
Abstract
Menangle Virus (MenV) is a zoonotic pathogenic virus that can penetrate humans, causing infection in them. It is an essential threat to public health due to the possibility of cross-species transmissions. Traditional vaccine development methods are time-consuming and resource-intensive, highlighting the need for innovative approaches. This study uses computational techniques to develop a multiepitope vaccine for MenV. We employed in silico tools to find potential B-cell, T-cell, and MHC-binding epitopes in the viral proteome. To ensure their safety and efficacy, these epitopes were evaluated for antigenicity, allergenicity, and toxicity. The selected epitopes were assembled into a multiepitope construct optimized for molecular stability and immunogenicity. Studies of molecular docking and MD simulations were considered to assess the interaction of vaccine candidates with human receptors, indicating a strong immune response. The goal of developing this vaccine is to prepare for potential future outbreaks. This in-silico vaccine design proposes a promising, efficient path to developing effective preventative measures against MenV, potentially expediting vaccine production and contributing to world health security.
Similar content being viewed by others

Introduction
The Menangle Virus (MenV) outbreak began in Menangle, New South Wales, Australia, when a female pig gave birth to congenital deceased piggeries. Subsequently, this outbreak also caused infection in workers working in pig farms. Infected individuals showed red spotty rash, chills, malaise, severe headache and rigours1. MenV is an RNA virus belonging to the Paramyxoviridae family and the Rubulavirus genus. Its structure comprises three key components: an envelope that encases its cytoplasm, a negative-sense single-stranded RNA and pleomorphic proteins, which appear in elongated and spherical forms on its surface. These spike-like proteins are crucial for the virus’s penetration into the host cell2. MenV is present among flying foxes and fruit bats in Australia and remains asymptomatic. Despite the absence of reported cases in recent years, developing a vaccine against MenV remained imperative to prevent future outbreaks. Furthermore, preliminary studies indicate that MenV is closely related to highly transmissible viruses such as Mumps, HPIV-4, Hendra Virus, and Measles, which frequently cause outbreaks. Consequently, there is a significant risk that MenV could also lead to an endemic3.
Moreover, despite attempts to isolate the virus in cell cultures was done, unfortunately, these efforts remained unsuccessful. The virus was isolated from tissues of the lungs, brain, and heart and seems to replicate efficiently in culture using cells such as HmLu-1 and BHK2. During virus isolation, it was found that this virus’s multiplication rate was relatively high. The complex nature of the virus demands innovative approaches to silicon vaccine development3. This study uses computational bioinformatics tools to demonstrate the construction of an in-silico multiepitope vaccine candidate against MenV. Additionally, computational vaccines have several advantages over conventional vaccines, such as efficient identification of viral genomes, specific antigenic sites, and reduced costs and time savings4. At the time, there was no vaccine available against this virus.
No preventive measures or defined treatments and therapeutics are available for this virus. Menangle virus (MenV) has a high potential for multiplication and can spread over large pig farms, posing a continuous risk to farm workers5. Given the virus’s limited human cases, the current risk to the general population is considered low. However, the potential for future outbreaks and the close relation to other highly transmissible viruses underscores the importance of monitoring and developing preventive measures, including vaccines, to mitigate potential risks4.
The development of a vaccine is crucial for mitigating the risks associated with MenV which include severe respiratory illness and potentially fatal outcomes. A vaccine would protect vulnerable populations, such as those in close contact with infected animals, and enhance global health security by preventing potential outbreaks and their socioeconomic consequences6. The persistent rarity of the virus cannot be guaranteed, as viruses can mutate and cross species barriers rapidly, leading to varying morbidity and potential pandemics. This study involves the development of multi-epitope vaccine candidate to protect world from another deadly pandemic, which could be occured because of MenV7.
Materials and methods
In the following study, researchers utilized computational bioinformatics tools to design a promising vaccine candidate against MenV leveraging advanced computational analyses and predictive modelling techniques to identify antigenic epitopes and structural features conducive to inducing protective immune responses.
Retrieval of targeted protein sequence
The process of developing a multiepitope vaccine candidate for MenV began with the retrieval of protein sequences from the UniProt database (https://www.uniprot.org/) in a specific FASTA (canonical) format. UniProt is an extensive source of protein sequence and functional information of proteins. It allows retrieval of protein sequence, the first step of vaccine development. A transmembrane protein named fusion glycoprotein F0 (K9MY70) was selected for indicating subcellular location of proteins TMHMM webserver https://services.healthtech.dtu.dk/services/TMHMM-2.0/ was used8.
Selection of targeted protein
The selected protein’s antigenicity, allergenicity and toxicity analysis is mandatory before proceeding further. Therefore, the web server VAXIJEN (http://www.ddg-pharmfac.net/vaxijen/) was used to predict the protein’s antigenicity. for predicting the allergenicity of the selected protein, ALLERTOP V2.0 (https://www.ddg-pharmfac.net/AllerTOP/) was used, and TOXIPRED (https://webs.iiitd.edu.in/raghava/toxinpred/) was used to predict the toxicity of the selected protein.
Identification of B-cell epitopes
B-cell epitopes are required to produce antibodies that expressly represent antigens and identify epitopes of B-cells. For the development of adaptive immunity, isolation of B-cell epitopes needed to be in the sequence of vaccine construct. We predicted B-cell epitopes for selected transmembrane proteins from IEDB. B-Cell Prediction Server (https://www.iedb.org/). The antigenicity and allergenicity of selected epitopes were predicted using tools like VAXIJEN and ALLERTOP V 2.0. After analysis, it was found that all selected epitopes are antigenic in nature and non-allergen, respectively9.
Prediction of MHC class I epitopes
MHC class I cells are located on the surface of every nucleated cell and present peptides for CD8 + cytotoxic T lymphocytes; for the prediction of MHC Class I epitopes IEDB server was used (http://tools.iedb.org/main/tcell/) by utilizing the ANN4.0 method. This method recognized diverse databases, distinguishing epitopes from non-epitopes, resulting in accurate prediction. IC50 score and epitope start and end points were also considered10.
Identification of MHC class II epitopes
MHC Class II cells are on APCs, presenting peptides to CD4 + helper T cells. For MHC Class II epitope identification, the IMMUNE EPITOPE DATABASE (IEDB) ANALYSIS RESOURCE (http://tools.iedb.org/mhcii/) was employed, along with the NN-align 2.3 (NetMHCII 2.3) method. The antigenicity, allergenicity and IC50 scores and rankings of the chosen epitopes were predicted. Prediction of such variables ensures that the selected epitopes are safe to use10.
Vaccine construction
To form a vaccine construct, the aid of linkers and adjuvant combined epitopes of B-cells, MHC-Class I and MHC-Class II was identified. We used the linkers in this vaccine construct: EAAAK, GPGPG, GGGGS and AAY. These linkers prevent steric hindrance and allow proper folding of epitopes so that they can be correctly recognized by an immune response to boost immunization; an adjuvant of amino acid sequence fifties ribosomal L7/L12 (UniProt ID: P9WHE3) was inserted at N-terminal, respectively by using EAAAK linker. This adjuvant is used because it is highly immunogenic and helps stimulate CD4 + helper T cells and CD8 + cytotoxic T cells. Furthermore, inserting this specific adjuvant facilitates the uptake of antigenic epitopes by antigen-presenting cells (APCs)11.
Population coverage of vaccine
An online web server, IEDB (https://www.iedb.org/), was used to check the population coverage of both MHC Class I and MHC Class II. We analyze the population coverage of both classes separately, and each class show population coverage of more than 60%, indicating that vaccine candidates are suitable worldwide12.
Vaccine allergenicity and antigenicity prediction
The allergenicity of the vaccine was predicted by the AllerTOP v.2.0 server (https://www.ddg-pharmfac.net/AllerTOP/). This approach transforms protein sequences into standardized vectors of equal length by employing auto cross-covariance. Their result shows whether the protein is non-allergenic or allergenic. The vaccine’s antigenicity was determined using the VaxiJen web server (http://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html). The server implements a methodology that does not require alignment but focuses on amino acids’ primary characteristics, with a defined threshold of 0.413.
Solubility prediction
The solubility of the vaccine construct was determined by SOLUPROT (https://loschmidt.chemi.muni.cz/soluprot/). SOLUPROT estimates the solubility of proteins based on their amino acid sequences and uses algorithms to predict whether a given protein sequence will be soluble or insoluble under certain conditions14.
Physiochemical properties
A tool known as Expasy ProtParam was considered to assess physicochemical properties. This tool offers details concerning the length of the vaccine’s intended the struct, including aliphatic index, solubility, molecular weight, and half-life. Other variables of equal importance, like theoretical PI and GRAVY, were also analyzed via this tool. The Grand Average of Hydropathy (GRAVY) is calculated by adding up all amino acids’ hydrophobic values and dividing by the total number of amino acids in the resulting protein15.
Secondary structure of vaccine prediction
The 2D structure of the vaccine construct was determined by PsiPRED (http://bioinf.cs.ucl.ac.uk/psipred/). The PSIPRED server utilizes position-specific prediction Psi-BLAST to identify and arrange sequences that exhibit significant similarity to the vaccine protein8.
Tertiary structure prediction of vaccine
TrRosetta server (https://yanglab.qd.sdu.edu.cn/trRosetta/) was utilized for the prediction of the 3D structure of the vaccine. To forecast the separation and direction of amino acid residues within a protein. TrRosetta can create precise three-dimensional models of protein structures by using these projected inter-residue distances and orientations12.
Validation of protein structure
The 3d structure of our vaccine model was verified through PROCHECK (http://services.mbi.ucla.edu/SAVES/). ERAAT value was predicted to check the reliability value of the selected 3D protein’s structure. Ramachandran plot was predicted to assess the functionality of the protein model based on dihedral angles -phi (φ)and psi (ψ)- of the amino acids10.
Molecular docking and deep interaction study
An online docking tool called Cluspro2.0 https://cluspro.org/tut_dock.php was used to execute molecular docking of vaccine and human receptors. The proposed vaccine was docked with TLR4 (PDBID: 2Z65) obtained from RCSB PDB https://www.rcsb.org/. The docking tool Cluspro2.0 calculates interaction complexes based on interaction energies and centres between proteins9. Deep interaction studies was carried out by Ligplot + https://www.ebi.ac.uk/thornton-srv/software/LigPlus/. This tool provides a detail overview of no. of chains present in the structure and interactions among vaccine and given receptor. Presence of large number of hydrogens bonds indicates that the vaccine have strong immune response in respect with the human receptor.
Molecular dynamics simulations
The AMBER software was employed to perform MD simulations to study the dynamic properties of two docked systems. Protein structures were modeled using the AMBER ff19SB force field, while ligand structures were parameterized with the General AMBER Force Field 2 (GAFF2). A TIP3P water model was used to solvate the systems within a cubic simulation box, ensuring a 12 Å buffer around the solute. To mimic physiological conditions and maintain charge neutrality, sodium and chloride ions (NaCl) were introduced into the system. Energy minimization was carried out for 20,000 steps to resolve any steric clashes or unfavorable interactions. This was followed by equilibration for 5 nanoseconds under constant temperature (298 K) and pressure (1 bar), with a 2 femtosecond (fs) integration timestep. For production MD runs, identical temperature and pressure conditions were maintained, and trajectory snapshots were recorded every 10 picoseconds, yielding 2,000 frames16.
Expression analysis
EMBOSS Backtranseq (https://www.ebi.ac.uk/jdispatcher/st/emboss_backtranseq) was utilized to convert our vaccine’s protein sequence to a nucleotide sequence. This nucleotide sequence was further optimized using the online Java Codon Adaption Tool. ( https://www.jcat.de/) A program called SnapGene, available offline, was employed for the in-silico cloning of the vaccine candidate. PET28a + plasmid was used to clone the vaccine construct7.
Immune stimulation
The immunological activity of the designed vaccine was analyzed using the tool C-ImmSim https://kraken.iac.rm.cnr.it/C-IMMSIM/index.php, which predicts a response similar to the human body’s natural immune response based on algorithms and models. This prediction is an estimation, not replicating a human body’s natural response. However, these Immune simulations can identify potential epitopes that are likely to elicit strong immune responses, aiding in the design of more effective vaccines. Additionally, we added comparison of immune response when only adjuvant is inserted, immune responses when only vaccine candidate is inserted and immune response when adjuvant linked with vaccine is inserted into the host’s body.
Results
Retrieval of targeted protein sequence
There were 159 proteins of this virus present on UniProt DATABASE, of which two were transmembrane proteins. The fusion glycoprotein (F0) (K9MY70) was selected based on its antigenicity and allergenicity. This protein sequence is based on 554 amino acids.
Selection of targeted protein
The selected protein was examined using ALLERTOP V 2.0, VAXIJEN and TOXIPRED. However, analysis shows that the following transmembrane protein is non-toxic, non-aland ergen, and its antigenicity is more significant than 0.5, indicating dating indicates probable and tighten. This protein’s function and sub-cellular locations are shown in Table 1; Fig. 1.

Graphical representation of subcellular locations of targeted protein by TMHMM server. (A) Fusion glycoprotein (F0).
Prediction of B-cell epitopes
We used the IEDB website to obtain B-cell epitopes. B-cell epitopes are important because they primarily bind to B-cell receptors, initiating the immune response by producing antibodies. They also result in the release of memory B-cells. B-cells also play a fundamental role in recognising and eliminating viral pathogens. For further consideration, ALLERTOP V 2.0, VAXIJEN and TOXIPRED were used to identify perfect epitope. Analysis of these tools indicates that every epitope was antigenic, non-allergen and non-toxic, as mentioned in Table 2.
Prediction of MHC class 1 epitopes
Though important, MHC Class I cells induce adaptive immunity against targeted viruses. They play a role in identifying viruses and destruction of viral pathogens. IC50 value and percentile score were analyzed, and based on these values, four epitopes of selected protein were selected. For each epitope, all alleles were selected. Further consideration was done by tools TOXIPRED, ALLERTOP V 2.0 and VAXIJEN. The results of these tools revealed that every single epitope is non-toxic, non-allergen and antigenic. Then, the IC50 value, Percentile Rank, and antigenicity value are shown in Table 2.
Prediction of MHC class II epitopes
MHC Class II epitopes are fundamental for the destruction of pathogens at a large scale. The selected protein has four chosen epitopes, each with six to seven alleles. Selection of epitopes is done based on antigenicity and allergenicity. It resulted that all epitopes chosen are non-toxic with the lowest IC50 value. Table 3 provides information on all the following parameters.
Vaccine construction
Linkers, epitopes, and adjuvants were used in constructing a vaccine construct. Linkers are important for stable protein folding and preserving vaccine protein structure. Three B cell epitopes, four MHC class I, and four MHC class II epitopes were joined using EAAAK, GPGPG, GGGS and AAY linkers to create a vaccine construct. The adjuvant fifties ribosomal L7/L12 sequence of proteins was added to the N-terminal with the aid of the EAAAK linker to increase the immunological response of the vaccine construct. An amino acid sequence of 296 was obtained as the final vaccine construct, shown in Fig. 2. Antigenicity, allergenicity and toxicity of this vaccine construct were predicted by Vaxijen, Allertop 2.0 and ToxinPred, and after analysis, it was found that the vaccine construct is non-toxic, non-allergen and has antigenicity greater than 0.5.

Vaccine construct of MenV comprises of 296 amino acids, Linker (Blue), 50s ribosomal L7/L12 protein adjuvant (Yellow), MHC-I epitopes (Green), B-cell epitopes (Grey), MHC-II epitopes (Purple), Histidine chain (red).
Population coverage of vaccine construct
We assess population coverage by using the web server IEDB. After analysis of MHC-I and MHC-II, the result shows that more than 70% of the population coverage by following the vaccine construct. The resultant percentage identity of population coverage is shown in Fig. 3.

Graphical representation of Population Coverage around the world. (A) World population coverage of MHC Class I. (B) World population coverage of MHC Class II.
Vaccine allergenicity and antigenicity prediction
Vaccine allergenicity and antigenicity prediction are important to ensure the vaccine is safe, non-allergenic, and capable of eliciting a strong immune response. Subsequently, ALLERTOP V2.0 predicts that the vaccine construct is non-allergenic, and the VAXIJEN web server was used to predict the antigenicity of the vaccine construct, which was 0.5, probable antigen.
Solubility prediction of vaccine candidate
The solubility of the vaccine is demonstrated to know whether our protein is soluble in the host cells. Vaccines must be soluble for better immunity, and their value must be greater than 0.5. the solubility value of our vaccine is 0.887.
Physiochemical properties of vaccine
The physicochemical properties of the vaccine construct were comprehensively analyzed using the Expasy ProtParam web server. This server provides a range of important parameters, including molecular weight, theoretical pI (isoelectric point), amino acid composition, atomic composition, extinction coefficient, estimated half-life, instability index, aliphatic index, and grand average of hydropathicity (GRAVY). The detailed results of these analyses are presented in Table 4.
Secondary structure prediction
The vaccine construct’s secondary structure, predicted by the PSIPRED V4.0 server, revealed that a substantial portion of the multiepitope vaccine comprises the alpha-helix, with the coil and beta-strand making up the remainder (Fig. 4A). Type of PsiPRED shows that protein sequence contains polar, small nonpolar, hydrophobic, and aromatic plus cystine residues in Fig.B. Structures obtained in PSIPRED is shown in Fig. 4.

2D structure prediction through PsiPRED (A) shows coil, helix and strands (B) AAtype results show types of amino acids present in the vaccine (C) PsiPRED cartoon image of the protein sequence.
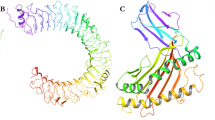
Tertiary structure prediction and validation of vaccine construct
The tertiary structure of the vaccine was constructed by utilizing the trRosetta server. This resulted in a four-domain model based on the provided amino acid sequence. The composition of the 3D structure included helix, Beta-sheet, and Coils. The TM score of the 3D structure is 0.482. 3D structure of the vaccine construct is shown in Fig. 5. The refined tertiary structure of the vaccine candidate was validated using the PROCHECK Ramachandran Plot and ERAAT value prediction as illustrated in Fig. 5 B. ERAAT value of 86.49 states that the structure is hydrophilic and highly stable while the Ramachandran plot analysis revealed that 95.5% of the residues are in the most favorable regions, 4.1% are in the additionally allowed regions, 0.4% are in the generously allowed regions and no residues are present in the disallowed areas. A high-quality model is expected to have at least 90% of residues in the most favored regions.

prediction and validation of MenV vaccine candidates. (A) Refined tertiary structure of MenV vaccine candidate. (B) Validation of 3D structure of vaccine candidate.
Molecular docking and deep interactions studies
Molecular docking of vaccine candidate was performed with TLR4 and Cluspro 2.0 predicted ten models for the receptor to analyze the interaction between the vaccine construct and the receptor in the human body. The finest zero model was selected for both TLR4. Docked complex of vaccine candidate and human receptor TLR4 reveals that the 0th model contain 74 members and − 913.4. Deep interaction studies were carried out by using Ligplot + which indicates a strong association of 7 hydrogen bonds between vaccine candidate and human receptor, as shown in Fig. 6.

(A) Molecular interaction of vaccine with TLR5. (B) Ligplot + interaction analysis between vaccine candidate and human receptor.
Molecular dynamics simulation
-
Root mean square fluctuation (RMSF)
The RMSF graph provided details of the stability of individual residues in the protein-ligand complex. Residues with RMSF values below 2 Å indicate structurally stable regions, likely forming the core of the protein or critical interaction sites with the ligand. These stable regions contribute to the overall structural integrity of the complex and are indicative of robust interactions that are maintained throughout the simulation as shown in Fig. 7 (A).
-
Root mean square deviation (RMSD)
The RMSD graph showed the overall stability of the protein-ligand complex during the simulation. After an initial equilibration phase, the RMSD plateaus around a stable value (e.g., ~ 1.5-2.0 Å), suggesting the complex achieves and maintains stability as shown in Fig. 7 (B). A consistent RMSD over the simulation duration highlights the ability of the complex to retain its structural integrity under simulated conditions, indicating reliable and stable interactions between the protein and ligand.
-
Radius of gyration (RoG)
The RoG graph measured the compactness of the protein structure throughout the simulation. A stable RoG value (e.g., ~ 26 Å) over time implies the protein retains its compactness without significant structural expansion or collapse as shown in Fig. 7 (C). This stability in compactness suggests the protein maintains its functional conformation, further supporting the overall structural robustness observed in the simulation.
-
Principal component analysis (PCA)
PCA provided a visualization of the dominant motions within the protein complex. Clusters of points in the PC1 vs. PC2 graph indicate regions of structural stability, where the protein samples specific conformations consistently as shown in Fig. 7 (D). The results here suggest the protein explores a limited conformational space, underscoring the stable nature of the complex as observed in the RMSD and RoG analyses.

Molecular simulations by using GROMACS (A) RMSF graph of docked complex of MenV vaccine candidate and TLR (B) Radius of Gyration graph of docked complex of MenV vaccine candidate and TLR (C) RMSD graph of docked complex of MenV vaccine candidate and TLR (D) PCA graph of docked complex of MenV vaccine candidate and TLR.
In-silico cloning with snapgene
The pET28(a) plasmid vector sequence was retrieved using the SnapGene database. As seen in Fig. 8, the vaccine was cloned into the pET-28(a) vector by cutting the vector and vaccine construct for expression in Escherichia coli using restriction enzymes NcoI and SphI.

(A) The Cloned vector has 5560 base pairs and is cut by restriction enzymes NcoI and SphI. (B) The clone and the red line represent the vaccine in the plasmid.
Immune simulation analysis
The comparative study represents the results of immune simulation and indicates that the addition of important adjuvants has an amplification effect on the vaccine candidate’s immune response (Fig. 9). In adjuvant-only simulations (panels A and B), we see modest effective antigen clearance and immune activation with less immunoglobulin (IgM, IgG1, and IgG2), and cytokines/interleukins produced. Vaccine-only simulations (panels C and D) show an enhanced immunoglobulin response that includes an increased IgG1 titer, typically associated with a stronger and more prolonged immune response than that induced by the adjuvant alone. Adding the adjuvant linked to the vaccine candidate by a linker (panels E and F) reveals synergism; antigen clearance is faster and more pronounced, and immunoglobulin titers, especially IgG2, peak at greater heights, thus portraying better humoral immunity. Likewise reported increases occur in the cytokines IFN-γ and IL-2, which present clear evidence for a stronger Th1-type cellular immune response. The inset plot of IL-2 presented in panels D and F shows a marked increase in danger signal and leukocyte growth factor production when the adjuvant was in conjunction with the vaccine, indicating very good overall activation of the immune system and increased proliferation of leukocyte production. Thus, all these results indeed confirm that the addition of an adjuvant could much amplify both humoral and cellular immune responses and therefore help to render a candidate vaccine far more effective in terms of achieving immunity.

Immune simulation results for the adjuvant, vaccine candidate, and adjuvant-linked vaccine candidate compared in respect to their effects. The immune simulations shown in (A, B) are attributed to the effect of the adjuvant regimen. The immune profiles for the vaccination candidate are shown in (C, D), where it has been noticed to boost both immunoglobulin and cytokine productions over and above that of the adjuvant. (E, F) show the immune responses when the adjuvant is conjugated to the vaccine candidate through a linker to enhance response.
Discussion
MenV is a zoonotic virus that causes infection in pigs and humans. This virus primarily affects pigs and causes different reproductive diseases like reproductive disease, with symptoms including increased fetal death, fetal abnormalities and stillborn piglets17. The clinical manifestations of MenV infection in pigs encompass a noticeable rise in the number of shrivelled and deceased piglets, some revealing severe congenital abnormalities which include arthrogryposis, hydranencephaly, brachygnathia, kyphosis and also porencephaly18. Additionally, deceased piglets may present with severe non-suppurated encephalomyelitis. In humans, the symptoms were comparatively milder and typically involved a sudden onset of malaise, chills, and fever, succeeded by profuse sweats, intense headaches, myalgia, lymphadenopathy, weight loss, and a non-itchy macular rash19.
There is no specific drug or vaccine for MenV, but some drugs that are available for viruses of the family paramyxovirus can be helpful for this virus. Novel antigens and vaccination strategies must be devised to achieve the goal of eradicating MenV infection20. Recent advances in immunoinformatics highlight the importance of developing dependable, inexpensive, successful, and dependable vaccine candidates for various infectious diseases. Subunit immunizations comprise protein fragments and a potent adjuvant that effectively stimulates the immune system and provides immunity to the host, respectively21. Priority is given to subunit-based vaccines in developing potential vaccine constructs targeting the disease-causing agents. Numerous investigations have been reported on multiepitope vaccines employing an immuno-informatics strategy against diverse pathogens22.
This study aimed to develop a multiepitope vaccine against MenV infection. This vaccine was designed by employing the identification of three epitopes of B-cell, four MHCI epitopes, and four epitopes of MHC-II, which were subsequently linked via selected linkers (EAAAK, GPGPG, AAY, and GGGGS) and an adjuvant, facilitating the elicitation of both humoral and cellular responses in the vaccine recipient. Different types of linkers, including EAAAK, GPGPG, AAY, and GGGGS, were employed in the vaccine construction to develope its structural and functional characteristics; each linker has specific contributions: EAAAK confers rigidity to structures to help in folding and provide space between functional domains; GPGPG increases immunogenicity as a spacer between epitopes; AAY is known to promote processing and presentation of the antigen that is particularly MHC-I epitopes; and GGGGS provides flexibility while maintaining structural integrity in the linked moieties. We thus established optimal spatial position, stability, and effective antigen presentation that altogether improved overall immune response by mixing these linkers14,23,24. A total of 554 amino acids engage in the Intended vaccine construct. The formulated vaccine’s physicochemical properties demonstrated that it is capable antigenicity, non-allergenic, and probable soluble. Moreover, the secondary and tertiary structures are predicted using PsiPRED and I-TASSER/tr-ROSETTA. The combined analyses of RMSF, RMSD, RoG, and PCA highlighted the stability of the protein-ligand complex under simulated conditions. Stable RMSF values correspond to robust core regions, while consistent RMSD and RoG values demonstrate overall structural integrity and compactness. PCA further confirms limited conformational variability, emphasizing the stable and reliable nature of the complex throughout the simulation16.
Furthermore, the refinement of the vaccine construct utilizing GalaxyRefine contributed to an improvement in the structural quality. The reliability of our vaccine was predicted via the Ramachandran plot. Analysis of this tool discovers that most residues are subsequently located in the preferred zone, and a minute quantity of residues were in the undesirable region. Model 0 of the vaccine-receptor (TLR4) complex was selected for interaction studies25. This study of interaction additionally revealed the most interactions with the lowest energy values, as illustrated by Clustpro2. After obtaining the docked complex, interaction analysis revealed that vaccine candidate shows strong bond with human receptor. Based on the recognized literature, it is anticipated that the designed structure of the vaccine in this study can effectively prevent MenV infection with 90.97% coverage worldwide.
Developing a novel multiepitope vaccine against MenV that targets its transmembrane proteins using in-silico methods indicates an intriguing prospect for future research and development. The next critical step is to validate our computational predictions with thorough in vitro and in vivo experiments to ensure the immunogenicity and safety of the designed vaccine. Following successful laboratory validation, the vaccine candidate should enter human clinical trials to assess its efficacy and safety in various populations. The approach demonstrated in this study has broader applicability and could be adapted for other viral pathogens with similar structural characteristics, potentially accelerating vaccine development for emerging or re-emerging viruses26. Exploring the integration of the MenV multiepitope vaccine with other vaccines could provide broader protection and improve vaccination coverage, particularly in regions burdened by multiple viral infections. Successful development and implementation of this vaccine could significantly reduce the incidence and transmission of MenV infections, leading to decreased disease burden and healthcare costs.
Conclusion
The primary objective of this study was to use computational analysis to produce an effective vaccination against the MenV. The demonstrated in-silico approach will benefit future experimental investigation since it reduces the expense and time required to find epitopes and target candidates. Strong immunological response, over 90% population coverage, and a proven computational approach are considered important requirements for clinical trials, and these are all followed in this work. Within the first five days following injection, the antigen was effectively elicited and eliminated from the host system, as predicted by the immune response. Even if verified, these results are only estimates, and experimental validation is required to confirm the vaccine’s effectiveness. Still, the design is advantageous for eradicating MenV.
Data availability
Data availability statement: The datasets generated and/or analyzed during the current study are available as follows:1. The protein sequence fasta of fusion glycoprotein F0 was retrieved from the UNIPROT database under accession number “K9MY70” accessible via this link: https://www.uniprot.org/uniprotkb/K9MY70/entry2. The TLR4 was used for docking with vaccine candidate of ID: 2Z65, accessible via this link: https://www.rcsb.org/structure/2Z65 3. Supplementary files, including additional datasets and materials relevant to this study, are publicly available on OneDrive and can be accessed at the following link: https://drive.google.com/drive/folders/1EbHZWO1jUR1Xv4N1T-uKl15h0g8LL9mI? usp=drive_link.
References
Barr, J. A., Smith, C., Marsh, G. A., Field, H. & Wang, L. F. Evidence of Bat origin for menangle virus, a zoonotic paramyxovirus first isolated from diseased pigs. J. Gen. Virol. 93 (12), 2590–2594. https://doi.org/10.1099/vir.0.045385-0 (2012).
Webby, M. N. et al. Structural analysis of the menangle virus P protein reveals a soft boundary between ordered and disordered regions. Viruses 13 (9), 1737. https://doi.org/10.3390/v13091737 (2021).
Enayatkhani, M. et al. Reverse vaccinology approach to design a novel multi-epitope vaccine candidate against COVID-19: an in Silico study. J. Biomol. Struct. Dynamics. 39 (8), 2857–2872. https://doi.org/10.1080/07391102.2020.1756411 (2021).
Kardani, K., Bolhassani, A. & Namvar, A. An overview of in Silico vaccine design against different pathogens and cancer. Expert Rev. Vaccines. 19 (8), 699–726. https://doi.org/10.1080/14760584.2020.1794832 (2020).
Kirkland, P. D. Menangle virus: one of the first of the novel viruses from fruit bats. Microbiol. Australia. 38 (1), 22. https://doi.org/10.1071/MA17007 (2017).
Virtue, E. R., Marsh, G. A. & Wang, L. F. Paramyxoviruses infecting humans: the old, the new and the unknown. Future Microbiol. 4 (5), 537–554. https://doi.org/10.2217/fmb.09.26 (2009).
Martinelli, D. D. In silico vaccine design: A tutorial in immunoinformatics. Healthc. Anal. 2, 100044 (2022). https://doi.org/10.1016/j.health.2022.100044
Naveed, M. et al. An Aedes–Anopheles vaccine candidate supplemented with BCG epitopes against the Aedes and Anopheles genera to overcome hypersensitivity to mosquito bites. Acta Parasitol. 69 (1), 483–504. https://doi.org/10.1007/s11686-023-00771-1 (2024).
Naveed, M. et al. Designing a novel Peptide-Based Multi-Epitope vaccine to evoke a robust immune response against pathogenic Multidrug-Resistant Providencia Heimbachae. Vaccines 10 (8), 1300. https://doi.org/10.3390/vaccines10081300 (2022a).
Naveed, M. et al. Immunoinformatics approach to design Multi-Epitope-Based vaccine against Machupo virus taking viral nucleocapsid as a potential candidate. Vaccines 10 (10), 1732. https://doi.org/10.3390/vaccines10101732 (2022).
Muhammad Rehman, H. et al. In Silico investigation of a chimeric IL24-LK6 fusion protein as a potent candidate against breast cancer. Bioinform. Biol. Insights. 17, 11779322231182560. https://doi.org/10.1177/11779322231182560 (2023).
Naveed, M. et al. Designing a novel Peptide-Based Multi-Epitope vaccine to evoke a robust immune response against pathogenic Multidrug-Resistant Providencia Heimbachae. Vaccines 10 (8), 1300. https://doi.org/10.3390/vaccines10081300 (2022b).
Naveed, M. et al. An mRNA-based reverse-vaccinology strategy to stimulate the immune response against Nipah virus in humans using fusion glycoproteins. Acta Biochim. Pol. https://doi.org/10.18388/abp.2020_6721 (2023).
Aziz, T. et al. Designing a multiepitope vaccine against the foodborne pathogenic bacteria Listeria monocytogenes using subtractive immunoinformatics approaches. Front. Bioscience-Landmark. 29 (5), 176. https://doi.org/10.31083/j.fbl2905176 (2024).
Naveed, M. et al. A vaccine construction against COVID-19-Associated mucormycosis contrived with Immunoinformatics-Based scavenging of potential Mucoralean epitopes. Vaccines 10 (5), 664. https://doi.org/10.3390/vaccines10050664 (2022).
Sadia, H. et al. Natural AI-based drug designing by modification of ascorbic acid and Curcumin to combat buprofezin toxicity by using molecular dynamics study. Sci. Rep. 14 (1), 28445. https://doi.org/10.1038/s41598-024-79275-5 (2024).
Kirkland, P. et al. Epidemiology and control of menangle virus in pigs. Aust. Vet. J. 79 (3), 199–206. https://doi.org/10.1111/j.1751-0813.2001.tb14580.x (2001).
Halpin, K., Young, P. L., Field, H. & Mackenzie, J. S. Newly discovered viruses of flying foxes. Vet. Microbiol. 68 (1–2), 83–87. https://doi.org/10.1016/S0378-1135(99)00063-2 (1999).
Bowden, T. R., Bingham, J., Harper, J. A. & Boyle, D. B. Menangle virus, a pteropid Bat paramyxovirus infectious for pigs and humans, exhibits tropism for secondary lymphoid organs and intestinal epithelium in weaned pigs. J. Gen. Virol. 93 (5), 1007–1016. https://doi.org/10.1099/vir.0.038448-0 (2012).
Philbey, A., Ross, A., Kirkland, P. & Love, R. Skeletal and neurological malformations in pigs congenitally infected with menangle virus. Aust. Vet. J. 85 (4), 134–140. https://doi.org/10.1111/j.1751-0813.2007.00131.x (2007).
Michel-Todó, L. et al. In Silico design of an Epitope-Based vaccine ensemble for Chagas disease. Front. Immunol. 10, 2698. https://doi.org/10.3389/fimmu.2019.02698 (2019).
Yaiw, K. C. et al. Viral morphogenesis and morphological changes in human neuronal cells following Tioman and menangle virus infection. Arch. Virol. 153 (5), 865–875. https://doi.org/10.1007/s00705-008-0059-0 (2008).
Alizadeh, M. et al. Designing a novel multi–epitope vaccine against Ebola virus using reverse vaccinology approach. Sci. Rep. 12 (1), 7757. https://doi.org/10.1038/s41598-022-11851-z (2022).
Kumar, A., Misra, G., Mohandas, S. & Yadav, P. D. Multi-epitope vaccine design using in Silico analysis of glycoprotein and nucleocapsid of NIPAH virus. PLOS ONE. 19 (5), e0300507. https://doi.org/10.1371/journal.pone.0300507 (2024).
Srivastava, S. et al. Structural basis for designing multiepitope vaccines against COVID-19 infection: in Silico vaccine design and validation. JMIR Bioinf. Biotechnol. 1 (1), e19371. https://doi.org/10.2196/19371 (2020).
Saha, C. K., Hasan, M., Hossain, M. S., Jahan, M. A., Azad, A. K. & Md., & In Silico identification and characterization of common epitope-based peptide vaccine for Nipah and Hendra viruses. Asian Pac. J. Trop. Med. 10 (6), 529–538. https://doi.org/10.1016/j.apjtm.2017.06.016 (2017).
Acknowledgements
The authors express their gratitude to Research Supporting Project number (RSP2025R462) King Saud University Riyadh Saudi Arabia.
Author information
Authors and Affiliations
Contributions
Conceptualization, Muhammad Naveed; methodology, Adeeba Ali; software, Tariq Aziz; validation, Noor Fatima; formal analysis, Sonia Amjad, investigation, Furmein Fatima; resources, Thamer H Albekairi.; data curation, Abdullah F Alasmari.; writing—original draft preparation, Adeeba Ali.; writing—review and editing, Metab Alharbi; visualization, Ayaz Ali Khan; Supervision, Muhammad Naveed.; project administration, Tariq Aziz.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Naveed, M., Ali, A., Aziz, T. et al. Development of a novel multiepitope vaccine against Menangle virus (MenV) using in-silico approaches by targeting its transmembrane proteins. Sci Rep 15, 13715 (2025). https://doi.org/10.1038/s41598-025-98151-4
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-98151-4
