notably, A58365A was the only compound with RMSF values consistently below 4.0 Å, indicating greater receptor stability upon binding. Since higher RMSF values generally correlate with reduced protein stability and weaker binding interactions, while lower values indicate stronger ligand affinity, these suggest that A58365A provides superior stabilization of the μ-opioid receptor compared to the other tested compounds, including morphine78.
The atomic flexibility within the 5C1M-A58365A complex was further evaluated using the B-factor curve (pink line). Peaks in B-factor values indicate regions of high mobility, whereas valleys correspond to structurally rigid areas. The RMSF and B-factor values, Fig. 7A, align well in both the A58365A and morphine complexes (Fig. 7E). However, residues 145–155 and 225–235, in both complex (Fig. 7A and E), exhibit notable peaks, suggesting increased structural flexibility in these regions due to ligand binding. Results from RMSD and RMSF analyses indicate that A58365A exhibits strong and stable binding interactions with the target protein 5C1M, compared to morphine. Additionally, A58365A effectively stabilizes the μ-opioid receptor’s binding regions while maintaining natural flexibility in non-essential areas. Therefore, these findings highlight A58365A as a promising μ-opioid receptor agonist for further pharmacological investigation.
Figure 8 depicts the ligand characteristics inside the complex, including RMSD, radius of gyration (rGyr), molecular surface area (MolSA), intramolecular hydrogen bonds (intraHB), solvent-accessible surface area (SASA) and polar surface area (PSA), of all the three complexes76. These parameters provide valuable insights into various facets of ligand behavior, including structural stability, compactness, surface properties, conformational changes and interactions within the complex, respectively79.

Ligand properties for: (A) A58365A, (B) Berberifuranol, (C) Cyclo(phenylalanyl-prolyl), (D) Antimycin A4 and (E) Morphine, with 5C1M protein, such as RMSD, the radius of gyration (rGyr), intramolecular hydrogen bonds (intraHB), molecular surface area (MolSA), solvent accessible surface area (SASA), polar surface area (PSA) on interacting with protein during MD simulation.
Considering the results in Fig. 8, among the ligands, A58365A demonstrated the most favorable interaction profile during the 100 ns simulation. This is due to its small fluctuations, observed between 0.1 and 0.4 Å, which remained below 1 Å throughout the entire simulation period with minimal deviation. This indicates stability and minimal displacement from the initial binding pose, reflecting a strong and reliable interaction between the ligand and the protein71,76.
In contrast, the other four ligands exhibited significant fluctuations in RMSD, ranging between 0.8 and 1.8 Å, indicating relatively less stable binding interactions with the protein. However, regarding the reference compound, morphine, its RMSD values remained consistently lower (~ 0.15–0.35 Å), similar to A58365A, with relatively small fluctuations. This suggests that the ligand maintains a stable position throughout the simulation78. Additionally, A58365A exhibits a stable radius of gyration (rGyr), fluctuating around 3.3–3.5 Å, indicating a relatively compact conformation during the simulation. It also maintains balanced solvent exposure (SASA), a stable molecular surface area (MolSA), and consistent polar interactions (PSA). These attributes collectively indicate strong and stable binding to 5C1M.
MM/GBSA binding free energy calculations
The binding free energy of the complexes was computed using the Maestro prime module’s MM/GBSA approach, which is generally recognized for its efficacy in measuring ligand-binding affinities within protein systems80, the results are shown in Table 2.
Based on the MM/GBSA profiles of the MD simulations for the five complexes, the 5C1M-A58365A complex exhibits comparable interaction parameters to the reference morphine, indicating a well-balanced equilibrium between the different interaction forces30. This balance is reflected in its stable binding free energy (ΔG bind = − 46.66 kcal/mol), which is close to that of the reference complex 5C1M-morphine (− 53.55 kcal/mol). The ΔG bind Coulomb (− 6.24 kcal/mol) is also similar to that of morphine (− 6.64 kcal/mol), suggesting comparable electrostatic contributions to binding81,82.
Additionally, the 5C1M-A58365A complex demonstrates a balanced binding profile in other aspects. Its covalent binding energy (1.75 kcal/mol) is slightly higher than that of morphine (1.26 kcal/mol), ensuring stability without excessive rigidity. Furthermore, its van der Waals contribution (− 38.53 kcal/mol) is well-balanced, providing optimal hydrophobic interactions without being excessively strong or weak22. These results confirm the findings from molecular docking and molecular dynamics simulations, further suggesting that compound A58365A exhibits strong and stable interactions with the target 5C1M. This indicates that A58365A could potentially serve as a promising candidate for targeting 5C1M with favorable binding properties68,71.
Conclusion
This study highlights the potential of marine-derived bacterial compounds as promising alternatives to conventional opioid drugs. By investigating bacterial strains isolated from marine sediments along the Bejaia coastline (Algeria), 45 marine bacteria were obtained using six different culture media, with SCA proving to be the most effective. Molecular identification, based on 16S rRNA gene sequencing, revealed that 18 strains, selected for their potent antimicrobial activity against various pathogenic microorganisms, belonged to six genera: Streptomyces, Nocardiopsis, Alloalcanivorax, Pseudonocardia, Sinomicrobium, and Lysinibacillus. LC-HRESIMS analysis of secondary metabolites secreted by three selected strains (S5T2H1, S56T3J31, and S7T2H1) identified five known compounds—A58365A, antimycin A, cyclo(phenylalanyl-prolyl), niphimycin Ia, and berberifuranol—along with several unknown metabolites. Molecular docking and molecular dynamics simulations demonstrated that A58365A exhibited strong and stable interactions with the μ-opioid receptor (5C1M), comparable to morphine. These findings suggest that marine bacteria represent an untapped reservoir of opioid-like bioactive molecules with significant therapeutic potential. Further structural elucidation, in vitro and in vivo studies, and pharmacokinetic assessments are necessary to validate their efficacy and safety as novel analgesics. This research underscores the importance of marine microbial biodiversity in drug discovery and paves the way for developing safer opioid alternatives with reduced side effects.
Data availability
The datasets used and/or analysed during the current study available from the corresponding author on reasonable request.
References
Mazzeo, F., Meccariello, R. & Guatteo, E. Molecular and epigenetic aspects of opioid receptors in drug addiction and pain management in sport. Int. J. Mol. Sci. 24(9), 7831. https://doi.org/10.3390/ijms24097831 (2023).
Fürst, S. & Hosztafi, S. The chemical and pharmacological importance of morphine analogues. Acta Physiol. Hung. 95(1), 3–44. https://doi.org/10.1556/APhysiol.95.2008.1.1 (2008).
Benyhe, S. Morphine: New aspects in the study of an ancient compound. Life Sci. 55(13), 969–979. https://doi.org/10.1016/0024-3205(94)00631-8 (1994).
Noha, S. M., Schmidhammer, H. & Spetea, M. Molecular docking, molecular dynamics, and structure-activity relationship explorations of 14-oxygenated N-methylmorphinan-6-ones as potent μ-opioid receptor agonists. ACS Chem. Neurosci. 8(6), 1327–1337. https://doi.org/10.1021/acschemneuro.6b00460 (2017).
Meissner, K. et al. Morphine and hydromorphone effects, side effects, and variability: A crossover study in human volunteers. Anesthesiology 139(1), 16–34. https://doi.org/10.1097/ALN.0000000000004567 (2023).
Ngamcharungchit, C., Chaimusik, N., Panbangred, W., Euanorasetr, J. & Intra, B. Bioactive metabolites from terrestrial and marine actinomycetes. Molecules 28(15), 5915. https://doi.org/10.3390/molecules28155915 (2023).
Damiescu, R. et al. Aniquinazoline B, a fungal natural product, activates the μ-opioid receptor. ChemMedChem 19(19), e202400213. https://doi.org/10.1002/cmdc.202400213 (2024).
Messaoudi, O. et al. Characterization of silver carbonate nanoparticles biosynthesized using marine actinobacteria and exploring of their antimicrobial and antibiofilm activity. Mar. Drugs. 21(10), 536. https://doi.org/10.3390/md21100536 (2023).
Feknous, N., Branes, Z., Rouabhia, K., Batisson, I. & Amblard, C. Isolation characterization and growth of locally isolated hydrocarbonoclastic marine bacteria (eastern Algerian coast). Environ. Monit. Assess. 189(2), 49. https://doi.org/10.1007/s10661-016-5758-5 (2017).
Quinn, G. A. & Dyson, P. J. Going to extremes: progress in exploring new environments for novel antibiotics. npj Antimicrob. Resist. 2, 8. https://doi.org/10.1038/s44259-024-00025- (2024).
Schumacher, B. A. Methods for the determination of total organic carbon (TOC) in soils and sediments. 1–23 (US Environmental Protection Agency, Office of Research and Development, Ecological Risk Assessment Support Center, 2003).
Jensen, P. R., Gontang, E., Mafnas, C., Mincer, T. J. & Fenical, W. Culturable marine actinomycete diversity from tropical Pacific Ocean sediments. Environ. Microbiol. 7(7), 1039–1048. https://doi.org/10.1111/j.1462-2920.2005.00785.x (2005).
Messaoudi, O., Wink, J., & Bendahou, M. Diversity of Actinobacteria Isolated from Date Palms Rhizosphere and Saline Environments: Isolation, Identification and Biological Activity Evaluation. Microorganisms. 8, 1853. https://doi.org/10.3390/microorganisms8121853 (2020).
Mincer, T. J., Jensen, P. R., Kauffman, C. A. & Fenical, W. Widespread and persistent populations of a major new marine actinomycete taxon in ocean sediments. Appl. Environ. Microbiol. 68(10), 5005–5011. https://doi.org/10.1128/AEM.68.10.5005-5011 (2002).
Magarvey, N. A., Keller, J. M., Bernan, V., Dworkin, M. & Sherman, D. H. Isolation and characterization of novel marine-derived actinomycete taxa rich in bioactive metabolites. Appl. Environ. Microbiol. 70(12), 7520–7529. https://doi.org/10.1128/AEM.70.12.7520-7529.2004 (2004).
Matan, N. & Matan, N. Antifungal activities of anise oil, lime oil, and tangerine oil against molds on rubberwood (Hevea brasiliensis). Int. Biodeterior. Biodegrad. 62, 75–78. https://doi.org/10.1016/j.ibiod.2007.07.014 (2008).
Yahla, S. et al. Exploring Teucrium Aureo-Candidum Essential Oil as a Promising Alternative to Triclosan for Targeting Enoyl-Acyl Carrier Protein Reductase: Chemical Composition, Antibacterial Activity, and Molecular Docking Study. Chem Biodivers. 22, e202401945. https://doi.org/10.1002/cbdv.202401945 (2024).
Patel, J. J. & Brown, M. E. Interactions of azotobacter with rhizosphere and root-surface microflora. Plant Soil 31, 273–281. https://doi.org/10.1007/BF01373570 (1969).
Messaoudi, O. Contribution à la Caractérisation des Souches D’Actinomycètes Productrice des Métabolites Antibactériennes Isolée de la Sebkha de Kendasa (Bechar). Master’s Thesis, Université of Tlemcen, Tlemcen, Algeria, (2013).
Lampert, Y. et al. Phylogenetic diversity of bacteria associated with the mucus of Red Sea corals. FEMS Microbiol. Ecol. 64(2), 187–198. https://doi.org/10.1111/j.1574-6941.2008.00458.x (2008) (Epub 2008 Mar 18).
Shirling, E. B. & Gottlieb, D. Methods for characterization of Streptomyces species. Int. Syst. Bacteriol. 16, 313–340 (1966).
Genheden, S. & Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 10(5), 449–461. https://doi.org/10.1517/17460441.2015.1032936 (2015).
Glättli, A., Daura, X. & van Gunsteren, W. F. Derivation of an improved simple point charge model for liquid water: SPC/A and SPC/L. J. Chem. Phys. 116(22), 9811–9828 (2002).
Jorgensen, W. L., Maxwell, D. S. & Tirado-Rives, J. Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids. J. Am. Chem. Soc. 118(45), 11225–11236 (1996).
Jorgensen, W. L. & Tirado-Rives, J. The OPLS [optimized potentials for liquid simulations] potential functions for proteins, energy minimizations for crystals of cyclic peptides and crambin. J. Am. Chem. Soc. 110(6), 1657–1666 (1988).
Bowers, K. J. et al. Scalable algorithms for molecular dynamics simulations on commodity clusters. In Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, Florida, 2006, November 11–17 (2006).
Desmond Molecular Dynamics System, D. E. Shaw Research, 2024. Maestro-Desmond Interoperability Tools, Schrödinger, (2024).
Mark, P. & Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 105(43), 9954–9960 (2001).
Shivakumar, D. et al. Prediction of absolute solvation free energies using molecular dynamics free energy perturbation and the OPLS force field. J. Chem. Theory Comput. 6, 1509–1519. https://doi.org/10.1021/ct900587b (2010).
Patel, C. N., Mall, R. & Bensmail, H. AI-driven drug repurposing and binding pose meta dynamics identifies novel targets for monkeypox virus. J. Infect. Public Health 16(5), 799–807 (2023).
Ghare, P. M. & Kumbhar, A. P. Study on physico chemical parameters of soil sample. Iarjset 8(9), 171–187. https://doi.org/10.17148/iarjset.2021.8930 (2021).
Okafor, U. C. Evaluation of the impact of crude oil contamination on soil’s physicochemical characteristics, micro-flora and crop yield. Trop. Aquat. Soil Pollut. 3(1), 24–35. https://doi.org/10.53623/tasp.v3i1.132 (2023).
Zhou, Z., Wang, C. & Luo, Y. Meta-analysis of the impacts of global change factors on soil microbial diversity and functionality. Nat. Commun. https://doi.org/10.1038/s41467-020-16881-7 (2020).
Sudha, S. & Masilamani, S. M. Characterization of cytotoxic compound from marine sediment derived actinomycete Streptomyces avidinii strain SU4. Asian Pac. J. Trop. Biomed. 2(10), 770–773. https://doi.org/10.1016/S2221-1691(12)60227-5 (2012).
Priyanka, S. et al. Characterisation and identification of antibacterial compound from marine actinobacteria: In vitro and in silico analysis. J. Infect. Public Health. 12(1), 83–89. https://doi.org/10.1016/j.jiph.2018.09.005 (2019).
Ramasamy, V., Durairaj, T., Alagappan, C. & Sudalaimuthu, R. S. S. Cultivation techniques of rare actinobacteria. In Methods in Actinobacteriology. Springer Protocols Handbooks (ed Dharumadurai, D.) https://doi.org/10.1007/978-1-0716-1728-1_25 (Humana, 2022).
Yu, J. et al. Isolation and characterization of actinobacteria from Yalujiang coastal wetland, North China. Asian Pac. J. Trop. Biomed. 5(7), 555–560. https://doi.org/10.1016/j.apjtb.2015.04.007 (2015).
Huang, Z. et al. A simple culture method enhances the recovery of culturable actinobacteria from coastal sediments. Front. Microbiol. 14(12), 675048. https://doi.org/10.3389/fmicb.2021.675048 (2021).
Messaoudi, O., Steinmann, E., Praditya, D., Bendahou, M. & Wink, J. Taxonomic characterization, antiviral activity and induction of three new kenalactams in Nocardiopsis sp. CG3. Curr. Microbiol. 79(9), 284. https://doi.org/10.1007/s00284-022-02954-x (2022).
Crawford, D. L., Lynch, J. M., Whipps, J. M. & Ousley, M. A. Isolation and characterization of actinomycete antagonists of a fungal root pathogen. Appl. Environ. Microbiol. 59(11), 3899–3905. https://doi.org/10.1128/aem.59.11.3899-3905.1993 (1993).
Boudemagh, A. et al. Isolation and molecular identification of actinomycete microflora, of some saharian soils of south east Algeria (Biskra, EL-Oued and Ourgla) study of antifungal activity of isolated strains. Journal de Mycologie Médicale 15(1), 39–44 (2005).
Messaoudi, O. Isolement et Caractérisation de Nouvelles Molécules Bioactives à Partir D’Actinomycètes Isolées du sol Algérien. Ph.D. Thesis, Université de Tlemcen, Tlemcen, Algeria, 2020.
Srinivasan, R., Kannappan, A., Shi, C. & Lin, X. Marine bacterial secondary metabolites: A treasure house for structurally unique and effective antimicrobial compounds. Mar. Drugs. 19(10), 530. https://doi.org/10.3390/md19100530 (2021).
Debbab, A., Aly, A. H., Lin, W. H. & Proksch, P. Bioactive compounds from marine bacteria and fungi. Microb. Biotechnol. 3(5), 544–563. https://doi.org/10.1111/j.1751-7915.2010.00179.x (2010).
Bharathi, D. & Lee, J. Recent advances in marine-derived compounds as potent antibacterial and antifungal agents: A comprehensive review. Mar. Drugs. 22(8), 348. https://doi.org/10.3390/md22080348 (2024).
Johnson, T. A. et al. Identification of the first marine-derived opioid receptor “balanced” agonist with a signaling profile that resembles the endorphins. ACS Chem. Neurosci. 8(3), 473–485. https://doi.org/10.1021/acschemneuro.6b00167 (2017).
Chakraborty, S. et al. A novel mitragynine analog with low-efficacy mu opioid receptor agonism displays antinociception with attenuated adverse effects. J. Med. Chem. 64(18), 13873–13892 (2021).
Fadhilah, Q. G., Santoso, I., Maryanto, A. E., Abdullah, S. & Yasman, Y. Evaluation of the antifungal activity of marine actinomycetes isolates against the phytopathogenic fungi Colletotrichum siamense KA: A preliminary study for new antifungal compound discovery. Pharmacia 68(4), 837–843 (2021).
Sangkanu, S., Rukachaisirikul, V., Suriyachadkun, C. & Phongpaichit, S. Antifungal activity of marine-derived actinomycetes against Talaromyces marneffei. J. Appl. Microbiol. 130(5), 1508–1522. https://doi.org/10.1111/jam.14877 (2021).
Thawabteh, A. M. et al. Antifungal and antibacterial activities of isolated marine compounds. Toxins 15(2), 93. https://doi.org/10.3390/toxins15020093 (2023).
Ravikumar, S., Thajuddin, N., Suganthi, P., Inbaneson, S. J. & Vinodkumar, T. Bioactive potential of seagrass bacteria against human bacterial pathogens. J. Environ. Biol. 31(3), 387–389 (2010).
Djinni, I., Defant, A., Kecha, M. & Mancini, I. Actinobacteria derived from algerian ecosystems as a prominent source of antimicrobial molecules. Antibiotics 8(4), 172. https://doi.org/10.3390/antibiotics8040172 (2019).
Lai, Q., Wang, J., Gu, L., Zheng, T. & Shao, Z. Alcanivorax marinus sp. nov., isolated from deep-sea water. Int. J. Syst. Evol. Microbiol. 63(Pt 12), 4428–4432. https://doi.org/10.1099/ijs.0.049957-0 (2013).
Xu, Y. et al. Sinomicrobium oceani gen. nov., sp. nov., a member of the family Flavobacteriaceae isolated from marine sediment. Int. J. Syst. Evol. Microbiol. 63(Pt 3), 1045–1050. https://doi.org/10.1099/ijs.0.041889-0 (2013).
Oliveira, M. M. et al. Biofilms of Pseudomonas and Lysinibacillus marine strains on high-density polyethylene. Microb. Ecol. 81(4), 833–846. https://doi.org/10.1007/s00248-020-01666-8 (2021).
Cherian, T. et al. Green chemistry based gold nanoparticles synthesis using the marine bacterium Lysinibacillus odysseyi PBCW2 and their multitudinous activities. Nanomaterials 12(17), 2940. https://doi.org/10.3390/nano12172940 (2022).
Jung, M. Y. et al. Description of Lysinibacillus sinduriensis sp. nov., and transfer of Bacillus massiliensis and Bacillus odysseyi to the genus Lysinibacillus as Lysinibacillus massiliensis comb. nov. and Lysinibacillus odysseyi comb. nov. with emended description of the genus Lysinibacillus. Int. J. Syst. Evol. Microbiol. 62(Pt 10), 2347–2355. https://doi.org/10.1099/ijs.0.033837-0 (2012).
Kyoung Kwon, K., Hye, Oh. J., Yang, S. H., Seo, H. S. & Lee, J. H. Alcanivorax gelatiniphagus sp. nov., a marine bacterium isolated from tidal flat sediments enriched with crude oil. Int. J. Syst. Evol. Microbiol. 65(7), 2204–2208. https://doi.org/10.1099/ijs.0.000244 (2015).
Kim, M., Oh, H. S., Park, S. C. & Chun, J. Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. https://doi.org/10.1099/ijs.0.059774-0 (2014).
Hunt, A. H. et al. Structure elucidation of A58365A and A58365B, angiotensin converting enzyme inhibitors produced by Streptomyces chromofuscus. J. Antibiot. 41(6), 771–779. https://doi.org/10.7164/antibiotics.41.771 (1988).
Ma, X. et al. Mitochondrial electron transport chain complex III is required for antimycin A to inhibit autophagy. Chem. Biol. 18(11), 1474–1481. https://doi.org/10.1016/j.chembiol.2011.08.009 (2011).
Belal, A., Gouda, A. M., Ahmed, A. S. & Abdel Gawad, N. M. Synthesis of novel indolizine, diazepinoindolizine and Pyrimidoindolizine derivatives as potent and selective anticancer agents. Res. Chem. Intermed. 41, 9687–9701 (2015).
Gautschi, M. et al. Chemical characterization of diketopiperazines in beer. J. Agric. Food Chem. 45(8), 3183–3189 (1997).
Kannabiran, K. Bioactivity-guided extraction and identification of antibacterial compound from marine actinomycetes strains isolated from costal soil samples of Rameswaram and Dhanushkodi, Tamil Nadu, India. Asian J. Pharm. 10(04) (2016).
Starr, A. M., Zabet-Moghaddam, M. & San, F. M. Identification of a novel secreted metabolite cyclo(phenylalanyl-prolyl) from Batrachochytrium dendrobatidis and its effect on Galleria mellonella. BMC Microbiol. 22(1), 293. https://doi.org/10.1186/s12866-022-02680-1 (2022).
Ström, K., Sjögren, J., Broberg, A. & Schnürer, J. Lactobacillus plantarum MiLAB 393 produces the antifungal cyclic dipeptides cyclo(L-Phe-L-Pro) and cyclo(L-Phe-trans-4-OH-L-Pro) and 3-phenyllactic acid. Appl. Environ. Microbiol. 68(9), 4322–4327. https://doi.org/10.1128/AEM.68.9.4322-4327.2002 (2002).
Chen, Y. et al. Discovery of niphimycin C from Streptomyces yongxingensis sp. nov. as a promising agrochemical fungicide for controlling banana fusarium wilt by destroying the mitochondrial structure and function. J. Agric. Food Chem. 70(40), 12784–12795. https://doi.org/10.1021/acs.jafc.2c02810 (2022).
Messaoudi, O., Sudarman, E., Patel, C., Bendahou, M. & Wink, J. Metabolic profile, biotransformation, docking studies and molecular dynamics simulations of bioactive compounds secreted by CG3 strain. Antibiotics 11(5), 657. https://doi.org/10.3390/antibiotics11050657 (2022).
Ali, M. & Sharma, S. K. Heterocyclic constituents from Berberis lycium roots. Ind. J. Hetero. Chem. 6, 127–130 (1996).
Dumitrascuta, M. et al. Synthesis, pharmacology, and molecular docking studies on 6-desoxo-N-methylmorphinans as potent μ-opioid receptor agonists. J. Med. Chem. 60(22), 9407–9412. https://doi.org/10.1021/acs.jmedchem.7b01363 (2017).
Bouzidi, A. et al. Phytochemical analysis, biological activities of methanolic extracts and an isolated flavonoid from Tunisian Limoniastrum monopetalum (L.) Boiss: An in vitro and in silico investigations. Sci. Rep. 14(1), 3281. https://doi.org/10.1038/s41598-024-53821-7.10.1038/s41598-023-46457-6 (2024).
Huang, W. et al. Structural insights into µ-opioid receptor activation. Nature 524(7565), 315–321. https://doi.org/10.1038/nature14886 (2015) (Epub 2015 Aug 5. Erratum in: Nature. 2020;584(7820):E16. doi: 10.1038/s41586-020-2542-z).
Dumitrascuta, M. et al. N-phenethyl substitution in 14-methoxy-N-methylmorphinan-6-ones turns selective µ OPIOID receptor ligands into dual µ/Δ opioid receptor agonists. Sci. Rep. 10(1), 5653. https://doi.org/10.1038/s41598-020-62530-w (2020).
Hassan, A. M. et al. Evaluating the binding potential and stability of drug-like compounds with the monkeypox virus VP39 protein using molecular dynamics simulations and free energy analysis. Pharmaceuticals. 17(12), 1617. https://doi.org/10.3390/ph17121617 (2024).
Moschovou, K. et al. Exploring the binding effects of natural products and antihypertensive drugs on SARS-CoV-2: An in silico investigation of main protease and spike protein. Int. J. Mol. Sci. 24(21), 15894. https://doi.org/10.3390/ijms242115894 (2023).
Tahira, S. et al. N-aroyl-N′-(1-Naphthyl)-N′′-aryl guanidines as a new entry to urease inhibitors: Synthesis, kinetic mechanism, molecular docking and MD simulation studies. ChemistrySelect 8(15), e202300339. https://doi.org/10.1002/slct.202300339 (2023).
Messaoudi, O. et al. Berries anthocyanins as potential SARS-CoV–2 inhibitors targeting the viral attachment and replication; molecular docking simulation. Egypt. J. Pet. 30(1), 33–43 (2021).
Adebayo-Gege, G. et al. Molecular modeling of the interactions of Curcuma longa compounds with VEGFR towards colorectal cancer drug development. Inform. Med. Unlocked 43, 101376 (2023).
Kaur, G., Bansal, M., Rehman, H. M., Kaur, M. & Kaur, A. Synthesis and studies of new purines/pyrimidine derivatives as multi-targeted agents involving various receptor sites in the immune system. Mol. Divers. 28(1), 97–110. https://doi.org/10.1007/s11030-023-10616-8 (2024).
Defant, A., Innocenti, N. & Mancini, I. 3a-(4-Chlorophenyl)-1-methyl-3a,4-dihydroimidazo[1,5-a]quinazolin-5(3H)-one: Synthesis and in silico evaluation as a ligand in the µ-opioid receptor. Molbank 2023, M1622. https://doi.org/10.3390/M1622 (2023).
Zhou, F., Yao, H., Ma, Z. & Hu, X. Investigating small molecule compounds targeting psoriasis based on cMAP database and molecular dynamics simulation. Skin Res. Technol. 29(4), e13301. https://doi.org/10.1111/srt.13301 (2023).
Podlewska, S., Bugno, R., Kudla, L., Bojarski, A. J. & Przewlocki, R. Molecular modeling of µ opioid receptor ligands with various functional properties: PZM21, SR-17018, morphine, and fentanyl-simulated interaction patterns confronted with experimental data. Molecules 25(20), 4636. https://doi.org/10.3390/molecules25204636 (2020).
Author information
Authors and Affiliations
Contributions
D.B., M.K and O.M. conceptualized and designed the project. J.W and O.M. developed methodology. D.B and S.B. acquired data. C.N.P and O.M analyzed and interpreted data. D.B., O.M and C.N.P. wrote manuscript. M.K and S.B. provided executive support. M.K. supervised the study. The whole manuscript was approved by all authors.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Boudrahem, D., Messaoudi, O., Balit, S. et al. Exploring marine-derived bacterial compounds targeting the μ-opioid receptor agonists through metabolic profiling to molecular modeling. Sci Rep 15, 17196 (2025). https://doi.org/10.1038/s41598-025-96411-x
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-96411-x
Keywords
This article is cited by
-
Decoding cross-talk between Mpox and HIV: insights from transcriptomics and network-based analyses
Virology Journal (2025)
-
Revealing Lactiplantibacillus Plantarum K25 Derived (Z)-18-Octadec-9-Enolide in Modulating Aquaporin-8 in Colorectal Cancer by Bridging the Gut Microbiome and Membrane Biology
Cell Biochemistry and Biophysics (2025)
