
Abstract
Effective treatment options for Mycobacterium abscessus (MAB) pulmonary diseases (PD) are limited due to inadequate drug efficacy, rising drug resistance, and genetic mutations. New compounds are urgently needed to treat MAB-PD. The MAB Enoyl Acyl Carrier Protein (ACP) Reductase InhA (MAB-InhA) plays a crucial role in mycobacterial cell death and mycolic acid (MA) biosynthesis, making it a potential drug target for new lead identification. The purpose of this study was to identify new potential inhibitors of MAB-InhA in MAB-PD by using structure-based virtual screening, docking, molecular mechanics-based generalized born surface area (MM/GBSA), Absorption, Distribution, Metabolism, and Excretion (ADME), and molecular dynamics (MD) simulations. The Enamine antibacterial library containing 32,000 compounds was prepared using phase to create the database. The identified hits were analysed using the phase score, which combines vector alignments, volume score, and root-mean-square deviation (RMSD) site matching. Based on the docking results and obtained scores of the Glide docking tool, we identified Z2378320480 (Z1), Z1188959831 (Z2), Z5292493137 (Z3), Z2437620504 (Z4), Z2440336150 (Z5), and Z3390516726 (Z6) ligand molecules as potential hits. MD simulations (200 ns) were conducted on the best-docked poses of potential hits Z4, Z5, and Z6 to analyse stability and interaction at the MAB-InhA active site. The MD simulation trajectories, including RMSD, root mean square fluctuation (RMSF), ligand-protein interaction, 2D principal component analysis (PCA), and molecular dynamics secondary structure analysis (SSE), were analysed to interpret the stability.
Similar content being viewed by others
Introduction
Computational methods in drug discovery play a crucial role in identifying potential drug candidates from vast compound libraries1. The integration of both structure-based and ligand-based approaches not only optimises the process but also enhances accuracy. This combined strategy is promising, particularly in identifying natural product-based compounds2.
Mycobacterium abscessus(MAB) is a group of rapidly growing mycobacteria (RGM) responsible for various infections, particularly in individuals with chronic lung conditions and immunocompromised patients3. Genome sequencing data revealed and classified MAB into three subspecies: M. abscessus subsp. abscessus (MASA), M. abscessus subsp. bolletti (MASB), and M. abscessus subsp. massiliense (MASM). Among these, the two major subspecies are MASA and MASM4. Pulmonary disease (PD) attributable to MAB typically presents symptoms resembling those observed in nodular/bronchiectasis Mycobacterium aviumcomplex (MAC) disease. The onset of MAB disease is gradual, characterized by symptoms such as cough and fatigue. High-resolution CT scans frequently indicate anomalies such as cylindrical bronchiectasis accompanied by multiple small nodules5,6. Our previous work examined that MAB pulmonary infection is most likely caused by contact with infected individuals or environmental sources. Aerosol from contaminated water and fomites from soil serve as reservoirs7. Host-related risk, i.e., cystic fibrosis, increases susceptibility, while lifestyle factors may also play a role8. The ability of MAB to resist the phagosomal defense systems initiates the recruitment of immune cells and the formation of granulomas. Subsequently, the breakdown of these granulomas leads to the development of numerous extracellular bacterial cords9.
The long-term treatment of MAB-PD is extremely complicated due to the strong drug resistance and lack of in vitro activity against most of the oral antibiotics. However, the 2020 guidelines of the American Thoracic Society (ATS), European Society for Clinical Microbiology and Infectious Diseases (ESCMID), European Respiratory Society (ERS), and Infectious Diseases Society of America (IDSA) suggest a treatment regimen that includes an oral macrolide (such as azithromycin or clarithromycin) combined with intravenous aminoglycosides (like amikacin) and β-lactams (such as imipenem or cefoxitin). Additional companion drugs, such as tigecycline and clofazimine, are often included in the regimen to address severe side effects or inadequate responses to treatment10. Despite the intensity of chemotherapy, treatment success rates remain very low, especially when there is resistance to macrolides due to erm(41) mutations, which can occur in 40–60% of clinical strains11. The complexities in treating these subspecies are partly attributed to the presence and absence of a functional erm(41) gene, which encodes an rRNA methylase responsible for inducible macrolide resistance12. Therefore, more effective drugs are required due to the unsatisfactory performance of the current regimens.
Enoyl Acyl Carrier Protein (ACP) Reductase (InhA) is an essential enzyme in fatty acid synthesis, especially mycolic acid (MA) biosynthesis. It is classified within the Tyrosine-dependent oxidoreductase family as an NADH-dependent enoyl ACP reductase. InhA’s primary function is to catalyse the reduction of trans double bonds linked to a carbonyl group of an intermediate that is covalently bound to an acyl carrier protein in the fatty acid synthase- II (FAS-II) pathway13. Inhibiting InhA disrupts mycolic acid synthesis, causing cell wall defects and bacterial death14. Recent studies highlight the potential of direct InhA inhibitors like NITD- 916, which bypass the activation of the compromised KatG enzyme in drug-resistant strains, showing efficacy against MAB clinical isolates and reducing bacterial burden in vitro and cystic fibrosis-derived lung organoids. These findings support the development of new antimicrobials targeting InhA for treating MAB infections15.
In the current study, we analyse bioactive inhibitors targeting the InhA protein against MAB-PD, aiming to develop a combined protocol for virtual screening using ligand- and structure-based approaches. This protocol emphasises an approach that involves generating a multi-ligand pharmacophore and internally validating it using a decoy set. Additionally, the study includes molecular docking analysis, free energy calculation, and ADME (Absorption, Distribution, Metabolism, and Excretion) analysis of the top six hits. After that, a molecular dynamics (MD) simulation was performed on the top three hits of protein/ligand complexes at 200 ns to determine their stability. Interactions, stability, and affinity between proteins and ligands were thoroughly investigated using root mean square deviation (RMSD), root mean square fluctuation (RMSF), protein-ligand contact analysis, principal component analysis (PCA), 2D PCA-based free energy surface mapping, secondary structure elements (SSE) analysis, and molecular mechanics/generalised Born surface area (MM/GBSA) calculations. The schematic workflow of the overall study is shown in (Fig. 1).
Methodology

Schematic representation of the methodology.
Pharmacophore hypothesis development and validation
The pharmacophore hypothesis was developed using Indole- 2-carboxamide (IC25), a ligand with a potent minimum inhibitory concentration (MIC) value of 0.03 µM against MAB16. This ligand was selected due to its structural and functional alignment with known active compounds, ensuring the inclusion of key pharmacophoric features necessary for activity17. The hypothesis was generated using the Phase module in Schrödinger Suite18, which leverages advanced molecular alignment tools to identify critical interaction features such as hydrogen bond acceptors/donors, hydrophobic regions, and aromatic rings. The query was validated by calculating the area under the curve (AUC) before proceeding to virtual screening. The decoy dataset was generated to validate the study from the ChEMBL database (https://www.ebi.ac.uk/chembl/) using physicochemical properties filters matching such as molecular weight, LogP, and hydrogen bond donor/acceptor counts of the IC25 ligand. The pharmacophore models were evaluated based on Align, Vector, Volume, and Phase Scores, which consider molecular alignment accuracy, feature vector matching, spatial volume fit, and overall fitness to the pharmacophore query19. The model with the highest scores, ensuring both sensitivity and specificity, was selected for subsequent virtual screening.
Database Preparation and virtual screening
We retrieved the antibacterial library of the Enamine database (https://enamine.net/) containing 32,000 compounds. Phase is used to prepare and organise these compounds into a database18. Twenty conformers were generated for each ligand by utilizing all suitable parameters. Various potential states at pH 7 were generated using Epik20, while high-energy tautomeric states were removed from the database. The constructed database was subsequently used to conduct a virtual screening process. Hits from the screening were evaluated using the phase screen score, which integrates vector alignment, volume score, and RMSD site matching. Additionally, molecular docking studies were performed on the potential screened hits having phase scores exceeding 1.8.
Preparation of the protein and molecular docking
Enoyl-ACP reductase InhA (PDB: 7L6 C) crystal structures were carefully chosen after an extensive literature review due to their significance in developing novel molecules against MAB-PD14,15,21. The protein structure exhibits an R-value of 0.190, a resolution of 1. 85 Å and, showing no mutations, and a sequence length of 277 amino acids. The structure was subjected to Protein Preparation Wizard of Maestro22for preparation. During receptor preparation, several stages were performed, including the generation of disulfide bonds, assignment of zero-order metal bonds, and addition of hydrogens. The additional ligands and crystal water were also removed. In the optimization step, the pKa values of the ionisable group were optimised at pH 7.0, utilising the PROPKA program23. Finally, the OPLS_2005 forcefield was used for energy minimisation. The grid was created by selecting the natural substrate and setting the coordinates to X: 34.65, Y: 49.39, and Z: 40.79, followed by ligand preparation in Maestro using the LigPrep tool24. Various ionisation states were created with Epik at pH 7.0, and stereoisomers were constructed with OPLS_2005 20. Using the Glide docking tool, we docked the prepared ligands to the receptor and evaluated binding poses based on Glide GScores.
Analysis of drug-likeness and ADME profiles
The drug-likeness and ADME profiles of the top six candidate molecules were rigorously analyzed to identify compounds with the most favorable pharmacokinetic properties for further evaluation. The analysis used admetSAR, a tool designed to predict compounds’ pharmacokinetic and toxicological properties25. The Lipinski Rule of Five was also applied to assess drug-likeness, focusing on molecular weight, the number of hydrogen bond donors and acceptors, and lipophilicity (logP). Furthermore, these evaluations analyzed oral bioavailability by assessing gastrointestinal (GI) absorption and water solubility. Blood-brain barrier (BBB) permeation was carefully examined for compounds targeting the central nervous system (CNS) to assess their potential for crossing this critical barrier. The likelihood of active transport by P-glycoprotein (P-gp), which can impact bioavailability and resistance, was also investigated. Skin permeability (log Kp) was assessed for compounds with potential topical or transdermal applications26. The compounds displaying better ADMET properties were carefully chosen for MD simulation, while the remaining were discarded.
Molecular dynamics simulation
The top three receptor-ligand complexes (Z4, Z5, and Z6) were simulated using Desmond software of the Schrodinger suit at 200 ns22, beginning with energy minimization to relieve steric clashes, employing the steepest descent algorithm followed by conjugate gradient until the root mean square gradient reached 0.01 kcal/mol/Å. The system was then gradually heated from 0 K to 300 K over 100 ps using a Nosé-Hoover thermostat27,28. During the simulation, constraints were applied to all hydrogen bonds via the SHAKE algorithm29, allowing for larger time steps while maintaining bond lengths. Long-range electrostatic interactions were calculated using the Particle Mesh Ewald (PME) method30with a cut-off distance of 10 Å for van der Waals interactions. Finally, binding free energies were assessed using the MM/GBSA approach31 over the last 10 ns of the trajectory, with snapshots taken every 01 ns to ensure adequate sampling and improve statistical reliability.
Analysis of binding energy using the MM_GBSA approach
After the structure-based virtual screening, the six leading compounds were subjected to free binding energy (ΔG) calculations using the MM-GBSA method through the Prime application32. This aimed to determine ligand-receptor binding affinity based on the GBSA theory. The calculation used the VSGB 2.0 solvation model33and OPLS4_force field24, with the equation: ΔGBind = ΔGcomplex - (ΔGprotein + ΔGligand).
The free energy components employed in the calculations included: ΔG_coulomb, ΔG_covalent, ΔG_Hbond, ΔG_Lipo, ΔG_Packing, ΔG_vdW, Δg_straing-energy, and ΔG_Solv-Gb.
Results
Data retrieval
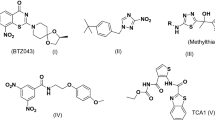
In this study, the pharmacophore hypothesis was developed based on previously documented inhibitor IC25 of MAB16. An overview of reported inhibitors, including their MIC values and structures, is presented in (Table 1).
Pharmacophore query generation and validation
The pharmacophore hypothesis was generated based on common features by importing IC25 inhibitor into Maestro’s workspace among all compounds. Before progressing to the next stage, the validity of the generated query was confirmed by assessing its area under the curve (AUC) metric34. In (Fig. 2a), the X-axis represents the percentage of the screen, indicating the proportion of data points or compounds screened, while the Y-axis denotes the percentage of active compounds or hits found during the screening process. The validated query differentiated the active ligands from decoys by an AUC value of 0.88. The Enamine database was screened using a pharmacophore model, which requires five features in a compound to qualify as a hit (Fig. 2b). The phase fitness score was determined by ranking the final hits from screening using the combination of vector, volume scores, and site matching of RMSD. The vector score varies between − 1.0 and 1.0, with higher values indicating better alignment. Conversely, the volume score ranges from 0 to 1; a higher volume score suggests that the aligned ligand and the reference ligand have a greater overlap of volumes. Volume score is determined by taking ligand volume overlap divided by their total volume. A score of zero indicates the absence of a reference ligand. A threshold phase screen score of 1.8 was employed to identify potential hits, resulting in the selection of 86 compounds (Table S1).

(a) AUC plot displays the validated query’s robust predictive performance in distinguishing active ligands from decoys. (b) Pharmacophore hypothesis with enhanced predictive capability showing essential features and spatial arrangement needed for ligand binding.
Molecular docking
Evaluating the glide gscore provides valuable insights into ligand-target protein interactions. InhA docking poses were selected using a glide gscore cut-off of − 8 kcal/mol, resulting in six selected hits. The hits selected and their glide and phase screen scores are detailed in (Table 2). The analysis of the selected hits revealed that ligand Z1 formed three hydrogen bonds with ILE194, SER20, and LYS165, along with two Alkyl bonds, one Pi-Sigma bond, one Carbon-Hydrogen bond, and one Halogen bond. Z2 formed hydrogen bonds with residues SER94 and GLY14 and six Pi-Alkyl bonds. Z3 made four hydrogen bonds with GLY14, ILE21, LYS65, and ASP148, one Carbon-Hydrogen bond with the residue GLY96 and three Pi-Alkyl bonds. The ligand Z4 forms various interactions with the binding site residues involving 5 Hydrogen bonds with ILE194, GLY14, ALA22, ILE21, and SER94 and two Carbon-Hydrogen bonds with the residues GLY192 and LYS165 and four Pi-Alkyl bonds. Z5 established two Conventional Hydrogen bonds, one with ILE194 and another with TYR158, and four Pi-Alkyl and two Carbon-Hydrogen bonds. However, the ligand Z6 shows various interactions with the binding site residues. The notable ones are two Conventional Hydrogen bonds with GLY96, SER94, and Pi-Donor Hydrogen bonds with PHE41. The molecular interactions are shown in (Fig. 3). Furthermore, the possible binding modes were studied and illustrated in (Fig. 4).

The interactions between hit compounds and 7 l6c protein binding pocket, with conventional-hydrogen bonds highlighted in green, Pi-Alkyl bonds outlined in purple, and carbon-hydrogen interactions illustrated in light green.

The possible binding modes of the top six hit compounds in the 7L6c binding pocket. Hit ligands are in different colors while binding site residues are in light blue sticks.
Calculation of ADMET properties
A combined ligand-based and structure-based virtual screening result was used to predict drug-likeness and ADME of the six best molecules with the admetSAR25. Toxicity evaluation of all six compounds hepatotoxicity, carcinogenicity, immunotoxicity, mutagenicity, cytotoxicity, predicted LD50, and toxicity class are summarised in (Table 3).
Using the Lipinski rule of five violations, we analysed the property of drug-likeness. Moreover, other ADME properties, such as water solubility and pharmacokinetics, were assessed using the admetSAR tool within the Maestro interface, which was used to analyse drug-likeness and Rule of Five violations. The drug-likeness and Rule of Five properties for the top six compounds are summarised in (Table 4).
Molecular dynamic simulation
Protein-ligand complexes are studied using MD simulations that examine the flexible interactions between the protein (biological target) and the ligand. Furthermore, this examination encompasses an assessment of the overall energy minimum to gain insights into the stability of the complex. The MD model aims to closely mimic the biological environment by examining the stability and binding interactions between proteins and ligands. It accurately represents the dynamic state within a physiologically relevant context by including water as a solvent. This allows for a deeper insight into protein-ligand interaction behaviour. After ADMET analysis, the top three hits (Z4, Z5, and Z6) exhibited the most optimal binding modes, and ADMET properties were selected to analyse their stability with InhA protein.
Root mean square deviation (RMSD)
The RMSD of carbon alpha atoms of the protein and ligand atoms was calculated to assess the stability of the protein-ligand complex35. Initially, the protein RMSD values for Z4 were around 0.5 Å, fluctuating between 1.5 Å and 3.5 Å during the first 50 ns before stabilising between 2.5 Å and 3.5 Å. Similarly, the ligand RMSD started low and fluctuated between 1.5 Å and 3.0 Å in the first 50 ns, then stabilised between 2.0 Å and 3.0 Å. RMSD values below 4.0 Å for both indicate a stable interaction throughout the simulation (Fig. 5a).

The RMSD plots of protein and ligands of top three hits during 200 ns simulation.
For complex Z5, minor deviations occur between 50 ns and 150 ns, with protein RMSD fluctuating between 2.5 Å and 3.5 Å and ligand RMSD ranging from 2.5 Å to 4.5 Å. Both stabilise after 150 ns, indicating structural integrity (Fig. 5b). For Z6, protein RMSD starts at 1.5 Å, stabilising around 3.0 Å after 75 ns, while ligand RMSD remains below 1.0 Å (Fig. 5c). These findings suggest stable protein-ligand interactions during MD simulation, offering new insights into their dynamic behaviour.
Root mean square fluctuation (RMSF)
The fluctuating behaviour of proteins bound to their ligands was assessed by calculating root mean square fluctuation (RMSF) values36. Proteins with a high RMSF value represent flexible regions, i.e., loops, while proteins with a low RMSF value represent rigid regions, such as alpha helices and beta sheets. RMSF of most protein residues, except loop regions, were changed slightly (2Å) during the 200 ns simulation. Consequently, these residues retained their relative stability when ligands were present. The RMSF value in the loop regions of the protein, however, was higher at around 4.8Å (Fig. 6). The RMSF values indicate a stable protein-ligand interaction.

The protein and ligand residue fluctuations during the 200 ns simulations were analysed using the Cα atoms of the receptor protein.
Protein-ligand interaction
The protein-ligand complex of Z4 exhibited partial stability during the 200 ns MD simulation, primarily attributed to non-covalent interactions. These included hydrogen bonds involving GLY_14 (8%), ILE_21 (10%), ALA_22 (85%), SER_94 (10%), GLY_96 (2%), PHE_149 (1%), TYR_158 (2%), LYS_165 (48%) and ILE_194 (85%) as well as hydrophobic interactions including ILE_21, ILE_25, MET_147, PHE_149, TYR_158 and MET_161. Water bridges were also observed, including THR_13, GLY_14, ILE_16, THR- 17, SER- 20, ILE- 21, ALA_22, SER_94, GLY_96, SER_123, ASP_148, PHE_149, MET_161, THR_162, LYS_165, VAL_189, GLY_192, and ILE_194 along with ionic interactions, as depicted in (Fig. 7a). Likewise, the residues of complex Z5 involved in Hydrogen bonds are ILE_21 (32%), ALA_22 (30%), SER_94 (4%), MET_98 (65%), GLN_100 (49%), PHE_149 (60%), LYS_165 (10%), and LEU_197 (35%) and the residues involving in Hydrophobic interactions are ILE_16, ILE_21, ILE_25, PHE_97, MET_147, PHE_149, MET_161, LYS_165, ALA_191, ILE_194. Ionic Interactions are also noted, with the residues SER_19, SER_20, ALA_22, SER_94, GLY_96, GLN_100, ASP_148, MET_161, THR_196, LEU_197 and ALA_198, as illustrated in (Fig. 7b). The residues of the Z6 complex involved in Hydrogen bonds are GLY_14 (15%), SER_94 (37%), GLY_96 (77%), MET_98 (20%), and hydrophobic interaction is also observed in (Fig. 7c).

The contact and 2D structure of the top three protein-ligand complexes during MD simulation. (a) Z4 complex Protein-ligand interaction (b) Z5 complex Protein-ligand interaction. (c) Z6 complex protein-ligand interaction.
Ligand-properties
The stability of the ligand bounded to pocket during 200 ns simulation was evaluated by six properties (Fig. 8). The ligand’s RMSD is evaluated at time t = 0 (the initial frame) relative to its reference conformation. The radius of gyration (rGyr) is utilized to assess the ligand’s extendedness, providing insights into its compactness and overall dimensions. Intramolecular hydrogen bonds (intraHB) indicate the internal hydrogen bonds present in the ligand. Additionally, molecular (MolSA), solvent (SASA), and polar (PSA) surface areas, which represent the total surface area of the ligand accessible to water molecules, are also evaluated. (Fig. 8) shows that all three ligands had RMSD values below 2.5Å. The ligand’s rGyr values in the binding pocket ranged from 3.6 to 5.0 Å during the 200 ns simulation, indicating stability. The ligands Z4 and Z6 showed no intramolecular hydrogen bonding (Fig. 8a-c). MolSA remained stable without any fluctuations throughout the 200 ns simulation. In contrast, SASA showed an initial increase at the beginning of the simulation and then showed stability for the rest of the simulation time. All ligands demonstrated stable PSA throughout the 200 ns simulation.

Analysis of ligand properties through molecular simulation, highlighting the Intramolecular-Hydrogen Bonding (IntraHB), Molecular (MolSA), Solvent (SASA), and Polar (PSA) surface areas.
Principal component analysis (PCA) and 2D PCA
Analysis of the dynamic behaviour of the protein in all three complexes was conducted using PCA, which identifies the collective motions of MD trajectories. The proportion of variance is plotted against the eigenvalue, representing the dynamic motions in hyperspace. In (Fig. 9a), Z4 complex PC1 captures the highest proportion of variance, with a percentage of 31.79%, indicating that it significantly contributes to the data variability. PC2 and PC3 also contribute notably to the variance, with PC2 of 17.77% and PC3 explaining 5.22% of the total variance. PC1 in the complex Z5 had the highest variation at 35.98%. PC2 and PC3 had variations of 18.64% and 8.72%, respectively (Fig. 9b). Furthermore, the third complex, Z6 had the maximum variation in PC1 at 31.79%, whereas PC2 and PC3 showed 21.83% and 6.01%, respectively (Fig. 9c). The PCA analysis of the PC subspace revealed that blue regions had the greatest movement, white regions had intermediate movements, and red areas had less flexibility movements. A 2D PCA-based free energy surface was generated to identify the thermodynamically stable conformations between the protein and ligands. The energy surface calculated the fluctuation direction of energy in two PCs (PC1 and PC2) for carbon alpha atoms. Most of the clusters were found in the local minima well (purple colour), indicating the stable transition of one configuration to another in all three complexes (Fig. 10a-c).

Principal component analysis (PCA) analysis of the three complexes: (a) Z4 complex shows an overall flexibility of 54.78%; (b) Z5 complex exhibits 63.34% flexibility; (c) Z6 complex reveals 59.63% flexibility. These results are depicted across three distinct hyperspaces.

2D PCA-based free energy surface representation for the three complexes.
Secondary structure analysis (SSE)
SSE was performed on the protein and observed throughout the simulation. Beta strands are shown in blue, whereas alpha helices are depicted in orange. (Fig. 11a-c) illustrate the distribution of SSE across the protein structure by residue index. A summary of the SSE composition is shown in (Fig. 11d-f) for each trajectory frame throughout the simulation. It tracks each residue’s SSE assignment over time. Z4 contains 26.58% helix, the percentage of the strand is 12.75%, and the total percentage of SSE is 39.33%. However, in Z5, the percentage of helix is 24.77%, the percentage of the strand is 13.24%, and the total percentage of SSE is 39.33%, and in Z6, the percentage of helix is 24.72%, the percentage of the strand is 13.25%, and the total percentage of SSE is 37.96%.

The secondary structure elements of the top three hits are analysed, with alpha-helices illustrated in orange and beta-strands in blue.
Molecular mechanics-generalized born surface area (MM/GBSA)
The selected complexes’ total binding free energy (ΔGtotal) was calculated using the prime MM/GBSA module37. The overall ΔGtotal of complexes was derived from the Coulombic energy (Coulomb), the Covalent-Interactions (Covalent), the Hydrogen-bond (Hbond), the Lipophilic-Binding (Lipo), π-π Packing-Interaction (Packing), Strain Energy, Generalized Born electrostatic solvation-energy (Solv_GB) and van der-Waals Energy (vdW). (Table 5) shows the contribution of each item.
The ΔGtotal value was − 78.28 kcal/mol for the Z4, − 75.19 kcal/mol for the Z5, and − 56.52 kcal/mol for the Z6 complex. Out of the three complexes, Z4 showed the lowest total ΔGtotal. Complex Z4 was observed to form more stable hydrogen bonds with the residues of the InhA subunit binding pocket, as determined by the values of H-bond interaction for complexes Z4, Z5, and Z6, which were − 1.08, − 0.72, and − 0.76 kcal/mol, respectively. Among all the interactions, the contribution of the Lipo and Solv_GB energy was greater than that of other items. Consequently, the binding energies obtained from the simulation confirmed the binding affinities of the ligands determined through docking studies38.
Discussion
In this study we selected MAB-InhA as a target because it plays a key role in the fatty acid synthesis pathway of MAB, making it a critical focus for addressing the growing challenge of drug resistance in MAB infections15. InhA docking poses were selected using a glide gscore cut-off of − 8 kcal/mol, resulting in six selected hits. The compounds Z4, Z5, and Z6 emerged as promising candidates, demonstrating robust interactions with MAB-InhA as evidenced by binding free energies of − 78.28 kcal/mol, − 75.19 kcal/mol, and − 56.52 kcal/mol, respectively, and dynamic stability, suggesting their suitability for further preclinical evaluation (Table 5).
The identified hit compounds possess better drug-like properties than the natural compounds39,40. The ADMET analysis highlighted the favourable pharmacokinetic properties of Z4, Z5, and Z6. All three compounds adhered to Lipinski’s Rule of Five, indicating good oral bioavailability, with Z4 showing particularly low predicted hepatotoxicity and high solubility. These findings address key limitations reported for other inhibitors, such as NITD- 916, which faced challenges related to toxicity despite its strong efficacy against resistant MAB strains41. Additionally, the MAB-InhA inhibitor demonstrated promising in vitro activity but lacked comprehensive pharmacokinetic evaluations, limiting its development potential42.
The docking results revealed strong Z4, Z5, and Z6 interactions with the MAB-InhA active site. Z4 formed key hydrogen bonds with conserved residues ILE194, SER94, and ALA22, along with hydrophobic contacts at ILE21 and MET147 (Fig. 3). Similar docking studies have identified benzenesulfonohydrazides as effective M. tuberculosisInhA inhibitors, binding to ILE194 and TYR158 with an IC₅₀ of 0.91 µM43. Z5 established two Conventional Hydrogen bonds, one with ILE194 and another with TYR158, and four Pi-Alkyl and two Carbon-Hydrogen bonds. Similarly, quercetin analogs evaluated against InhA demonstrated hydrogen bonding with active site residues, including TYR158, highlighting the significance of these conserved interactions44. The Glide docking scores and MM/GBSA results further validated the strong binding affinities of Z4, Z5, and Z6. Compared to diphenyl ether derivatives, which also target InhA and demonstrated binding stability in earlier docking studies45,46, Z4’s broader interaction network, including water-mediated interactions, underscores its potential as a highly effective inhibitor47.
Our study’s MD simulation (200ns) trajectories showed fewer deviations and fluctuations with the alpha carbons of the targeted protein after binding. Z4, Z5, and Z6 demonstrated stable binding to MAB-InhA with low RMSD values (~ 2.5–3.0 Å). These findings extend insights from previous studies, where shorter simulations (e.g., 40 ns) on diphenyl ether derivatives48and other inhibitors highlighted the importance of key hydrogen bonds and hydrophobic contacts49. The extended simulation time here provides a more detailed understanding of ligand stability, emphasising the robust inhibitory potential of these compounds. Hydrogen bonding, hydrophobic interactions, and water bridges drove the stability of the protein-ligand complexes. Z4 formed strong hydrogen bonds with ALA_22 and ILE_194 (both 85%), while Z5 showed significant interactions with MET_98 (65%) and GLN_100 (49%). Z6 formed fewer interactions overall, with GLY_96 (77%) playing an important role in its binding stability (Fig. 7). These results align with previous studies highlighting the critical role of such interactions in stabilising InhA inhibitors48. The ligand’s stability within the binding pocket during simulation is consistent with recent studies on Mycobacterium targets49. M. tuberculosisPks13 and PknG exhibited ligand RMSD values stabilising below 2 Å, while our ligands demonstrated slightly higher RMSD values, remaining below 2.5 Å, indicating comparable stability50. Similarly, studies on MmpL3 inhibitors reported consistent rGyr values, aligned with our rGyr results ranging from 3.6 to 5.0 Å, reflecting stable ligand compactness51,52. PCA results, indicating that PC1 accounts for 31.79–35.98% of the variance across different complexes, align with recent studies employing PCA to elucidate protein dynamics53. Furthermore, our study’s 2D PCA-based free energy surfaces, identifying thermodynamically stable conformations, align with recent computational methods that utilise PCA to analyse protein-ligand interactions and detect stable inhibitor binding states54.
This study highlights Z4, Z5, and Z6 as promising MAB-InhA inhibitors demonstrating strong binding, stability, and favorable pharmacokinetics. Future research should focus on experimental MIC and IC₅₀ testing to validate these findings and assess their in vivo efficacy.
Conclusion
This research involved a comprehensive in-silico approach to identify potential inhibitors for Mycobacterium abscessus (MAB), focusing on the Enoyl Acyl Carrier Protein Reductase InhA (MAB-InhA), which is essential for the biosynthesis of mycolic acid and contributes to mycobacterial cell death. Initially, a structure-based pharmacophore model was established to screen the compounds, and subsequently, the selected hits were docked to the InhA protein to identify the plausible binding poses. The hits showed strong interactions with the InhA protein. Further, the dynamic behavior of the hits with protein was assessed by measuring the RMSD of alpha atoms of protein and ligands and residual fluctuations, which showed stable confirmations. The interaction contacts showed strong binding between protein and ligands, while the PCA indicated a high static number of hydrogen bonds. Lastly, the MMGBSA indicated the strong binding of the hits with the InhA protein. Thus, these compounds can be used as lead compounds to study their biophysical activity against the InhA protein.
Data availability
All data generated or analysed during this study are included in this published article (and its Supplementary Information files).
Abbreviations
- MAB:
-
Mycobacterium abscessus
- PD:
-
Pulmonary diseases
- ADME:
-
Absorption, Distribution, Metabolism, and Excretion
- RMSD:
-
Root-mean-square deviation
- RMSF:
-
Root mean square fluctuation
- PCA:
-
Principal component analysis
- SSE:
-
Secondary structure analysis
- RGM:
-
Rapidly growing mycobacteria
- MASA:
-
M. abscessus subsp. abscessus
- MASB:
-
M. abscessus subsp. bolletti
- MASM:
-
M. abscessus subsp. massiliense
- MAC:
-
Mycobacterium avium complex
- ATS:
-
American Thoracic Society
- ESCMID:
-
European Society for Clinical Microbiology and Infectious Diseases
- IDSA:
-
Infectious Diseases Society of America
- BBB:
-
Absorption, blood-brain barrier
- ERS:
-
European Respiratory Society
- ACP- InhA:
-
Enoyl Acyl Carrier Protein Reductase
- P-gp:
-
P-glycoprotein
- Log Kp:
-
Substrate potential, and skin permeation
- MIC:
-
Minimum inhibitory concentration
- intraHB:
-
Intramolecular hydrogen bonds
- rGyr:
-
Radius of gyration
- MolSA:
-
Molecular surface area
- SASA:
-
Solvent surface area
- PSA:
-
Polar surface area
- MM/GBSA:
-
Molecular mechanics-based generalised born surface area
- IC25:
-
Indole-2-carboxamide
- AUC:
-
Area under the curve
- logS:
-
Water solubility
- GI:
-
Gastrointestinal
- FAS-II:
-
Fatty acid synthase- II
References
Mak, K. K., Wong, Y. H. & Pichika, M. R. Artificial intelligence in drug discovery and development. In Drug Discovery and Evaluation: Safety and Pharmacokinetic Assays (eds Hock, F. J. & Pugsley, M. K.) 1–38 https://doi.org/10.1007/978-3-030-73317-9_92-1 (Springer International Publishing, 2022).
Jana, S., Ganeshpurkar, A. & Kumar Singh, S. Multiple 3D-QSAR modeling, e-pharmacophore, molecular docking, and in vitro study to explore novel ache inhibitors. RSC Adv. 8, 39477–39495 (2018).
Victoria, L., Gupta, A., Gómez, J. L. & Robledo, J. Mycobacterium abscessus complex: A review of recent developments in an emerging pathogen. Front. Cell. Infect. Microbiol. 11, 659997 (2021).
Heydari, H. et al. MabsBase: A Mycobacterium abscessus genome and annotation database. PLOS ONE. 8, e62443 (2013).
Koh, W. J., Stout, J. E. & Yew, W. W. Advances in the management of pulmonary disease due to Mycobacterium abscessus complex. Int. J. Tuberc. Lung Dis. 18, 1141–1148 (2014).
Philley, J. V. spsampsps Griffith, D. E. Disease Caused by Mycobacterium Abscessus and Other Rapidly Growing Mycobacteria (RGM). in Nontuberculous Mycobacterial Disease: A Comprehensive Approach to Diagnosis and Management (ed. Griffith, D. E.) 369–399 https://doi.org/10.1007/978-3-319-93473-0_13 (Springer International Publishing, Cham, 2019).
Abbas, M. et al. Sources, transmission and hospital-associated outbreaks of nontuberculous mycobacteria: a review. Future Microbiol. https://doi.org/10.2217/fmb-2023-0279 (2024).
Ruis, C. et al. Dissemination of Mycobacterium abscessus via global transmission networks. Nat. Microbiol. 6, 1279–1288 (2021).
Johansen, M. D., Herrmann, J. L. & Kremer, L. Non-tuberculous mycobacteria and the rise of Mycobacterium abscessus. Nat. Rev. Microbiol. 18, 392–407 (2020).
Daley, C. L. et al. Treatment of nontuberculous mycobacterial pulmonary disease: an official ATS/ERS/ESCMID/IDSA clinical practice guideline. Clin. Infect. Dis. 71, e1–e36 (2020).
Roux, A. L. et al. Comparing Mycobacterium Massiliense and Mycobacterium abscessus lung infections in cystic fibrosis patients. J. Cyst. Fibros. 14, 63–69 (2015).
Richard, M., Gutiérrez, A. V. & Kremer, L. Dissecting erm(41)-Mediated Macrolide-Inducible Resistance in Mycobacterium abscessus. Antimicrob. Agents Chemother. https://doi.org/10.1128/aac.01879-19 (2020).
de Souza, M. V. N. et al. Synthesis and biological aspects of mycolic acids: an important target against Mycobacterium tuberculosis. Sci. World J. 8, 720–751 (2008).
Alcaraz, M., Edwards, T. E. & Kremer, L. New therapeutic strategies for Mycobacterium abscessus pulmonary diseases – untapping the mycolic acid pathway. Expert Rev. Anti-infective Therapy. 21, 813–829 (2023).
Alcaraz, M. et al. Efficacy and mode of action of a direct inhibitor of Mycobacterium abscessus InhA. ACS Infect. Dis. 8, 2171–2186 (2022).
Franz, N. D. et al. Design, synthesis and evaluation of indole-2-carboxamides with Pan anti-mycobacterial activity. Bioorg. Med. Chem. 25, 3746–3755 (2017).
Bhattarai, P. et al. Structural determinants of Indole-2-carboxamides: identification of lead acetamides with Pan antimycobacterial activity. J. Med. Chem. https://doi.org/10.1021/acs.jmedchem.2c00352 (2022).
Dixon, S. L. et al. PHASE: a new engine for pharmacophore perception, 3D QSAR model development, and 3D database screening: 1. Methodology and preliminary results. J. Comput. Aided Mol. Des. 20, 647–671 (2006).
Szwabowski, G. L., Daigle, B. J., Baker, D. L. & Parrill, A. L. Structure-based pharmacophore modeling 2. Developing a novel framework for structure-based pharmacophore model generation and selection. J. Mol. Graph Model. 122, 108488 (2023).
Shelley, J. C. et al. Epik: a software program for pKaprediction and protonation state generation for drug-like molecules. J. Comput. Aided Mol. Des. 21, 681–691 (2007).
Prasad, M. S., Bhole, R. P., Khedekar, P. B. & Chikhale, R. V. Mycobacterium Enoyl acyl carrier protein reductase (InhA): A key target for antitubercular drug discovery. Bioorg. Chem. 115, 105242 (2021).
Schrödinger. Schrödinger Release Notes - Release 2022. Schrödinger https://www.schrodinger.com/life-science/download/release-notes/release-2022-4/ (2022).
Kieseritzky, G. & Knapp, E. W. Optimizing pKA computation in proteins with pH adapted conformations. Proteins Struct. Funct. Bioinform. 71, 1335–1348 (2008).
Kb, S. et al. Structure based pharmacophore modelling approach for the design of Azaindole derivatives as DprE1 inhibitors for tuberculosis. J. Mol. Graph. Model. 101, 107718 (2020).
Yang, H. et al. AdmetSAR 2.0: web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 35, 1067–1069 (2019).
Daina, A., Michielin, O. & Zoete, V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717 (2017).
Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 81, 511–519 (1984).
Hoover, W. G. Canonical dynamics: equilibrium phase-space distributions. Phys. Rev. A. 31, 1695–1697 (1985).
Ryckaert, J. P., Ciccotti, G. & Berendsen, H. J. C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 23, 327–341 (1977).
Darden, T., York, D. & Pedersen, L. Particle mesh Ewald: an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 (1993).
Genheden, S. & Ryde, U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin. Drug Discov. 10, 449–461 (2015).
Schrödinger. Life Science: Prime. https://www.schrodinger.com/platform/products/prime/ (2021).
Li, J. et al. The VSGB 2.0 model: A next generation energy model for high resolution protein structure modeling. Proteins Struct. Funct. Bioinform. 79, 2794–2812 (2011).
Kohlbacher, S. M., Langer, T. & Seidel, T. QPHAR: quantitative pharmacophore activity relationship: method and validation. J. Cheminform. 13, 57 (2021).
Sargsyan, K., Grauffel, C. & Lim, C. How molecular size impacts RMSD applications in molecular dynamics simulations. J. Chem. Theory Comput. 13, 1518–1524 (2017).
Martínez, L. Automatic identification of mobile and rigid substructures in molecular dynamics simulations and fractional structural fluctuation analysis. PLOS ONE. 10, e0119264 (2015).
Godschalk, F., Genheden, S., Söderhjelm, P. & Ryde, U. Comparison of MM/GBSA calculations based on explicit and implicit solvent simulations. Phys. Chem. Chem. Phys. 15, 7731–7739 (2013).
Decherchi, S. & Cavalli, A. Thermodynamics and kinetics of Drug-Target binding by molecular simulation. Chem. Rev. 120, 12788–12833 (2020).
Jayaraman, M., Gosu, V., Kumar, R. & Jeyaraman, J. Computational insights into potential marine natural products as selective inhibitors of Mycobacterium tuberculosis InhA: A structure-based virtual screening study. Comput. Biol. Chem. 108, 107991 (2024).
Abbas, M. et al. Antimicrobial properties and therapeutic potential of bioactive compounds in Nigella sativa: A review. Molecules 29, 4914 (2024).
Khaleel, E. F. et al. Identification of new anti-mycobacterial agents based on quinoline-isatin hybrids targeting Enoyl acyl carrier protein reductase (InhA). Bioorg. Chem. 144, 107138 (2024).
Sabt, A. et al. Identification of 2-(N-aryl-1,2,3-triazol-4-yl) Quinoline derivatives as antitubercular agents endowed with InhA inhibitory activity. Front. Chem. 12, 1424017 (2024).
Al-Warhi, T. et al. Benzenesulfonohydrazide-tethered non-fused and fused heterocycles as potential anti-mycobacterial agents targeting Enoyl acyl carrier protein reductase (InhA) with antibiofilm activity. RSC Adv. 14, 30165–30179 (2024).
Pitaloka, D. A. E., Ramadhan, D. S. F., Arfan, Chaidir, L. & Fakih, T. M. Docking-Based virtual screening and molecular dynamics simulations of Quercetin analogs as Enoyl-Acyl carrier protein reductase (InhA) inhibitors of Mycobacterium tuberculosis. Sci. Pharm. 89, 20 (2021).
Kamsri, P. et al. Rational design of InhA inhibitors in the class of Diphenyl ether derivatives as potential anti-tubercular agents using molecular dynamics simulations. SAR QSAR Environ. Res. (2014).
Tiwari, A. P., Giliyar, V. B., Shenoy, G. G. & Eshwara, V. K. Identifying the structural features of Diphenyl ether analogues for InhA Inhibition: A 2D and 3D QSAR based study. Lett. Drug Des. Discovery. 17, 31–47 (2020).
Shekhar, Roquet-Banères, F., Anand, A., Kremer, L. & Kumar, V. Rational design and microwave-promoted synthesis of triclosan-based dimers: targeting InhA for anti-mycobacterial profiling. Royal Soc. Open. Sci. https://doi.org/10.1098/rsos.240676 (2024).
da Lima, C. H. Aqueous molecular dynamics simulations of the M. tuberculosis Enoyl-ACP Reductase-NADH system and its complex with a substrate mimic or Diphenyl ethers inhibitors. Int. J. Mol. Sci. 16, 23695–23722 (2015).
Zhang, Q. et al. Discovery of novel and potent InhA direct inhibitors by ensemble docking-based virtual screening and biological assays. J. Comput. Aided Mol. Des. 37, 695–706 (2023).
Chikhale, R. V. et al. Identification of novel hit molecules targeting M. tuberculosis polyketide synthase 13 by combining generative AI and physics-based methods. Comput. Biol. Med. 176, 108573 (2024).
Taniguchi, H., Kawamoto, S., Monobe, K. & Aoki, S. Data on molecular Docking and molecular dynamics targeting Mycobacterium tuberculosis Shikimic acid kinase. Data Brief. 54, 110370 (2024).
Dingiş Birgül, S. İ et al. In silico design, synthesis and antitubercular activity of novel 2-acylhydrazono-5-arylmethylene-4-thiazolidinones as enoyl-acyl carrier protein reductase inhibitors. J. Biomol. Struct. Dyn. https://doi.org/10.1080/07391102.2024.2319678 (2024).
Moradi, S. et al. A review on description dynamics and conformational changes of proteins using combination of principal component analysis and molecular dynamics simulation. Comput. Biol. Med. 183, 109245 (2024).
Nirwan, S., Chahal, V. & Kakkar, R. Structure-based virtual screening, free energy of binding and molecular dynamics simulations to propose novel inhibitors of Mtb-MurB oxidoreductase enzyme. J. Biomol. Struct. Dyn. 39, 656–671 (2021).
Acknowledgements
Authors extend their appreciation to researchers supporting project Number (RSPD2025R885) at King Saud University Riyadh Saudi Arabia for supporting this research.
Funding
Dong-Qing Wei is supported by grants from the Intergovernmental International Scientific and Technological Innovation and Cooperation Program of The National Key R&D Program(2023YFE0199200), the National Key R&D Program (2024YFA1306402), the National Science Foundation of China (32030063).
Author information
Authors and Affiliations
Contributions
M.A. and D.W. conceptualized the study. M.A. wrote the original manuscript. M.A., M.N., and K.I.S. performed the computational work. M.A., G.F., and K.I.S. prepared the figures. M.A., A.R.A., M.N., and G.F. handled the visualization. M.A., A.R.A., M.N., and G.F. edited and revised the manuscript. M.A., A.R.A., and K.I.S. curated the data. M.A., D.W., and K.I.S. conducted the formal analysis. D.W. acquired the funding. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Abbas, M., Alanzi, A.R., Sahibzada, K.I. et al. Identification of novel inhibitors targeting Mycobacterium abscessus InhA through virtual screening, docking, and molecular dynamic simulations. Sci Rep 15, 12795 (2025). https://doi.org/10.1038/s41598-025-97513-2
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-97513-2
Keywords
This article is cited by
-
In Silico Evaluation of Acalypha indica Phytochemicals as Potential Antifungal Agents Targeting Saccharomyces cerevisiae Lanosterol 14-Alpha Demethylase
Cell Biochemistry and Biophysics (2025)
-
Synthesis, molecular docking, molecular dynamic simulation and biological evaluation of novel 3,4-dihydropyridine derivatives as potent antituberculosis agents
Molecular Diversity (2025)
