
Abstract
Subarachnoid hemorrhage (SAH) is a fatal pathological condition in the central nervous system (CNS), characterized by severe clinical consequences. Its treatment remains a significant challenge, especially due to the incomplete understanding of its molecular mechanisms. In this study, we integrated comprehensive bioinformatics analyses with experimental validation to explore the potential pathogenic mechanisms and immune cell infiltration characteristics of SAH, aiming to identify novel diagnostic biomarkers and therapeutic targets. We selected relevant gene expression data from the gene expression omnibus (GEO) database and obtained a gene set associated with SAH from the GeneCards database. Through bioinformatics analysis, we constructed a protein-protein interaction (PPI) network and performed functional enrichment analysis using gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) databases. The analysis revealed 11 key genes and indicated 3 main signaling pathways. Additionally, Drug target prediction and molecular docking analyses revealed that Isorhynchophylline (IRN) exhibits a strong binding affinity to these hub proteins. Importantly, Western blot (WB) experiments confirmed that IRN significantly downregulates the expression of CCL20, IL6, TLR4, and MMP9 in LPS-induced microglial cells, validating its anti-inflammatory effects. In conclusion, our findings not only elucidate the molecular mechanisms underlying SAH but also provide robust bioinformatics and experimental evidence supporting IRN as a promising therapeutic candidate, offering novel insights for future intervention strategies in SAH.
Similar content being viewed by others


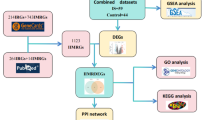
Introduction
SAH is a life-threatening condition in the central nervous system (CNS), characterized by bleeding in the subarachnoid space, often leading to severe neurological dysfunction and high mortality rates1. The pathogenesis of SAH is complex and not yet fully understood, posing significant challenges to its treatment2. The pathophysiology of SAH is closely associated with neuroinflammation, blood-brain barrier disruption, and neuronal injury, which together trigger a series of cellular and molecular events, resulting in brain damage and functional impairment3. Although considerable research has unveiled potential mechanisms of SAH, there is still a lack of in-depth understanding regarding the roles of different cell types in disease progression4.
During the course of SAH, immune cell infiltration and dysfunction are considered key factors exacerbating brain injury and inflammation5. Microglia and astrocytes play important roles in the CNS immune response, particularly in modulating neuroinflammation and responding to tissue damage6. However, the functional changes and interactions of these cells in SAH remain insufficiently explored7. Traditional histological techniques, while useful for assessing tissue damage, have limitations in identifying the cellular heterogeneity involved in SAH pathology. As a result, our understanding of the cellular composition and microenvironment in SAH remains incomplete8.
Advances in bioinformatics provide powerful tools to analyze large-scale gene expression data and understand complex biological processes. By integrating transcriptomic data with immune cell infiltration analysis, we can gain a deeper understanding of the molecular mechanisms underlying SAH and identify potential key genes as therapeutic targets9,10. Previous studies have indicated that inflammation, oxidative stress, and cell death play crucial roles in the development of SAH, processes that are often closely associated with changes in specific cell types and immune responses11.
The goal of this study is to explore the molecular mechanisms of SAH, with a focus on immune cell infiltration and its role in disease progression. We aim to identify key genes and signaling pathways associated with SAH and investigate potential therapeutic strategies. Specifically, we will examine Isorhynchophylline (IRN) as a potential treatment for SAH and analyze its effects on relevant targets and signaling pathways. Through a comprehensive understanding of the pathological features of SAH, this study aims to provide valuable insights into future clinical interventions and treatment strategies.
Materials and methods
Microarray data source and limma differential analysis
We retrieved the GSE36791 dataset from the Gene Expression Omnibus (GEO) database12. This dataset includes 43 samples from patients with SAH and 18 control samples, all sourced from human peripheral blood. The sequencing of this dataset was carried out on the GPL10558 platform. To identify genes with differential expression, we applied the “Limma” method, which is based on a generalized linear model13. The analysis was conducted using the R software package “limma” (version 3.40.7). Initially, we loaded the gene expression data and performed multiple linear regression using the lmFit function. Next, we utilized the eBayes function to apply empirical Bayes moderation to the standard errors, which allowed us to obtain the adjusted t-statistics, F-statistics, and the log odds of differential expression for each gene. In the final step, we identified the genes with statistically significant differential expression.
Weighted gene co-expression network analysis (WGCNA)
We applied the WGCNA method to explore the relationships between genes14. Initially, we calculated the median absolute deviation (MAD) for each gene and removed the 50% of genes with the lowest MAD values. Next, we filtered the differentially expressed genes (DEGs) expression matrix using the sample genes function, excluding poor-quality genes and samples, and constructed a scale-free co-expression network. We then determined an optimal “soft” threshold power (β) to calculate the adjacency relationships between genes, derived from their co-expression similarity. In the following step, the adjacency relationships were converted into a topological overlap matrix to evaluate gene co-expression and differences, which provided a more accurate representation of the gene network. Using hierarchical clustering and dynamic tree cutting functions, we detected co-expression modules. The genes with similar expression patterns were grouped into multiple modules using average linkage hierarchical clustering. Finally, we calculated the dissimilarity of module feature genes, selected the appropriate cut-off for the module tree, and merged related modules for further analysis. Each module represented genes with similar expression patterns. To identify key genes potentially involved in the pathology of SAH, we used the VennDiagram tool to find overlaps between core module genes and DEGs. Through these steps, we successfully built a gene co-expression network and identified gene modules that may play crucial roles in SAH.
Collection of subarachnoid hemorrhage disease dataset
To investigate potential disease targets and their roles, we performed a search using the keyword “Subarachnoid Hemorrhage” in the GeneCards database (https://www.genecards.org/) to identify relevant disease-associated protein targets15. The results from these searches were combined and duplicates were removed, resulting in a curated gene set for SAH.
Protein–protein interaction(PPI)network analysis
The STRING database (https://cn.string-db.org/) is a tool used to analyze protein-protein interaction (PPI) networks and their functional relationships15,16,17. We uploaded the intersecting genes from the WGCNA core modules, DEGs, and GeneCards-related genes into the STRING database. Genes with a calculated interaction score higher than 0.4 were considered significant. The resulting PPI network was visualized with Cytoscape software (version 3.9.0) (www.cytoscape.org/)18. To assess the importance of the genes within the network, we used CytoHubba to evaluate the topological properties and centrality indices of the nodes. In this study, we employed the Degree centrality algorithm, which measures the number of direct connections (edges) a gene has in the network. Genes with higher degrees are considered to play more significant roles in the network. We also used the Betweenness Centrality metric, which evaluates how often a gene acts as a bridge or connector between other genes within the network, and Closeness Centrality, which reflects how close a gene is to all other genes in the network in terms of shortest paths. These three centrality measures (Degree, Betweenness, and Closeness) were calculated to rank the genes based on their importance in the PPI network.We considered genes with higher scores across these centrality metrics to be the most crucial in the network, as they are likely to be involved in key biological processes. The most critical genes were highlighted in red, representing their prominent roles within the network. Based on this analysis, we identified the top four genes, marked in red, as the hub genes for SAH.
Functional enrichment analysis
To explore the biological roles of the identified genes, we performed functional enrichment analyses using Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. The GO analysis covered three main categories: biological processes (BP), cellular components (CC), and molecular functions (MF)19. In parallel, KEGG analysis was conducted to identify the signaling pathways relevant to SAH20. Functional enrichment was carried out using the R package ClusterProfiler, and the significantly enriched GO terms and KEGG pathways were identified based on an adjusted P-value (FDR < 0.05).
Identification of immune-infiltrating cells in disease
The immune microenvironment consists of various cell types, including immune cells, inflammatory cells, fibroblasts, along with numerous cytokines and chemokines. Analyzing immune cell infiltration is essential for assessing disease progression and predicting the patient’s response to therapy. To investigate this, we used the CIBERSORT method, which applies linear support vector regression to deconvolve gene expression data. By analyzing RNA sequencing data, we estimated the immune cell proportions in the samples and conducted immune cell infiltration analysis using the CIBERSORT (version 0.1.0) tool21,22.
Single sample gene set enrichment analysis (ssGSEA)
The identified hub genes were analyzed using ssGSEA to explore the key signaling pathways associated with their regulation. We obtained GSEA software (version 3.0) from the official website (http://software.broadinstitute.org/gsea/index.jsp) and categorized the samples into two groups based on the expression levels of the selected hub genes: high-expression (≥ 50%) and low-expression (< 50%). We performed the analysis using gene sets from the Molecular Signatures Database (http://www.gsea-msigdb.org/gsea/downloads.jsp), specifically focusing on the c2.cp.kegg.v7.4.symbols.gmt subset. The analysis was conducted to identify relevant pathways and molecular mechanisms linked to gene expression patterns and phenotypic groupings. A minimum gene set size of 5 and a maximum of 5000 genes were applied, with 1000 permutations. Pathways with a P-value < 0.05 and a false discovery rate (FDR) < 0.25 were considered statistically significant23.
ROC curve analysis of hub genes
To ensure the accuracy of each candidate hub gene, we performed evaluation using Receiver Operating Characteristic (ROC) curves. The “pROC” package was employed for ROC curve analysis24, a widely used method for assessing the performance of classification models, especially in terms of accuracy and sensitivity. The ROC curve plots the relationship between the true positive rate (TPR) and false positive rate (FPR), which helps determine the model’s effectiveness at various threshold values. In our study, ROC analysis was used to evaluate the diagnostic accuracy of each candidate hub gene for the disease. The “pROC” package was chosen because it offers comprehensive tools to compute the area under the curve (AUC), which is a key metric for classifier performance. A higher AUC value indicates a better-performing classifier. In our research, genes with an AUC greater than 0.70 were considered to potentially have diagnostic value for the disease. This threshold was selected based on prior research and experimental experience, though it may vary depending on the specific disease and research context. By identifying genes with a higher AUC, we were able to highlight their diagnostic significance and provide a solid foundation for further investigation.
Hub genes expression and protein localization analysis
To investigate the expression of hubs genes across various cell types, we utilized R language packages to create UMAP plots, showcasing the expression profiles of specific genes. The identified key genes were cross-referenced with the Human Protein Atlas (THPA) (https://www.proteinatlas.org/), which provides information on the expression of these proteins in brain-related cells and immune cells. This step helped validate our gene expression findings by matching them against a comprehensive database that compiles the expression of common human proteins across various tissues, cell lines, and immune cell types25,26. To better interpret the gene expression data, we employed Uniform Manifold Approximation and Projection (UMAP), a dimensionality reduction technique27. UMAP plots for subcellular localization were generated using a large collection of confocal microscopy images, which visually represent the spatial distribution of human proteins within cells. This method, integrated into THPA, offers a novel approach to visualize and analyze high-dimensional protein localization data at the subcellular level. To explore the potential cellular functions and localization of the hub genes, we examined subcellular 3D images and confocal microscopy data to pinpoint their precise locations and propose possible mechanisms of action.
Drug prediction
In this study, we employed bioinformatics techniques to predict potential therapeutic drugs targeting 11 key genes. Initially, we screened existing drugs from public databases such as DrugBank, PubChem, and CTD based on the functions and relevance of these genes. Our focus was primarily on drugs associated with SAH, and we ultimately identified IRN as a promising candidate. To further evaluate its potential, we applied molecular docking techniques to assess the binding affinity between IRN and the target genes.
Small molecule agents screening and molecular docking analysis
The Connectivity Map (CMap) database (https://clue.io/) is a tool used to investigate the relationships between genes, small molecule drugs, and diseases, facilitating drug discovery by analyzing differential gene expression. To conduct molecular docking, we first retrieve the 3D structure of the target molecule in mol2 format from the PubChem database. Using AutodockTools 1.5.6, we prepare the ligand by adding hydrogen atoms, calculating charge distributions, identifying the root and rotatable bonds, and saving the ligand as a pdbqt file. The 3D structure of the target protein is sourced from the RCSB Protein Data Bank (www.rcsb.org/), and is processed by adding Gasteiger charges, including nonpolar hydrogens, defining it as a receptor, and saving it in pdbqt format. For docking using Autodock Vina, parameters like exhaustiveness are set to 15, with defined box size and coordinates, while other settings are left unchanged. The docking is performed using semi-flexible docking, and the conformation exhibiting the highest binding affinity is chosen as the final docked result28.
Experimental validation by WB analysis
To validate the expression of the core genes identified through bioinformatics analysis, we conducted Western blot experiments using BV2 microglial cells (SaiBeikang Biotechnology Co., Ltd., China). BV2 cells were cultured in DMEM supplemented with 10% fetal bovine serum (FBS; Gibco, USA) and 1% penicillin-streptomycin at 37 °C in a humidified incubator containing 5% CO₂. For the inflammatory model, BV2 cells were stimulated with 100 ng/mL lipopolysaccharide (LPS; Sigma-Aldrich, USA) for 24 h. After LPS treatment, cells in the treatment group were exposed to 50 µM of IRN (MedChemExpress, USA) for another 24 h.Cells were then lysed in RIPA buffer containing protease and phosphatase inhibitors (Beyotime, China). The total protein concentration was measured using a BCA assay kit (Thermo Scientific, USA). Equal amounts of protein (30 µg) were separated on 10% SDS-PAGE gels and transferred onto PVDF membranes (Millipore, USA). Membranes were blocked with 5% non-fat milk for 1 h at room temperature and incubated overnight at 4 °C with primary antibodies against CCL20 (1:1000, Abcam, UK), IL6 (1:1000, Cell Signaling Technology, USA), TLR4 (1:1500, Abcam, UK), and MMP9 (1:1000, Proteintech, China). After washing, membranes were incubated with HRP-conjugated secondary antibodies (1:5000, CST, USA) for 1 h at room temperature.Protein bands were visualized using enhanced chemiluminescence (ECL) detection reagents (Thermo Scientific, USA), and images were captured using a chemiluminescence imaging system (Bio-Rad, USA). Band intensities were quantified with ImageJ software (NIH, USA).All experiments were repeated three times independently.
Results
Identification of differentially expressed genes
We began by acquiring the dataset and proceeded to identify DEGs through the Limma rapid differential analysis method. This process revealed 372 DEGs, with 193 genes showing a down-regulation expression and 179 genes displaying up-regulation. To facilitate the visualization and interpretation of these gene expression patterns, we generated volcano plots and heatmaps. These graphical representations of the DEGs in SAH, shown in (Fig. 1A,B, and Supplement Table S1), highlight the distinct expression profiles and statistical significance of the differences. These findings provide a strong foundation and clear direction for our subsequent investigations.

Heatmap and volcano plot for the DEGs identified from the integrated SAH dataset. (A) Red and green plot triangles represent DEGs with upregulated and downregulated gene expression. (B) Each row displays DEGs and each column refers to one of the SAH case or control group samples. Red and blue colors represent DEGs with up- and down-regulated gene expression.
Weighted gene co-expression network analysis and key module identification
For the weighted gene co-expression network analysis, we first determined the appropriate parameter settings, selecting a β-value of 10 as the “soft” threshold, based on the scale-free R² of 0.86 and average connectivity measures (Fig. 2A,B). The gene clustering dendrograms for both the normal and SAH groups are shown in Fig. 2C. From this, we identified 14 distinct gene co-expression modules (GCMs), each represented by a unique color (Fig. 2D).

WGCNA analysis. (A, B) Screening for soft thresholds based on scale independence and average connectivity. (C) Gene co-expression modules indicated by different colors under the gene tree. (D) Heatmap of gene adjacency. (E) Heatmap of the association between modules and SAH. The turquoise module is shown to be correlated significantly with SAH. Numbers at the top and bottom brackets represent the correlation coefficient and p-value. (F) Correlation plot between module membership and gene significance of genes included in the green module.
To identify the module most strongly correlated with SAH, we assessed the correlation between each gene co-expression module and the disease status (Fig. 2E). The green module showed the strongest correlation with SAH, with a correlation coefficient of 0.59 and a P-value of less than 0.01. As a result, this module, containing 1833 hub genes, was selected for further investigation (Supplement Table S2).
Next, we analyzed the relationships between genes within the green module and SAH. Our analysis revealed a significant positive correlation between the module’s gene members and their individual gene significance, with an r-value of 0.73 and a P-value of less than 0.01 (Fig. 2F). This suggests that the green module is closely associated with SAH and warrants further exploration.
Screening of subarachnoid hemorrhage hub genes
To uncover the hub genes associated with SAH, we constructed a protein-protein interaction (PPI) network by integrating the hub genes identified from the WGCNA analysis with those retrieved from the GeneCards disease database (Supplement Table S3) and the DEGs. This resulted in 18 intersecting genes (Fig. 3A). We then visualized the PPI network using the STRING database (v11.5) and Cytoscape software, which showed interactions among 11 genes (Fig. 3B). These 11 genes were selected as potential key genes for SAH.

Identification and visualization of hub genes associated with SAH. (A) Venn diagram showing the overlap of 18 genes derived from WGCNA, GeneCards database, and DEGs. (B) PPI network of the 18 overlapping genes visualized using the STRING database and Cytoscape software, demonstrating the interactions among critical genes related to SAH. (C) Assessment of node significance within the PPI network using CytoHubba, with node shading indicating the relative importance of the top 11 genes based on topological properties and centrality metrics. (D) Expression levels of the final top 4 hub genes across different experimental groups.
To further assess the importance of these genes within the network, we utilized CytoHubba to evaluate their topological features and centrality. The significance of each node was reflected by the shading, with darker shades indicating higher importance (Fig. 3C). After further analysis, we identified the top 4 hub genes, which were prioritized for subsequent study. The expression patterns of these genes across different groups are presented in Fig. 3D.
Enrichment analysis
We performed functional enrichment analysis on the 11 key genes to uncover their biological roles and pathways in SAH. GO enrichment analysis indicated that these genes were enriched in 325 GO terms (Supplement Table S4), including 235 biological processes (BP), 48 cellular components (CC), and 42 molecular functions (MF). In the category of biological processes, the top 10 significantly enriched terms included -biological adhesion, biological regulation, cellular component organization or biogenesis, cellular process, developmental process, establishment of localization, immune system process, localization, locomotion, metabolic process. For cellular components, significant terms included cell junction, cell part, extracellular matrix, etc. In terms of molecular functions, notable terms were antioxidant activity, binding, catalytic activity among others (Fig. 4A,B).

Functional enrichment analysis of key genes in SAH. (A) GO enrichment analysis showing the top enriched BP, CC, and MF. (B) Detailed terms associated with enriched processes. (C) KEGG pathway analysis highlighting significant pathways related to SAH. (D) Visualization of key pathways involved, including TNF signaling pathway. (E) Overview of enrichment results for the TNF signaling pathway, illustrating its role in SAH pathology. The KEGG pathway diagram was adapted from www.kegg.jp/kegg/kegg1.html (Kanehisa Laboratory, with permission).
KEGG pathway analysis revealed that these genes are involved in 114 signaling pathways (Supplement Table S5), with significant enrichment observed in pathways such as TNF signaling pathway, HIF-1 signaling pathway, and IL-17 signaling pathway (Fig. 4C,D). These pathways are crucial in the development and progression of SAH.
Figure 4E presents detailed enrichment results of the key genes in the TNF signaling pathways. The pathways play pivotal roles in the development and progression of SAH. This visualization aids in understanding the specific roles and mechanisms of these genes and pathways in SAH pathology. The enrichment findings offer valuable insights into the molecular mechanisms of SAH and provide direction for future research.
Immune infiltration analysis of hub genes in SAH
To explore the immune landscape associated with the hub genes in SAH, we applied the CIBERSORT algorithm to assess immune cell infiltration scores. Figure 5A presents the immune infiltration scores in a stacked bar plot, illustrating the distribution of various immune cell types across SAH and control samples. The analysis revealed that major immune cell types, including NKT cells, monocytes, B cells, Th17 cells, and macrophages, were significantly correlated with the expression of SAH hub genes.

Immune infiltration analysis of SAH hub genes. (A) Stacked bar plot showing the distribution of immune cell types in SAH and control samples, with significant correlations between major immune cells (NKT cells, monocytes, B cells, Th17 cells, and macrophages) and SAH hub gene expression. (B) Correlation matrix of immune cell populations, highlighting complex interactions within the SAH immune microenvironment. (C) Statistical differences in immune cell infiltration between SAH and control groups. (D) Ridge plot comparing the distribution patterns of immune cells (CD8 naïve, Tr1, nTreg, Th17, and Tfh) between SAH and control groups, demonstrating significant differences in immune cell expression.
Furthermore, Fig. 5B displays the correlations between different immune cell populations, emphasizing the complex interactions within the immune microenvironment of SAH. Statistical differences in immune cell infiltration between the SAH and control groups were also analyzed and are shown in Fig. 5C.
A ridge plot (Fig. 5D) further compares the distribution patterns of immune cell types, highlighting significant differences in the expression of immune cells such as CD8 naïve, Tr1, nTreg, Th17, and Tfh between the SAH and normal groups. These findings suggest that these immune cells are markedly infiltrated during the progression of SAH, contributing to the disease pathogenesis.
ssGSEA and ROC analysis of SAH hub genes and their diagnostic potential
To gain insight into the biological pathways associated with the SAH hub genes, ssGSEA on the four key hub genes identified earlier. The analysis revealed that these genes were significantly enriched in the Apoptosis, Cell Cycle, MAPK signaling, and p53 signaling pathways (Fig. 6A–D). These findings suggest that these pathways may play a critical role in the pathogenesis of SAH.

Pathway enrichment and diagnostic potential of SAH hub genes. (A–D) Single gene set enrichment analysis (ssGSEA) showing significant enrichment of the hub genes (CCL20, IL6, TLR4, MMP9) in the Apoptosis, Cell Cycle, MAPK signaling, and p53 signaling pathways. (E–H) Receiver Operating Characteristic (ROC) curve analysis of each hub gene’s diagnostic potential. The area under the curve (AUC) values with 95% confidence intervals (CI) indicate high diagnostic accuracy for CCL20 (AUC = 0.85, CI = 0.75–0.95), IL6 (AUC = 0.70, CI = 0.56–0.84), TLR4 (AUC = 0.79, CI = 0.67–0.91), and MMP9 (AUC = 0.86, CI = 0.75–0.97).
Additionally, we assessed the diagnostic specificity and sensitivity of each hub gene using Receiver Operating Characteristic (ROC) curve analysis. We calculated the area under the curve (AUC) and 95% confidence intervals (CI) to evaluate their diagnostic potential. The results showed that CCL20 (AUC = 0.85, CI = 0.75–0.95), IL6 (AUC = 0.70, CI = 0.56–0.84), TLR4 (AUC = 0.79, CI = 0.67–0.91), and MMP9 (AUC = 0.86, CI = 0.75–0.97) all demonstrated high diagnostic value for SAH (Fig. 6E–H).
UMAP analysis reveals distribution patterns of four hub genes in brain tissues and cell subpopulations
We utilized UMAP (Uniform Manifold Approximation and Projection) to analyze the expression patterns of the four hub genes in various brain tissues and cell subpopulations. The results revealed distinct distribution patterns for each gene across different regions. Specifically, CCL20 showed higher expression levels in the cerebellum and white matter (Fig. 7A). IL6 was widely distributed across vesicles (Fig. 7B). TLR4 exhibited a predominant expression in the Golgi apparatus and plasma membrane (Fig. 7C), while MMP9 was mainly localized to the cytosol (Fig. 7D).

UMAP analysis of the expression patterns of SAH hub genes in brain tissues and cell subpopulations. (A) CCL20 expression is predominantly higher in the cerebellum and white matter. (B) IL6 is widely distributed across vesicles. (C) TLR4 exhibits strong expression in the Golgi apparatus and plasma membrane. (D) MMP9 is mainly localized in the cytosol.These results suggest that the differential expression and localization of these genes may play specific roles in the pathophysiology of SAH.
These findings suggest that the four hub genes are differentially expressed in specific brain regions and cellular compartments, which may imply their distinct roles in the pathophysiology of SAH. The differential localization of these genes could contribute to their involvement in various molecular mechanisms, such as inflammation and cell signaling, which are central to the development and progression of SAH.
Results of molecular docking and experimental verification by Western blot analysis
In this study, molecular docking was utilized to predict the potential of IRN as a therapeutic agent. Initially, bioinformatics analysis identified four key target genes: CCL20, IL6, TLR4 and MMP9. Subsequently, molecular docking simulations were conducted using AutoDock Vina to assess the binding potential of IRN with these target proteins. Figure 8A illustrates the chemical structure of IRN. Based on the screening outcomes, docking simulations were performed to evaluate the interactions between IRN and the four hub genes (CCL20, IL6, TLR4 and MMP9). Binding energies lower than 0 kJ/mol suggest that spontaneous binding is likely, while values under − 5.0 kJ/mol indicate a stronger binding affinity. The results of the docking simulations revealed favorable binding interactions, with three out of four docking energies falling below − 5 kJ/mol, suggesting a strong affinity between IRN and the target proteins. The docking conformations are visualized in Fig. 8B–E using PyMOL software.

Molecular docking analysis of IRN with SAH hub genes. (A) Chemical structure of IRN. (B–E) Docking interactions between IRN and the four hub genes (CCL20, IL6, TLR4, and MMP9). The binding energies are displayed, with values below − 5.0 kJ/mol indicating strong binding affinity. Three of the docking simulations show binding energies below − 5 kJ/mol, suggesting a favorable interaction between IRN and these target proteins. The docking conformations are visualized using PyMOL software. (F) WB Analysis of hub genes.
To validate the expression of the core genes CCL20, IL6, TLR4, and MMP9 identified from our bioinformatics analysis, WB analysis demonstrated that LPS stimulation significantly upregulated the expression levels of CCL20, IL6, TLR4, and MMP9 in BV2 microglial cells, compared to the untreated control group (P < 0.05). Treatment with 50 µM IRN significantly downregulated these proteins’ expression (P < 0.01). Quantitative densitometric analysis further confirmed the inhibitory effect of IRN on inflammatory gene expression in BV2 cells (Fig. 8F).These findings validate the bioinformatics predictions and molecular docking results, highlighting the potential of IRN as a therapeutic agent for SAH.
Discussion
SAH is a life-threatening acute neurological disorder, typically characterized by sudden onset of severe headache, vomiting, and altered consciousness. The disease is associated with high mortality and long-term disability, significantly burdening both patients and society. Epidemiological studies show that the annual incidence of SAH is approximately 6–8 cases per 100,000 people, with 25–50% of patients dying within the first month after onset. Among those who survive, around 50% experience varying degrees of neurological dysfunction29. SAH is most commonly caused by the rupture of cerebral aneurysms, although in some cases it may result from cerebrovascular malformations, trauma, or other factors30,31. Despite advances in early diagnosis and treatment, the prognosis for SAH remains poor, primarily due to acute brain injury, secondary cerebral ischemia, brain edema, and neuroinflammation32,33. Therefore, a deeper exploration of the molecular mechanisms of SAH, along with the identification of early diagnostic biomarkers and potential therapeutic targets, is of great importance for improving the clinical prognosis of SAH patients34.
This study systematically investigated the key genes, signaling pathways, and potential therapeutic strategies associated with SAH through bioinformatics analysis, integrating DEGs, immune cell infiltration analysis, and molecular docking simulations. Our approach has revealed the complex biological mechanisms underlying SAH and has proposed new diagnostic and therapeutic strategies.
Our ssGSEA results indicate that four key hub genes—CCL20, IL6, TLR4, and MMP9—are predominantly enriched in the apoptosis, cell cycle, MAPK pathway, and p53 signaling pathway. These pathways play crucial roles in cellular responses to damage and stress. In particular, the apoptosis pathway, which regulates programmed cell death, is a key mechanism in brain injury following SAH. Increased apoptotic activity may exacerbate neuronal loss and hinder the repair process after brain injury. The MAPK signaling pathway is widely involved in inflammatory responses, cell proliferation, and stress reactions, and it is activated in SAH, further promoting neuronal damage. The p53 signaling pathway, through its regulation of the cell cycle and apoptosis, may contribute to neuronal death in SAH.
Of particular note is the interaction between these pathways. CCL20, IL6, TLR4, and MMP9 are not only involved in separate signaling pathways but also interact with each other to regulate critical cellular processes. For example, TLR4 activation can induce inflammatory responses through the MAPK pathway, while IL6 plays a pivotal role in both inflammation and apoptosis. Additionally, MMP9 is involved in extracellular matrix degradation, which, in conjunction with IL6 and CCL20, could exacerbate blood-brain barrier disruption and neuronal injury in SAH. These genes form a complex regulatory network that modulates the balance between cell survival and death, influencing the overall severity of the brain injury.Understanding how these genes interact within these signaling networks provides valuable insights into the mechanisms underlying SAH and suggests that targeting these pathways could offer novel therapeutic strategies. Modulating the apoptosis and immune response pathways, in particular, may help alleviate neuronal damage and improve outcomes for SAH patients. Through ROC curve analysis, we assessed the diagnostic ability of the four hub genes. The results showed that the AUC values for CCL20, IL6, TLR4, and MMP9 were all above 0.7, with CCL20 reaching an AUC of 0.85, indicating high diagnostic accuracy for SAH. These findings suggest that these genes can serve as reliable biomarkers for the early diagnosis of SAH. Given the challenges in the early diagnosis of SAH, where symptoms overlap with those of other conditions, these biomarkers can assist clinicians in more efficiently identifying high-risk patients. Due to the strong diagnostic potential of these hub genes, future research should validate their expression levels in clinical samples and further explore their application in assessing disease severity and predicting patient prognosis.
Immune cell infiltration plays a critical role in the progression of SAH. Our immune infiltration analysis revealed that immune cells such as NKT cells, monocytes, B cells, Th17 cells, and macrophages were significantly associated with the hub genes in SAH. These immune cells are known to play key roles in the inflammatory response following brain injury, with particular emphasis on the activation of Th17 cells and macrophages, which may be crucial in the immune response during SAH. The increased presence of immune cells suggests an imbalance in the chronic inflammatory response in SAH patients. While immune activation may have protective effects during the early stages of the disease, excessive immune activation can exacerbate neuronal damage and cell death. These findings further underscore the dual-edged role of the immune system in the pathological process of SAH. Future studies should explore strategies to regulate the immune response to enhance neuroprotection and improve patient prognosis.
To explore potential therapeutic interventions for SAH, we conducted molecular docking analysis to assess the binding affinity between IRN and the four hub genes. Our docking results showed that three of the hub genes (CCL20, IL6, and MMP9) exhibited binding energies below − 5 kJ/mol, indicating strong binding affinity between these genes and IRN. These findings suggest that IRN may exert its therapeutic effects by binding to these key genes. Further experimental validation is required to confirm IRN’s regulatory effects on the expression of these genes and its therapeutic potential in SAH. So our in vitro WB validation further confirmed that IRN effectively suppresses the LPS-induced overexpression of CCL20, IL6, TLR4, and MMP9 in BV2 microglial cells. These findings suggest that IRN may mitigate neuroinflammation in SAH by modulating these critical inflammatory mediators. This experimental validation supports our molecular docking findings and highlights the translational potential of IRN for SAH treatment.
In conclusion, this study systematically identified key genes and signaling pathways associated with SAH, highlighting the critical role of the immune system in disease progression. Among these, CCL20, IL6, TLR4, and MMP9 were identified as not only potential diagnostic biomarkers but also promising therapeutic targets. Through comprehensive molecular docking analysis, we demonstrated a strong binding affinity between IRN and these hub proteins, supporting its potential therapeutic efficacy in SAH. Additionally, WB validation confirmed that IRN significantly downregulates the expression of CCL20, IL6, TLR4, and MMP9 in LPS-induced microglial cells, providing robust experimental evidence for its anti-inflammatory and neuroprotective effects. Overall, this research not only enhances our understanding of the molecular mechanisms underlying SAH but also provides important theoretical foundations for the development of new diagnostic tools and therapeutic strategies, paving the way for future advances in the clinical treatment of SAH.
Data availability
All data analysed during this study are publicly available in the GEO database under accession number GSE36791 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE36791). Additional data supporting the findings of this study are included in this published article and its supplementary information files.
References
Claassen, J. & Park, S. Spontaneous subarachnoid haemorrhage. Lancet 400(10355), 846–862 (2022).
Osgood, M. L. Aneurysmal subarachnoid hemorrhage: Review of the pathophysiology and management strategies. Curr. Neurol. Neurosci. Rep. 21(9), 50 (2021).
Thilak, S. et al. Diagnosis and management of subarachnoid haemorrhage. Nat. Commun. 15(1), 1850 (2024).
Kane, S. F., Butler, E. & Sindelar, B. D. Nontraumatic subarachnoid hemorrhage and ruptured intracranial aneurysm: Recognition and evaluation. Am. Fam Physician 108(4), 386–395 (2023).
Wang, X. et al. Comprehensive analysis of immune cell infiltration and role of MSR1 expression in aneurysmal subarachnoid haemorrhage. Cell. Prolif. 56(6), e13379 (2023).
Mohme, M. et al. Immune characterization in aneurysmal subarachnoid hemorrhage reveals distinct monocytic activation and chemokine patterns. Transl Stroke Res. 11(6), 1348–1361 (2020).
Cheng, L., Zhao, Y. & Ke, H. Screening for Immune-Related RNA biomarkers of aneurysmal subarachnoid hemorrhage. Clin. Invest. Med. 45(2), E28–38 (2022).
Weiland, J. et al. Neuroprotective strategies in aneurysmal subarachnoid hemorrhage (aSAH). Int. J. Mol. Sci. 22(11) (2021).
Wang, X. et al. Identification of the key immune-related genes in aneurysmal subarachnoid hemorrhage. Front. Mol. Neurosci. 15, 931753 (2022).
Liu, J. et al. Screening of immune-related biological markers for aneurysmal subarachnoid hemorrhage based on machine learning approaches. Biochem. Biophys. Rep. 36, 101564 (2023).
Ye, F. et al. Bioinformatic analysis of coexpressed differentially expressed genes and potential targets for intracerebral and subarachnoid hemorrhage. World Neurosurg. 159, e442–e452 (2022).
Gao, H. et al. Identification of hub genes significantly linked to subarachnoid hemorrhage and epilepsy via bioinformatics analysis. Front. Neurol. 14, 1061860 (2023).
Ritchie, M. E. et al. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43(7), e47 (2015).
Langfelder, P. & Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 9, 559 (2008).
Vizcaíno, J. A. et al. ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32(3), 223–226 (2014).
Szklarczyk, D. et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 49(D1), D605–d612 (2021).
von Mering, C. et al. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res. 31(1), 258–261 (2003).
Shannon, P. et al. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 13(11), 2498–2504 (2003).
The Gene Ontology Resource. 20 Years and still going strong. Nucleic Acids Res. 47(D1), D330–d338 (2021).
Ogata, H. et al. Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 27(1), 29–34 (1999).
Kim, Y. et al. Novel deep learning-based survival prediction for oral cancer by analyzing tumor-infiltrating lymphocyte profiles through CIBERSORT. Oncoimmunology 10(1), 1904573 (2021).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12(5), 453–457 (2015).
Canzler, S. & Hackermüller, J. MultiGSEA: A GSEA-based pathway enrichment analysis for multi-omics data. BMC Bioinform. 21(1), 561 (2020).
Robin, X. et al. pROC: An open-source package for R and S + to analyze and compare ROC curves. BMC Bioinform. 12, 77 (2011).
Uhlén, M. et al. Transcriptomics resources of human tissues and organs. Mol. Syst. Biol. 12(4), 862 (2016).
Chen, W. et al. Shared diagnostic genes and potential mechanism between PCOS and recurrent implantation failure revealed by integrated transcriptomic analysis and machine learning. Front. Immunol. 14, 1175384 (2023).
Becht, E. et al. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. (2018).
Morris, G. M. et al. AutoDock4 and AutoDockTools4: Automated Docking with selective receptor flexibility. J. Comput. Chem. 30(16), 2785–2791 (2009).
Goertz, L. et al. Impact of aneurysm morphology on aneurysmal subarachnoid hemorrhage severity, cerebral infarction and functional outcome. J. Clin. Neurosci. 89, 343–348 (2021).
Tschiderer, L. et al. Sex differences in risk factor relationships with subarachnoid haemorrhage and intracranial aneurysms: A Mendelian randomisation study. Eur. J. Prev. Cardiol. 31(Suppl 1) (2024).
Hostettler, I. C. et al. Duration between aneurysm rupture and treatment and its association with outcome in aneurysmal subarachnoid haemorrhage. Sci. Rep. 13(1), 1527 (2023).
Balança, B., Bouchier, B. & Ritzenthaler, T. The management of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Rev. Neurol. (Paris) 178(1–2), 64–73 (2022).
Ader, J. Guidelines in action: Volume and blood pressure management after aneurysmal subarachnoid hemorrhage. Stroke 55(2), e39–e41 (2024).
Lei, Z. et al. Treatment and clinical analysis of cerebral vasospasm after aneurysmal subarachnoid hemorrhage. Minerva Med. 115(1), 102–104 (2024).
Acknowledgements
We are grateful to the contributors to the public databases used in this study.
Funding
This work was supported by the Science and Technology Department of Sichuan Province (No: 2023NSFSC0655), Sichuan Science and Technology Program (No:2022YFS0621), and Technology and Human resources’ Bureau of Luzhou (No: 2022-SYF-36).
Author information
Authors and Affiliations
Contributions
Writing—original draft preparation, Y.L., Y.Z. and H.W.; methodology, software, and validation, Y.L., Y.Z. and H.W.; data curation, L.L.; writing—review and editing, L.W. and L.L. All authors have read and agreed to the published version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Liu, Y., Zhang, Y., Wei, H. et al. Exploring the molecular mechanisms of subarachnoid hemorrhage and potential therapeutic targets: insights from bioinformatics and drug prediction. Sci Rep 15, 12861 (2025). https://doi.org/10.1038/s41598-025-97642-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-97642-8
