
Abstract
Tick-borne bacterial pathogens from animals play a significant role in the (re)emergence of human diseases. Rhipicephalus sanguineus sensu lato, a globally prevalent tick, predominantly parasitises domestic dogs but can also feed on humans. We characterised temporal changes in the bacterial microbiome of the midgut and salivary gland tissues of R. sanguineus s.l. ticks and analysed their potential as reservoirs for pathogenic bacteria. A 16 S microbiome and amplicon sequence variant (ASV) approach was used to study the bacteria present in the tissues of R. sanguineus s.l. ticks collected from dogs in Hluvukani, a village in a rural community in Bushbuckridge, Mpumalanga, South Africa, in 2016, 2017 and 2019. Post processing, we obtained 43,161 total sequence reads which were clustered into ASVs by sample year. The final ASVs dataset consisted of seven genera: Coxiella, Anaplasma, Escherichia/Shigella, Ehrlichia, Borrelia, Rickettsia and Wolbachia. No differences in the microbiome profiles of the MG and SG tissues were noted. Coxiella endosymbionts dominated the microbiome in all years. Anaplasma was first detected in 2017, and an increase in Anaplasma levels was detected in 2019, when compared to 2017. All other genera were present at low levels. With the exclusion of Wolbachia, the other detected genera could have pathogenic potential, highlighting the role that R. sanguineus s.l. might play as a reservoir of pathogens.
Similar content being viewed by others

Introduction
Zoonotic pathogens from domestic and wild animals are a leading cause of emerging and re-emerging diseases in humans1. Ticks, with their hematophagous feeding behaviours, wide host range, and global distribution are ideal vectors of zoonotic pathogens among animals and humans2. Analysing the bacterial microbiome of specific tissues like the midgut (MG) and salivary glands (SG) is an effective method for identifying tick-borne pathogens (TBPs) and endosymbionts3. Bacteria from a blood meal will invade the tick’s MG tissues, traverse through the tick cells, and finally infiltrate the SG cells where they can be transmitted during the next blood meal. Thus, the MG and SG are the organs of acquisition and transmission, respectively. Primary endosymbionts significantly impact tick fitness and survival and can influence TBP acquisition and transmission3,4.
TBPs pose a substantial risk to rural South African communities at the interface between wildlife, livestock, and humans1. Rhipicephalus sanguineus sensu lato (s.l.) is a species complex consisting of a tropical and temperate lineage. The tropical lineage is found in countries such as Mozambique, Zambia, Kenya and South Africa, while the temperate lineage is found in countries such as Argentina, Chile, and Italy5. However, there is still some uncertainty regarding the phylogenetic relationships within this species complex, thus we will refer to Rhipicephalus sanguineus s.l. throughout the study. Rhipicephalus sanguineus s.l., the dominant tick species on domestic dogs, is commonly found around human dwellings and can parasitize humans, as well as most vertebrate hosts6. This tick transmits Ehrlichia canis7, and several potentially zoonotic pathogens, including Babesia canis, Rickettsia conorii8, Rickettsia rickettsii9, and Rickettsia massiliae10. While R. sanguineus s.l. may transmit Anaplasma platys, which has been implicated in zoonotic infections, its vector competence for A. platys is still under investigation11. Furthermore, R. sanguineus s.l. is known to harbour a specific Coxiella endosymbiont (CE), which can influence tick fitness and bacterial acquisition or transmission12,13. Thus, it is possible that R. sanguineus s.l. can significantly contribute to the spread of zoonotic diseases in rural communities.
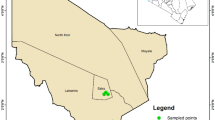
Hluvukani, a rural village situated within the eastern part of Bushbuckridge Local Municipality, Mpumalanga, South Africa, is at a wildlife-livestock-human interface, making it susceptible to tick infestations. Surrounded by conservation areas, community members rely on subsistence farming and most people own livestock14, which are kept in close proximity, often in kraals near their homes. Various animals, such as cows, goats, pigs, and chickens, are common, and domestic dogs roam freely, often assisting with hunting and herding. These factors, combined with the proximity to wildlife on neighbouring conservation areas, contribute to frequent contact between the community, their animals, and ticks. This creates an ideal situation for the spread of TBPs between wildlife, livestock and humans. Historically, research in this area has focused on the impact of tick-borne pathogens (TBPs) on livestock, with less attention on human health15.
The study analysed temporal changes in the bacterial microbiome of the midgut and salivary gland tissues of R. sanguineus s.l. ticks and assessed its potential role in human and animal health in this rural community. A Pacific Biosciences (PacBio) circular consensus sequencing (CCS) approach was used to sequence the 16 S rRNA gene and an amplicon sequence variant (ASV) pipeline was used for the analysis of the microbiome data.
Materials and methods
Ethical approvals
The following committees approved this project: The Faculty of Veterinary Science, University of Pretoria (UP) Research Ethics Committee (REC010-18), the UP Animal Ethics Committee (V012-18 and V064-16), and the UP Faculty of Humanities Research Ethics Committee (GW20180719HS). Permission was obtained to conduct the research, in terms of Section 20 of the Animal Diseases Act of 1984, from the Department of Agriculture, Land Reform and Rural Development (DALRRD) South Africa, with reference numbers 12/11/1/1/8 and 12/11/1/1. All experiments in this manuscript were performed in accordance with the relevant guidelines and regulations. This study is reported in accordance with ARRIVE guidelines.
Sample collection and genomic DNA extraction
The study aimed to understand how the bacterial microbiome of R. sanguineus s.l. impacted human health in a rural community. Ticks were collected from dogs from areas surrounding Hluvukani (24.644397° S, 31.347644° E), a village in the eastern part of the Bushbuckridge local Municipality, Mpumalanga, South Africa, in 2016, 2017, 2018 and 2019. Dogs were sampled at random, with inclusion based on factors like the weight, health, and dog hostility as well as owner presence. Dogs weighing less than 500 g (or younger than 6 weeks) or showing clinical symptoms of disease or injury were not included, to avoid any additional stress during sampling, as indicated by the ethics committees. If multiple dogs were present at a household, all were examined for ticks. As a pilot study, a total of ten dogs were sampled in 2016 and a total of ten dogs were sampled in 2017. From 2018 to 2019, a total of 73 dogs were sampled. As sampling was random, and dogs within this community are usually able to roam freely, it is plausible that the same dog was sampled more than once. Ticks were collected using blunt forceps, focusing on male R. sanguineus s.l. ticks due to the degeneration of certain organs in engorged females16. Collected ticks were stored in a temperature and humidity-controlled chamber for 2 days to allow for the blood meal to be cleared from the midgut to prevent false-positive detection of tick infection3,17,18,19 at the Hans Hoheisen Research Centre. Ticks were morphologically identified using a tick identification key20 and surface sterilized using a triple tick rinse21.
Ten male R. sanguineus s.l. ticks per dog were dissected using surface sterilized tweezers and Hyde single-edged razor blades (Hyde, Massachusetts, United States)21. The ten SGs comprised one pool, while the ten MGs comprised a second pool. Each pool was placed into a storage solution with Cell Lysis Solution (Qiagen, Valencia, CA) and proteinase K (1.25 mg/mL) (Thermo Fisher Scientific, Massachusetts, USA), and genomic DNA was extracted using the PureGene Extraction kit (Qiagen) according to the manufacturer’s specifications. It was not always possible to identify ten R. sanguineus s.l. ticks from each dog, and SG and MG pools from fewer ticks did not yield sufficient good quality DNA.
PCR and sequencing
The 16S rRNA gene (V1-V8) was amplified in triplicate, using modified universal barcoded 16S rRNA primers; 27F (5’-AGA GTT TGA TCM TGG CTC AGA ACG-3’) and 1435R (5’-CGA TTA CTA GCG ATT CCR RCT TCA-3’)3, targeting an ~ 1300 bp region. In 2016–2017 amplification was performed with Platinum Pfx, following the protocol outlined by Gall3. In 2019, due to the discontinuation of Platinum Pfx, Phusion Flash High-Fidelity PCR Master Mix (Thermo Fisher Scientific) was used, following the manufacturer’s protocol. Cycling conditions were: initial denaturation at 98 °C for 30 s, 40 cycles of 98 °C for 10 s, 60 °C for 30 s, and 72 °C for 30 s, with a final extension at 72 °C for 10 min. Triplicate PCR products were pooled, and DNA concentration was measured with a bioanalyzer (Agilent, Santa Clara, CA, USA). Pooled PCR products were submitted to the Washington State University’s Sequencing Core where the samples were pooled in equimolar amounts and CCS was conducted on the PacBio Sequel system (Pacific BioSciences, Menlo Park, CA). The raw microbiome datasets were deposited in the National Centre for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under bioproject accession number PRJNA1176486.
Amplicon sequence variant analysis
The Dada2 PacBio pipeline, created specifically for long sequence reads was used for quality filtering and processing of the raw sequence data22. The analysis was implemented in R23 and R studio24, using the following R packages: Dada2 package25 (version 1.30.0), Biostrings package26 (version 2.70.1), ShortRead package27 (version 1.60.0), and Reshape2 package28 (version 1.44.4).
Raw sequencing reads from different runs were analysed individually on the Dada2 pipeline. Quality filtering removed low-quality sequences or reads with high expected errors, retaining sequences between 1250 bp and 1600 bp. Sequences were dereplicated, run through an error model, denoised and chimeras removed, to produce ASVs. Taxonomic and species assignments were made using the RefSeq + RDP taxonomic training dataset formatted for Dada229. The RefSeq + RDP dataset incorporates the RefSeq database from NCBI and supplements with the RDP dataset, to provide high taxonomic resolution. The output was further analysed using the R phyloseq package (version 1.46.0).
To identify possible contaminants, a database was generated using information from previous studies30,31,32,33,34,35,36 (Supplementary file 1: Table 1). The obtained ASVs were filtered to remove contaminant taxa and those not identified at the genus level. A rarefaction curve was performed using the iNEXT package37 (version 3.0.1), which was designed to perform rarefaction curves without needing to down-sample (rarefy) the data. The final ASV data was visualised on the R ggplot2 package38 (version 3.4.4) and R gridExtra package39 (version 2.3), to create a 100% stacked bar plot containing the abundance of each ASV per sample.
For ASVs not identified to the species level, a nucleotide NCBI BLAST search using blastn40 was performed to identify the closest match in GenBank. The association between the number of reads of Anaplasma and Coxiella in all samples was assessed using both the Kendall Correlation and Spearman Correlation tests using the R stats package (version 4.3.2). A comparison between the microbiome profiles of corresponding MG and SG tissues from the same host (where available) was performed and visualised with a heatmap using the R package pheatmap (version 1.0.12).
Phylogenetic analysis
ASVs from organisms of interest (i.e. Coxiella, Anaplasma, Ehrlichia, Rickettsia-like and Borrelia-like) were aligned with homologous sequences from GenBank using QIAGEN CLC Main Workbench 24.0 (QIAGEN, Aarhus, Denmark). Information for all 16 S rRNA gene sequences retrieved from GenBank is shown in Supplementary Tables 2–6. The 16 S sequences reported in this study were deposited in GenBank under the following accession numbers: Wolbachia-like endosymbiont: PQ508350; Ehrlichia canis: PQ508351; Borrelia-like: PQ508352; Rickettsiales: PQ508353; Coxiella endosymbiont: PQ508354, PQ508355; Anaplasma centrale: PQ508356, PQ508357, PQ508358, PQ508359, PQ508360, PQ508366, PQ508362, PQ508363, PQ508364, PQ508365; Anaplasma platys: PQ508361. Alignments were truncated as follows: Coxiella (1124 bp, 25 sequences), Anaplasma (1243 bp, 37 sequences), Ehrlichia (1251 bp, 21 sequences), Rickettsia (1251 bp, 24 sequences), and Borrelia (1297 bp, 23 sequences). Statistical selection of the best-fit model was performed using IQTREE ModelFinder41. Maximum likelihood (ML) analysis was performed on the alignments using the IQTREE web server42 with ultrafast bootstrap replicates (UF). The models used were: Coxiella – K2P + I + G4 (1000 UF), Anaplasma - TPM2u + F + I + G4 (3000 UF), Ehrlichia - TIM3 + F + I + G4 (1000 UF), Rickettsia - GTR + F + I + G4 (1000 UF) and Borrelia - TIM3 + F + I + G4 (1000 UF). The ML output was visualised using ITOL: Interactive Tree of Life (version 6.8.1)43,44 with ML bootstrap values above 75 shown on the tree, and values above 95 considered good support.
Results
Sample collection
The following tick genera were observed co-feeding on the dogs: Rhipicephalus (R. sanguineus s.l., R. simus, R. turanicus, R. microplus), Amblyomma (A. hebraeum), Haemaphysalis (H. leachi) and Hyalomma (Hy. truncatum). Tick species prevalence was not analysed, however, R. sanguineus s.l. was the most predominant tick observed on the dogs in all three years. All dog samples from 2018 and 17 dog samples from 2019 were excluded from further analysis, as they contained too few ticks to yield high quality DNA. R. sanguineus s.l. ticks from ten dogs in 2016 and ten dogs in 2017 were used for further analysis, while in 2019, R. sanguineus s.l. ticks from six dogs were used for further analysis.
Sequence analysis
A total of 20 tick pools (10 MG and 10 SG) were processed in 2016, 20 tick pools (10 MG and 10 SG) were processed in 2017, while 12 tick pools (six MG and six SG) were processed in 2019. A subset of samples failed amplification in 2016 (three MG samples and two SG samples), 2017 (six MG samples and five SG samples) and 2019 (one MG sample and one SG sample). These samples were subsequently removed from further analysis. A rarefaction curve analysis indicated that the sequencing effort was sufficient for most samples to capture the microbial diversity present in the samples as evidenced by the plateauing of the curves (Supplementary file 1: Fig. 1). This suggests that additional sequencing would likely yield few new species.
Samples from 2016 contained 38,241 raw sequence reads (Supplementary file 1: Table 7). After quality control, 58 ASVs were detected from 17,027 sequence reads, assigned to nine genera. After filtering and removing contaminants and two SG samples with low read counts, the microbiome profile included one Coxiella ASV (98.23%) found in all samples, and seven Escherichia/Shigella ASVs (1.77%), in six samples (Fig. 1A). Samples from 2017 contained 15,642 raw sequence reads (Supplementary file 1: Table 7). After quality control, 39 ASVs were detected from 8,556 sequence reads, assigned to 15 genera. After filtering and removing contaminants, the microbiome profile consisted of one Coxiella ASV (98.17%) in all samples, five Escherichia/Shigella ASVs (1.16%) in three samples, and one Anaplasma ASV (0.67%) in two samples (Fig. 1B). In 2019 samples contained 80,695 raw sequence reads (Supplementary file 1: Table 7). After quality control, 379 ASVs detected from 54,583 sequence reads, were assigned to 106 genera. After filtering, removing contaminants and two MG and one SG low read samples, the microbiome profile consisted of two Coxiella ASVs (54.21%) from all samples, five Escherichia/Shigella ASVs (0.63%) in four samples, 11 Anaplasma ASVs (27.99%) in all samples, two Ehrlichia ASVs (17.06%) in three samples, one Borrelia-like ASV (0.07%) in one sample and one Rickettsia-like ASV (0.04%) in one sample (Fig. 1C).

Prevalence and diversity of the bacterial genera of midgut and salivary gland pools of Rhipicephalus sanguineus s.l. ticks sampled from domestic dogs in the study site. (A) 2016, (B) 2017 and (C) 2019. Sample identification codes are indicated on the x-axis (M-Midgut pool, S-salivary gland pool, Y1-2016, Y2-2017, Y3-2019).
Species-level assignment indicated that Coxiella ASV 1, detected in all three years, was identical to the sequence of the 16 S rRNA gene of a known Coxiella-like endosymbiont (CLE) (MZ836861) identified from an R. sanguineus s.l. tick. Coxiella ASV 2, detected in 2019, was not identified to the species level but had 99.92% identity to a CE from an R. microplus tick (CP094229.1). Escherichia/Shigella ASVs indicated that all ASVs were similar to 16 S rRNA gene sequences from various known Escherichia coli strains (MT647245.1, CP053607.1, CP142044.1)45,46. Anaplasma ASVs were divided into two groups. Group one (Anaplasma ASV 11), detected in two samples from 2017 and one sample from 2019, was identical to a known A. platys sequence (CP046391.1), isolated from a dog47. Group two (Anaplasma ASV 1 to Anaplasma ASV 10) detected in 2019 matched A. centrale. Anaplasma ASV 1 was identical to the 16 S rRNA gene sequence of the Israel strain of A. centrale (CP001759.1)48. Anaplasma ASV 2 to Anaplasma ASV 10 were not assigned to species level. A BLAST search indicated that these ASVs matched A. centrale (CP001759.1) with an average of 99.68% identity.
Ehrlichia ASV 1 could not be assigned to species level using Dada2, however, a BLAST search identified it as identical to a known Wolbachia endosymbiont (MN383047.1) from mosquitoes. Ehrlichia ASV 1 will hereafter be referred to as Wolbachia ASV1. Ehrlichia ASV 2 was identical to E. canis (MK507008.1) isolated from a dog and an uncultured Ehrlichia (JN121380). Borrelia-like ASV 1 was not assigned to species level; the highest BLAST match (98.81% identity) was to an uncultured bacterium (EU137037.1)49, with 58% query coverage, and it also matched “Candidatus Borreliella tachyglossi” (CP025785.1) with 85.69% identity and 100% query coverage. Rickettsia-like ASV 1 was not assigned to species level, however a BLAST search indicated that the highest match (91.24% identity), was to an uncultured Rickettsiales bacteria (MK616428.1) from a nucleariid amoeba (Pompholyxophrys punicea) sampled from a lake in Zwönitz, Germany50.
Kendall Correlation and Spearman Correlation tests found a moderate positive association between the number of Anaplasma sequence reads and Coxiella sequence reads. This was, however, not statistically significant (Kendall: R = 0.24, p = 0.36. Spearman: R = 0.3, p = 0.37. (Supplementary file 1: Fig. 2). A comparison between the microbiome profiles of the tissue types (MG and SG) from the same host was visualised on a heatmap (Supplementary file 1: Fig. 3) and no difference could be seen between the tissue types. Due to the small sample size, robust statistical testing could not be performed.
Phylogenetic analysis
A ML analysis of the Coxiella ASVs (Fig. 2) indicated that Coxiella ASVs clustered in a monophyletic group with known CE sequences from Rhipicephalus ticks, but with low bootstrap support. This monophyletic group split into two subgroups with moderate bootstrap support (92). One subgroup contained Coxiella ASV 1 and sequences from known CEs isolated from R. sanguineus s.l. (CP024961, KU892220, MZ836861 and KP994843)51,52,53 and “Candidatus Coxiella mudrowiae” from R. turanicus (CP011126)54. The second subgroup contained Coxiella ASV 2 and sequences from known CEs isolated from R. decoloratus (KP994833), R. microplus (KP994839) and R. evertsi (KP994835)51. Within this group, Coxiella ASV 2 and the CE from R. microplus clustered together (99.92% identity), with good bootstrap support (100).
A ML tree (Fig. 3) of the 11 Anaplasma ASVs, indicated that Anaplasma ASV 1 to ASV 10 formed a monophyletic group with two previously characterised A. centrale sequences (A. centrale Uganda: KU686784 and A. centrale Israel: CP001759)48, with bootstrap support of 92. Anaplasma ASV 11 formed a monophyletic group with two known A. platys sequences: A. platys S3 (CP046391)47 and A. platys Apla1 from a dog in the study community (MK814419)55, with high bootstrap support of 99.

Maximum likelihood analysis indicating the relationships of Coxiella amplicon sequence variants detected in this study with known Coxiella sequences from GenBank. A maximum likelihood inference was conducted using the K2P + I + G4 model and 1000 ultrafast bootstrap replicates (alignment length 1124 bp). Maximum likelihood bootstrap values (> 75) are indicated below the branch. Coxiella amplicon sequence variants are indicated in bold. The Coxiella sequences associated with Rhipicephalus sanguineus s.l. and Rhipicephalus turanicus are indicated in a blue box, while the Coxiella sequences associated with Rhipicephalus microplus are indicated in a green box. Rickettsia rickettsii (U11021) was used as an outgroup.

Maximum likelihood tree indicating the relationships of the Anaplasma amplicon sequence variants detected in this study, with known Anaplasma sequences from GenBank. A maximum likelihood inference was conducted using the TPM2u + F + I + G4 model and 3000 ultrafast bootstrap replicates (alignment length 1243 bp). Maximum likelihood bootstrap values (> 75) are indicated below the branch. Anaplasma amplicon sequence variants are indicated in bold. The Anaplasma centrale group is indicated in a blue box, while the Anaplasma platys group is indicated in a green box. Ehrlichia ruminantium strain Welgevonden (NR074513) was used as an outgroup.
Wolbachia ASV 1 grouped with known Wolbachia endosymbionts (MN383047 and KX155506)56 and an uncultured bacterium (JX669531) with bootstrap support of 100. It was most closely related to a Wolbachia endosymbiont from Aedes aegypti (MN383047) (Supplementary file 1: Fig. 4). Ehrlichia ASV 2 grouped with known E. canis and uncultured Ehrlichia sequences (MK507008, NR118741 and JN121380)57,58 with bootstrap support of 100. ML phylogenetic analysis indicated that Rickettsia-like ASV 1 was ancestral to known Rickettsia, Orientia and Occidentia sequences and clustered together with uncultured bacterial sequences, but with low bootstrap support (Supplementary file 1: Fig. 5). Borrelia-like ASV 1 did not group with known Borrelia sequences or closely related spirochetes (Supplementary file 1: Fig. 6). Instead, it branched off from the Cristispira genus, with good bootstrap support of 96.
Discussion
This study showed that the bacterial microbiome of MG and SG tissues from R. sanguineus s.l. ticks changed over the study period, and highlighted the role that R. sanguineus s.l. ticks might play as reservoirs of potential pathogens.
Rhipicephalus sanguineus s.l. was the most abundant tick species observed on the dogs in all three years of our study, in agreement with previous findings59. As R. sanguineus s.l. ticks are well adapted to human dwellings and rely on dogs as their main host, infestations can become extremely high with a consequent increase in the risk of exposure to TBPs for animals and humans60. Dogs weighing less than 500 g or younger than 6 weeks (puppies) and showing signs of injury or clinical disease were not sampled, in accordance with ethical approvals. Exclusion of dogs with clinical symptoms could prevent the detection of potential canine bacterial pathogens, however exclusion of puppies should not significantly influence our findings.
In the MG and SG microbiomes of R. sanguineus s.l. ticks collected in the study, we found Coxiella, Anaplasma, Ehrlichia¸ Escherichia, Rickettsia-like and Borrelia-like 16 S rRNA gene sequences. The microbiomes were initially dominated by a CE in 2016 and 2017, consistent with other studies of R. sanguineus s.l. tick microbiomes from northern Spain, France, Senegal, Arizona and Oklahoma61,62,63. In 2017, A. platys was introduced into the microbiome. By 2019, Anaplasma species had increased, with the introduction of large numbers of A. centrale sequences. Other pathogens (Ehrlichia, Rickettsia-like and Borrelia-like) emerged in 2019, but at very low levels. Our findings align with Portillo et al.62, who also found low levels of Rickettsia, Borrelia, Ehrlichia and Wolbachia in R. sanguineus s.l. ticks. The difference in the microbiome between 2017 and 2019 could possibly be attributed to climatic differences with a long drought ending in 2019.
The low humidity conditions during a period of drought are unfavourable for tick populations64. Therefore, we surmise that the drought led to a decrease in the overall tick population and thus to decreased transmission of bacterial organisms within the community. Once the drought ended, conditions were favourable for tick population expansions allowing for increased transmission of bacterial organisms and changes in the bacterial microbiome.
Another potential explanation for the difference in the microbiome profile could be the change in reagents between 2017 and 2019. PCR reagents have varying levels of amplification efficiency and accuracy which might result in differential amplification of the bacteria present in the samples. The dog host could also potentially influence the changes seen in the microbiome profiles between the sampling years. Dogs were sampled randomly during each sampling excursion, and given that dogs can roam freely within the community, identification of individual dogs was not possible. This introduces variability in the host-related factors, such as diet and environmental exposures, which could all impact the microbiome of Rhipicephalus sanguineus s.l.
This study identified a sparse microbiome profile, with an average of 3.3 species in the MG samples and 3 species in the SG samples. These findings align with previous research65, which demonstrated that MG and SG samples exhibit lower alpha diversity compared to whole tick samples. This is likely because MG and SG contain tissue-specific microbiomes, whereas whole tick samples can include the MG, SG, Malpighian tubes, haemolymph, heart, tracheae, rectal sac, synganglion and other tissues, leading to a greater variety or total number of bacterial species. Additionally, MG and SG samples are characterized by low biomass, containing a smaller proportion of total bacterial DNA, making them more susceptible to the over amplification of environmental contaminant DNA. In 2019, we detected many potentially contaminating sequences. Given the sensitivity of next-generation sequencing, microbiome studies are prone to external and cross-contamination66, warranting the need for negative controls, however this was not part of our standard protocol when this study began. A list of bacterial genera identified as possible contaminants in microbiome studies were removed from the dataset (Supplementary file 1: Table 1). This list included Bacillus, which was previously detected in the R. sanguineus s.l. tick microbiome61, but because it has also been found in laboratory reagents, it was removed as a possible contaminant. This allowed for the 2019 dataset to be compared in a more meaningful way to the 2016 and 2017 dataset.
In this study, CE sequences were detected in every MG and SG sample over three years. Historically, the genus Coxiella included only Coxiella burnetii, the agent of Q-fever, which is endemic in South Africa, with a prevalence of up to 59% in vulnerable communities67. More recently, various CEs have been discovered, mainly in ticks, but also in the spleens of some wild mammals68. CEs have evolved with their tick hosts, causing different CEs to appear to be specific for different tick species53. These CEs benefit ticks by enhancing immunity and nutrition12, and can affect pathogen acquisition and transmission13. Molecular methods for detection of C. burnetii may cross-react with CEs, potentially overestimating C. burnetii infection rates69. As R. sanguineus s.l. can bite humans, CE transmission to humans might contribute to Q-fever positive results in South Africa, while not manifesting disease. Two Coxiella ASVs were present in the 2019 datasets: ASV 1, identical to the known CE from R. sanguineus s.l. ticks, and ASV 2, closely related (99.92% identity) to a CE from R. microplus ticks. The presence of Coxiella ASV 2 could be due to co-feeding of different tick species at the same site on the dog host, or potentially an R. microplus tick could have been erroneously included in one of our 2019 pools. To rule this out, we performed molecular typing on the 2019 ticks, which confirmed they were R. sanguineus s.l. (data not shown).
Anaplasma sequences were detected only in 2017 and 2019. Analysis revealed two species: A. platys and A. centrale. In 2017, A. platys was found in two samples, while in 2019, A. platys was found in a single sample, and A. centrale was detected in all nine samples. Anaplasma platys causes canine thrombocytopenia in dogs70, and has been reported in humans71,72. Arraga-Alvarado et al.72 provided the first clinical evidence of A. platys infection in a human patient from Venezuela who exhibited fever symptoms as well as headache and thrombocytopenia. Though studies on the vector competence of R. sanguineus s.l. for A. platys are limited, R. sanguineus s.l. is thought to transmit A. platys, since their geographical distributions overlap, and dogs are the primary hosts for both R. sanguineus s. l and A. platys73. Although Simpson, et al.11 found no detectable A. platys in R. sanguineus s.l. fed on laboratory-infected dogs, Snellgrove et al.74 showed that R. sanguineus s.l. could maintain A. platys through transovarial, transstadial, and horizontal transmission, under laboratory conditions. Detection of A. platys in one MG and two SG samples of R. sanguineus s.l. in our study highlights the possible role of R. sanguineus s.l. as a vector.
An unexpected finding was the detection of A. centrale in 2019, as dogs are not known hosts, nor is R. sanguineus s.l. a known vector. Anaplasma centrale has been shown to be transmitted by Rhipicephalus simus and Dermacentor andersoni75,76,77 and causes a less virulent form of bovine anaplasmosis78. A previous study identified A. centrale in the SG of R. sanguineus s.l. ticks but did not demonstrate transmission to calves77.
The introduction of A. platys and A. centrale into the R. sanguineus s.l. microbiome in 2017 and 2019, respectively, could be from wildlife or cattle. While there is little information regarding the role of wildlife in the epidemiology of Anaplasma in Africa, A. platys and A. centrale have been documented in African buffalo79,80,81,82, and A. centrale has also been documented in black and blue wildebeest, eland, waterbuck, zebra, warthog, and lion78,82. The introduction of these organisms into adult R. sanguineus s.l. ticks could occur through transstadial transmission from immature ticks feeding on smaller wildlife hosts. This emphasizes the significance of R. sanguineus s.l. at the wildlife-livestock-human interface. Another source could be the movement of cattle and dogs within the community. During sampling, we observed livestock being moved between residences, grazing areas, and dip tanks, with dogs often accompanying them as herding dogs. This activity could facilitate the exchange of ticks and thus TBPs among domestic animals throughout the community.
The introduction of Anaplasma coincided with a moderate increase in Coxiella, however a Spearman and Kendall Rank correlation indicated that this correlation was not statistically significant. These preliminary findings warrant further study into the correlation between these bacteria in R. sanguineus s.l. It is also important to mention that while the proportion of a given bacterium must change with the introduction of additional species, 16 S microbiome sequencing does not indicate whether the absolute numbers of the bacterium have changed significantly.
This study also detected Escherichia/Shigella sequences in various samples across all three years. Escherichia/Shigella has been detected in several bacterial tick microbiome studies83,84,85. All variants of Escherichia/Shigella were similar to known E. coli strains, however there is currently no evidence to suggest that E. coli is transmitted by R. sanguineus s.l.
In two 2019 samples, we detected Ehrlichia ASVs. One sequence present at low read numbers (Ehrlichia ASV 2) was classified as E. canis, the etiological agent of canine monocytic ehrlichiosis. Rhipicephalus sanguineus s.l. is a known vector of E. canis, and E. canis has been previously detected in R. sanguineus s.l. ticks as well as in dogs in the community86,87. While E. canis has not been documented in humans, it does infect wild canids88.
During the ASV-based taxonomy assignment, a second “Ehrlichia variant” (Ehrlichia ASV 1) was detected. However, BLAST search and phylogenetic analysis revealed it to be identical to a Wolbachia endosymbiont from a mosquito. The ASV-based taxonomic assignment utilizes an offline database that is not as robust and complete as an online method such as BLAST. No Wolbachia sequences were present in the offline database and thus identification was made as Ehrlichia, a very closely related genus.
While ticks harbour endosymbionts like Coxiella, little is known regarding Wolbachia in ticks. One study found Wolbachia in Ixodes ticks due to parasitism by Ixodiphagus hookeri (a tick wasp) harbouring a Wolbachia endosymbiont89. In our study we saw high Wolbachia read numbers in one MG sample with lower numbers in the corresponding SG sample, and a second positive MG sample, which suggests that this was not an accidental finding. Further investigation is needed to determine the origin of this Wolbachia variant.
In 2019, we detected very low read counts of a Borrelia-like sequence from a single MG sample. Neither the ASV method nor a manual NCBI BLAST search could identify the sequence to species level, and phylogenetic analysis also failed to determine its relationship to known Borrelia sequences. This sequence might belong to an unclassified genus within the Borreliaceae family (closest match is 85.69% identity to “Candidatus Borreliella tachyglossi”), or it could be a chimeric sequence, as each half of the sequence has a higher match (> 94%) to different uncultured bacteria. We detected the Borrelia-like sequence in a single midgut sample, which is in alignment with previous research which found that Borrelia species are “stored” in the midgut until a second blood meal is taken, upon which they move into the salivary glands90.
A Rickettsia-like sequence was detected in a single SG sample from 2019 at very low read counts. Further investigation could not identify the sequence to species level, using either the ASV method or a manual BLAST search. The highest match was to an uncultured Rickettsiales bacterium, an endosymbiont of the amoeba, Pompholyxophrys punicea50. Phylogenetic analysis indicated that it clustered with 16 S rRNA gene sequences from various uncultured Rickettsiales, some from amoebas and others from environmental samples. Further investigation is needed to determine the occurrence and importance of this Rickettsia-like organism.
This study had several limitations that should be taken into consideration. Firstly, no negative “blank” controls were included during sample preparation and sequencing, making it difficult to identify potential contaminants. Secondly, during the study certain PCR reagents became unavailable, which led to protocol alterations between years, potentially affecting the microbiome profile. Thirdly, the study had a small sample size due to the difficulty in collecting enough adult male R. sanguineus s.l. ticks from the community dogs. Lastly, samples were analysed from non-consecutive years (samples from 2018 did not yield high quality DNA), creating a data gap between 2017 and 2019, which may have resulted in valuable information on the introduction of Anaplasma not being analysed. Advantages of our study include the use of dissected organs (allows for a cleaner look at the actual tick microbiome rather than environmental species that cling to the surface of the tick), use of pools (which provide a better grasp of the overall population, and reduces the variation seen when analysing individual ticks) and our longitudinal study demonstrates how the tick microbiome varies over time.
Conclusion
Our study revealed the bacterial microbiome diversity of R. sanguineus s.l. MG and SG tissues from dogs from a rural community in Mpumalanga, South Africa. We identified R. sanguineus s.l. ticks as potential reservoirs for pathogens like A. platys and A. centrale, though further research is needed to confirm their reservoir role and vector competence. Our findings also suggest that R. sanguineus s.l. from the community hosts a species-specific CE, corroborating earlier findings in R. sanguineus s.l. ticks51. We observed a potential correlation between Coxiella and Anaplasma, warranting further investigation. Additionally, while other pathogenic bacteria were detected, their impact remain unclear. Overall, our research highlights the role of R. sanguineus s.l. in influencing pathogen diversity and distribution at the wildlife-livestock-human interface in rural South Africa.
Data availability
The raw microbiome datasets generated during the current study are available in the NCBI SRA (PRJNA1176486, https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA1176486). 16 S sequences reported in this study are available on GenBank under accession numbers: Wolbachia-like endosymbiont: PQ508350. Ehrlichia canis: PQ508351. Borrelia-like: PQ508352. Rickettsiales: PQ508353. Coxiella endosymbiont: PQ508354, PQ508355. Anaplasma centrale: PQ508356, PQ508357, PQ508358, PQ508359, PQ508360, PQ508362, PQ508363, PQ508364, PQ508365, PQ508366. Anaplasma platys: PQ508361.
Change history
17 June 2025
The original online version of this Article was revised: The Acknowledgments section in the original version of this Article was incomplete. “We acknowledge Luis Neves for verifying the tick identification. We also thank the staff of the Hans Hoheisen Wildlife Research Station for logistical support, environmental monitors, Handry Mathebula and Julia D. Sithole, for assistance in the Bushbuckridge-East community, and the tribal council for allowing us to work in their community. Appreciation is extended to Derek Pouchnik and Mark Wildung from the Genomics Sequencing Core at Washington State University for assistance with microbiome sequencing.” It now reads: "We acknowledge Luis Neves for verifying the tick identifications. We thank the staff of the Hans Hoheisen Research Centre for logistical support, environmental monitors, Handry Mathebula and Julia D. Sithole, for assistance in the Bushbuckridge-East community, and the tribal council for allowing us to work in their community. Appreciation is extended to Derek Pouchnik and Mark Wildung from the Genomics Sequencing Core at Washington State University for assistance with microbiome sequencing.
We thank the South African National Research Foundation (NRF) for grants 92739 and 110448, and 109350, NIH NIAID R01AI136832, and the Belgian Directorate General for Development Cooperation Framework FA4 (ITM-DGCD) awarded to Marinda Oosthuizen, as well as the Agricultural Sector Education Training Authority (AgriSETA; BU18UP124-18.2) Scholarship and the Meat Industry Trust for the bursary awarded to Rebecca E. Ackermann. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding bodies." The original Article has been corrected.
References
Chitanga, S., Gaff, H. & Mukaratirwa, S. Tick-borne pathogens of potential zoonotic importance in the Southern African region. J. S Afr. Vet. Assoc. 85, 1084. https://doi.org/10.4102/jsava.v85i1.1084 (2014).
de la Fuente, J. et al. Ticks as vectors of pathogens that cause disease in humans and animals. Front. Biosci. 13, 6938–6946. https://doi.org/10.2741/3200 (2008).
Gall, C. A. Functional Characterization of the Bacterial Microbiome of the Dermacentor andersoni Tick Exhibits Interactions with Pathogen Acquisition Doctor of philosophy thesis, Washington State Unviersity (2016).
Bonnet, S. I., Binetruy, F., Hernandez-Jarguin, A. M. & Duron, O. The tick microbiome: why non-pathogenic microorganisms matter in tick biology and pathogen transmission. Front. Cell. Infect. Microbiol. 7, 236. https://doi.org/10.3389/fcimb.2017.00236 (2017).
Chitimia-Dobler, L. et al. Genetic analysis of Rhipicephalus sanguineus sensu Lato ticks parasites of dogs in Africa North of the Sahara based on mitochondrial DNA sequences. Vet. Parasitol. 239, 1–6. https://doi.org/10.1016/j.vetpar.2017.04.012 (2017).
Gray, J., Dantas-Torres, F., Estrada-Pena, A. & Levin, M. Systematics and ecology of the brown dog tick, Rhipicephalus sanguineus. Ticks Tick. Borne Dis. 4, 171–180. https://doi.org/10.1016/j.ttbdis.2012.12.003 (2013).
Socolovschi, C. et al. Ehrlichia canis in Rhipicephalus sanguineus ticks in the Ivory Coast. Ticks Tick. Borne Dis. 3, 411–413. https://doi.org/10.1016/j.ttbdis.2012.10.005 (2012).
Dantas-Torres, F. & Otranto, D. Further thoughts on the taxonomy and vector role of Rhipicephalus sanguineus group ticks. Vet. Parasitol. 208, 9–13. https://doi.org/10.1016/j.vetpar.2014.12.014 (2015).
Byrd, R. P. Jr, Vasquez, J. & Roy, T. M. Respiratory manifestations of tick-borne diseases in the southeastern united States. South. Med. J. 90, 1–4. https://doi.org/10.1097/00007611-199701000-00001 (1997).
Garcia-Garcia, J. C. et al. Case report: A patient from Argentina infected with Rickettsia massiliae. Am. J. Trop. Med. Hyg. 82, 691–692. https://doi.org/10.4269/ajtmh.2010.09-0662 (2010).
Simpson, R. M. et al. Evaluation of Rhipicephalus sanguineus as a potential biologic vector of Ehrlichia platys. Am. J. Vet. Res. 52 (1991).
Kolo, A. O. & Raghavan, R. Impact of endosymbionts on tick physiology and fitness. Parasitology 150, 859–865. https://doi.org/10.1017/S0031182023000793 (2023).
Li, L. H., Zhang, Y., Zhu, D. & Zhou, X. N. Endosymbionts alter larva-to-nymph transstadial transmission of Babesia microti in Rhipicephalus haemaphysaloides ticks. Front. Microbiol. 9, 1415. https://doi.org/10.3389/fmicb.2018.01415 (2018).
Berrian, A. M. et al. One health profile of a community at the wildlife-domestic animal interface, Mpumalanga, South Africa. Prev. Vet. Med. 130, 119–128. https://doi.org/10.1016/j.prevetmed.2016.06.007 (2016).
Hotez, P. J. & Kamath, A. Neglected tropical diseases in sub-saharan Africa: Review of their prevalence, distribution, and disease burden. PLoS Negl. Trop. Dis. 3, e412. https://doi.org/10.1371/journal.pntd.0000412 (2009).
Wang, H. et al. Comprehensive analysis of the global protein changes that occur during salivary gland degeneration in female Ixodid ticks Haemaphysalis longicornis. Front. Physiol. 9, 1943. https://doi.org/10.3389/fphys.2018.01943 (2019).
Kocan, K. M. et al. Development of Anaplasma marginale in salivary glands of male Dermacentor andersoni. Am. J. Vet. Res. 54, 107–112 (1993).
De la Fuente, J. et al. Phylogeography of new world isolates of Anaplasma marginale based on major surface protein sequences. Vet. Microbiol. 88, 275–285. https://doi.org/10.1016/S0378-1135(02)00122-0 (2002). https://doi.org:.
Futse, J., Ueti, M. W., Knowles, D. P. & Palmer, G. H. Transmission of Anaplasma marginale by Boophilus microplus: retention of vector competence in the absence of vector-pathogen interaction. J. Clin. Microbiol. 41, 3829–3834. https://doi.org/10.1128/JCM.41.8.3829-3834.2003 (2003).
Walker, A. R. et al. Ticks of Domestic Animals in Africa: A Guide To Identification of Species (Bioscience Reports, 2003).
Scoles, G. A., Ueti, M. W. & Palmer, G. H. Variation among geographically separated populations of Dermacentor andersoni (Acari Ixodidae) in midgut susceptibility to Anaplasma marginale (Rickettsiales: Anaplasmataceae). J. Med. Entomol. 42, 153Ð162. https://doi.org/10.1093/jmedent/42.2.153 (2005).
Callahan, B. J. et al. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res. 47, e103. https://doi.org/10.1093/nar/gkz569 (2019).
R. A Language and Environment for Statistical Computing (R Foundation for Statistical Computing, 2023).
RStudio Integrated Development Environment for R (Posit Software, 2024).
Callahan, B. J. et al. DADA2: High-resolution sample inference from illumina amplicon data. Nat. Methods. 13, 581–583. https://doi.org/10.1038/nmeth.3869 (2016).
Biostrings Efficient manipulation of biological strings (2023).
Morgan, M. et al. ShortRead: A bioconductor package for input, quality assessment and exploration of high-throughput sequence data. Bioinformatics 25, 2607–2608. https://doi.org/10.1093/bioinformatics/btp450 (2009).
Wickham, H. Reshaping data with the reshape package. J. Stat. Softw. 21 https://doi.org/10.18637/jss.v021.i12 (2007).
DADA2 formatted 16S rRNA gene sequences for both bacteria & archaea. (2021).
Bedarf, J. R. et al. Much ado about nothing? Off-target amplification can lead to false-positive bacterial brain Microbiome detection in healthy and Parkinson’s disease individuals. Microbiome 9, 75. https://doi.org/10.1186/s40168-021-01012-1 (2021).
Laurence, M., Hatzis, C. & Brash, D. E. Common contaminants in next-generation sequencing that hinder discovery of low-abundance microbes. PLoS One. 9, e97876. https://doi.org/10.1371/journal.pone.0097876 (2014).
Lejal, E. et al. Taxon appearance from extraction and amplification steps demonstrates the value of multiple controls in tick microbiota analysis. Front. Microbiol. 11, 1093. https://doi.org/10.3389/fmicb.2020.01093 (2020).
Salter, S. J. et al. Reagent and laboratory contamination can critically impact sequence-based microbiome analyses. BMC Biol. 12 https://doi.org/10.1186/s12915-014-0087-z (2014).
Walker, S. P., Tangney, M. & Claesson, M. J. Sequence-based characterization of intratumoral bacteria - A guide to best practice. Front. Oncol. 10, 179. https://doi.org/10.3389/fonc.2020.00179 (2020).
Weyrich, L. S. et al. Laboratory contamination over time during low-biomass sample analysis. Mol. Ecol. Resour. 19, 982–996. https://doi.org/10.1111/1755-0998.13011 (2019).
Zhu, X., Li, X., Wang, W. & Ning, K. Bacterial contamination screening and interpretation for biological laboratory environments. Med. Microecol. 5 https://doi.org/10.1016/j.medmic.2020.100021 (2020).
iNEXT: Interpolation and extrapolation for species diversity v. R package version 3.0.1. (2024).
ggplot2: Elegant Graphics for Data Analysis. (2016).
gridExtra: Miscellaneous functions for. Grid graphics (2017).
Sayers, E. W. et al. Database resources of the National center for biotechnology information. Nucleic Acids Res. 50, D20–D26. https://doi.org/10.1093/nar/gkab1112 (2022).
Kalyaanamoorthy, S. et al. Fast model selection for accurate phylogenetic estimates. Nat. Methods. 14, 587–589. https://doi.org/10.1038/nmeth.4285 (2017).
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: A fast online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44, W232–235. https://doi.org/10.1093/nar/gkw256 (2016).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–296. https://doi.org/10.1093/nar/gkab301 (2021).
Letunic, I. & Bork, P. Interactive tree of life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics 23, 127–128. https://doi.org/10.1093/bioinformatics/btl529 (2007).
Anton, B. & Raleigh, E. A. Complete genome sequence of NEB 5-alpha, a derivative of Escherichia coli K-12 DH5α. Genome Announc. 4 https://doi.org/10.1128/genomea.01245-16 (2016).
El-Gendy, M. M. A. A. et al. Evaluation of carcinogenic activities and sperm abnormalities of gram-negative bacterial metabolites isolated from cancer patients after subcutaneous injection in albino rats. Antonie Leeuwenhoek. 114, 287–302. https://doi.org/10.1007/s10482-021-01522-w (2021).
Llanes, A. & Rajeev, S. First whole genome sequence of Anaplasma platys, an obligate intracellular rickettsial pathogen of dogs. Pathogens. 9 https://doi.org/10.3390/pathogens9040277 (2020).
Herndon, D. R., Palmer, G. H., Shkap, V., Knowles, D. P. & Brayton, K. A. Complete genome sequence of Anaplasma marginale subsp. Centrale. J. Bacteriol. 192, 379–380. https://doi.org/10.1128/jb.01330-09 (2010).
Jones, S. E., Newton, R. J. & McMahon, K. D. Potential for atmospheric deposition of bacteria to influence bacterioplankton communities. FEMS Microbiol. Ecol. 64, 388–394. https://doi.org/10.1111/j.1574-6941.2008.00476.x (2008).
Galindo, L. J. et al. Combined cultivation and single-cell approaches to the phylogenomics of Nucleariid amoebae, close relatives of fungi. Philos. Trans. R Soc. Lond. B Biol. Sci. 374, 20190094. https://doi.org/10.1098/rstb.2019.0094 (2019).
Duron, O. et al. The recent evolution of a maternally-inherited endosymbiont of ticks led to the emergence of the Q fever pathogen, Coxiella burnetii. PLoS Pathog. 11, e1004892. https://doi.org/10.1371/journal.ppat.1004892 (2015).
Oskam, C. L. et al. Molecular investigation into the presence of a Coxiella Sp. in Rhipicephalus sanguineus ticks in Australia. Vet. Microbiol. 201, 141–145. https://doi.org/10.1016/j.vetmic.2017.01.021 (2017).
Tsementzi, D., Castro Gordillo, J., Mahagna, M., Gottlieb, Y. & Konstantinidis, K. T. Comparison of closely related, uncultivated Coxiella tick endosymbiont population genomes reveals clues about the mechanisms of symbiosis. Environ. Microbiol. 20, 1751–1764. https://doi.org/10.1111/1462-2920.14104 (2018).
Gottlieb, Y., Lalzar, I. & Klasson, L. Distinctive genome reduction rates revealed by genomic analyses of two Coxiella-like endosymbionts in ticks. Genome Biol. Evol. 7, 1779–1796. https://doi.org/10.1093/gbe/evv108 (2015).
Kolo, A. O. et al. Anaplasma phagocytophilum and other Anaplasma spp. In various hosts In the Mnisi community, Mpumalanga Province, South Africa. Microorganisms. 8 https://doi.org/10.3390/microorganisms8111812 (2020).
Matsumoto, K., Dumon, I. A., Raoult, D. & Parola, P. First detection of Wolbachia spp., including a new genotype, in sand flies collected in Marseille, France. J. Med. Entomol. 45, 466–469. https://doi.org/10.1603/0022-2585(2008)45[466:fdowsi]2.0.co;2 (2008).
Anderson, B. E., Dawson, J. E., Jones, D. C. & Wilson, K. H. Ehrlichia chaffeensis, a new species associated with human ehrlichiosis. J. Clin. Microbiol. 29, 2838–2842. https://doi.org/10.1128/jcm.29.12.2838-2842.1991 (1991).
Díaz-Sánchez, A. A. et al. Molecular diagnosis, prevalence and importance of zoonotic vector-borne pathogens in Cuban shelter dogs—A preliminary study. Pathogens 9, 901–919. https://doi.org/10.3390/pathogens9110901 (2020).
Bryson, N. R., Horak, I. G., Höhn, E. W. & Louw, J. P. Ectoparasites of dogs belonging to people in resource-poor communities in North West Province, South Africa. J. S. Afr. Vet. Assoc. 71, 175–179. https://doi.org/10.4102/jsava.v71i3.709 (2000).
Horak, I. G., Fourie, L. J., Heyne, H., Walker, J. B. & Needham, G. R. Ixodid ticks feeding on humans in South Africa—with notes on preferred hosts, geographic distribution, seasonal occurrence and transmission of pathogens. Exp. Appl. Acarol. 27, 113–136. https://doi.org/10.1023/a:1021587001198 (2002).
Rene-Martellet, M. et al. Bacterial microbiota associated with Rhipicephalus sanguineus (s.l.) ticks from France, Senegal and Arizona. Parasit. Vectors. 10, 416. https://doi.org/10.1186/s13071-017-2352-9 (2017).
Portillo, A. et al. Exploring the bacteriome in anthropophilic ticks: to investigate the vectors for diagnosis. PLoS One. 14, e0213384. https://doi.org/10.1371/journal.pone.0213384 (2019).
Rounds, M. A. et al. Identification of endosymbionts in ticks by broad-range polymerase chain reaction and electrospray ionization mass spectrometry. J. Med. Entomol. 49, 843–850. https://doi.org/10.1603/me12038 (2012).
Gage, K. L., Burkot, T. R., Eisen, R. J. & Hayes, E. B. Climate and vectorborne diseases. Am. J. Prev. Med. 35, 436–450 (2008).
Wiesinger, A. et al. Revealing the tick microbiome: insights into midgut and salivary gland microbiota of female Ixodes ricinus ticks. Int. J. Infect. Dis. 24 https://doi.org/10.3390/ijms24021100 (2023).
Eisenhofer, R. et al. Contamination in low microbial biomass Microbiome studies: issues and recommendations. Trends Microbiol. 27, 105–117. https://doi.org/10.1016/j.tim.2018.11.003 (2019).
Quan, V. C. et al. Zoonotic infections in adults in the Bushbuckridge district of Mpumalanga Province. Int. J. Infect. Dis. 21 https://doi.org/10.1016/j.ijid.2014.03.808 (2014).
Brinkmann, A. et al. A cross-sectional screening by next-generation sequencing reveals Rickettsia, Coxiella, Francisella, borrelia, Babesia, theileria and hemolivia species in ticks from Anatolia. Parasit. Vectors. 12 (26). https://doi.org/10.1186/s13071-018-3277-7 (2019).
Jourdain, E., Duron, O., Severine, B., Gonzalez-Acuna, D. & Sidi-Boumedine, K. Molecular methods routinely used to detect Coxiella burnetii in ticks cross-react with Coxiella-like bacteria. Infect. Ecol. Epidemiol. 5, 29230. https://doi.org/10.3402/iee.v5.29230 (2015).
Harvey, J. W., Simpson, C. F. & Gaskin, J. M. Thrombocytopenia induced by a Rickettsia-like agent in dogs. J. Infect. Dis. 137, 182–188. https://doi.org/10.1093/infdis/137.2.182 (1978).
Breitschwerdt, E. B. et al. Intravascular persistence of Anaplasma platys, Ehrlichia chaffeensis, and Ehrlichia ewingii DNA in the blood of a dog and two family members. Parasit. Vectors. 7, 1–7. https://doi.org/10.1186/1756-3305-7-298 (2014).
Arraga-Alvarado, C. M. et al. Case report: molecular evidence of Anaplasma platys infection in two women from Venezuela. Am. J. Trop. Med. Hyg. 91, 1161–1165. https://doi.org/10.4269/ajtmh.14-0372 (2014).
Yuasa, Y., Tsai, Y. L., Chang, C. C., Hsu, T. H. & Chou, C. C. The prevalence of Anaplasma platys and a potential novel Anaplasma species exceed that of Ehrlichia canis in asymptomatic dogs and Rhipicephalus sanguineus in Taiwan. J. Vet. Med. Sci. 79, 1494–1502. https://doi.org/10.1292/jvms.17-0224 (2017).
Snellgrove, A. N. et al. Vector competence of Rhipicephalus sanguineus sensu stricto for Anaplasma platys. Ticks Tick. Borne Dis. 11, 101517. https://doi.org/10.1016/j.ttbdis.2020.101517 (2020).
Ueti, M. W. et al. Quantitative differences in salivary pathogen load during tick transmission underlie strain-specific variation in transmission efficiency of Anaplasma marginale. Infect. Immun. 77, 70–75. https://doi.org/10.1128/IAI.01164-08 (2009).
Potgieter, F. T. & van Rensburg, L. Tick transmission of Anapaslma centrale. Onderstepoort J. Vet. Res. 54, 5–7 (1987).
Shkap, V. et al. Experimental transmission of field Anaplasma marginale and the A. centrale vaccine strain by Hyalomma excavatum, Rhipicephalus sanguineus and Rhipicephalus (Boophilus) annulatus ticks. Vet. Microbiol. 134, 254–260. https://doi.org/10.1016/j.vetmic.2008.08.004 (2009).
Khumalo, Z. T. H. et al. Evidence confirming the phylogenetic position of Anaplasma centrale (ex theiler 1911) Ristic and Kreier 1984. Int. J. Syst. Evol. Microbiol. 68, 2682–2691. https://doi.org/10.1099/ijsem.0.002832 (2018).
Al-Hosary, A. et al. Epidemiology and genotyping of Anaplasma marginale and co-infection with Piroplasms and other Anaplasmataceae in cattle and buffaloes from Egypt. Parasit. Vectors. 13, 495. https://doi.org/10.1186/s13071-020-04372-z (2020).
Khumalo, Z. T. et al. Characterization of Anaplasma marginale subsp. centrale strains by use of msp1aS genotyping reveals a wildlife reservoir. J. Clin. Microbiol. 54, 2503–2512. https://doi.org/10.1128/JCM.01029-16 (2016).
Machado, R. Z. et al. Molecular diagnosis and genetic diversity of tick-borne Anaplasmataceae agents infecting the African Buffalo Syncerus caffer from Marromeu reserve in Mozambique. Parasit. Vectors. 9, 454. https://doi.org/10.1186/s13071-016-1715-y (2016).
Makgabo, S. M., Brayton, K. A., Oosthuizen, M. C. & Collins, N. E. Unravelling the diversity of Anaplasma species Circulating in selected African wildlife hosts by targeted 16S microbiome analysis. Curr. Res. Microb. Sci. 5, 100198. https://doi.org/10.1016/j.crmicr.2023.100198 (2023).
Gomez-Chamorro, A., Hodzic, A., King, K. C. & Cabezas-Cruz, A. Ecological and evolutionary perspectives on tick-borne pathogen co-infections. Curr. Res. Parasitol. Vector Borne Dis. 1, 100049. https://doi.org/10.1016/j.crpvbd.2021.100049 (2021).
Chigwada, A. D., Mapholi, N. O., Ogola, H. J. O., Mbizeni, S. & Masebe, T. M. Pathogenic and endosymbiotic bacteria and their associated antibiotic resistance biomarkers in Amblyomma and Hyalomma ticks infesting Nguni cattle (Bos spp). Pathogens. 11 https://doi.org/10.3390/pathogens11040432 (2022).
Khalaf, J. M., Mohammed, I. A. & Karim, A. J. The epidemiology of tick in transmission of Enterobacteriaceae bacteria in buffaloes in marshes of the South of Iraq. Vet. World. 11, 1677–1681. https://doi.org/10.14202/vetworld.2018.1677-1681 (2018).
Kolo, A. O. Detection of Zoonotic Bacterial Pathogens in Multiple Hosts Using a Microbiome Approach and Molecular Characterization of Anaplasma phagocytophilum Doctor Philosophiae thesis, University of Pretoria (2019).
Kolo, A. O. Identification and Molecular Characterization of Zoonotic Rickettsiae and Other Vector-borne Pathogens infecting dogs in Mpumulanga province South Africa Magister Scientiae thesis, University of Pretoria, (2014).
Woodroffe, R. et al. Contact with domestic dogs increases pathogen exposure in endangered African wild dogs (Lycaon pictus). PLoS One. 7, e30099. https://doi.org/10.1371/journal.pone.0030099 (2012).
Plantard, O. et al. Detection of Wolbachia in the tick Ixodes ricinus is due to the presence of the Hymenoptera endoparasitoid Ixodiphagus hookeri. PLoS One. 7, e30692. https://doi.org/10.1371/journal.pone.0030692 (2012).
Piesman, J. & Schneider, B. S. Dynamic changes in Lyme disease spirochetes during transmission by nymphal ticks. Exp. Appl. Acarol. 28, 141–145. https://doi.org/10.1023/A:1025351727785 (2002).
Acknowledgements
We acknowledge Luis Neves for verifying the tick identifications. We thank the staff of the Hans Hoheisen Research Centre for logistical support, environmental monitors, Handry Mathebula and Julia D. Sithole, for assistance in the Bushbuckridge-East community, and the tribal council for allowing us to work in their community. Appreciation is extended to Derek Pouchnik and Mark Wildung from the Genomics Sequencing Core at Washington State University for assistance with microbiome sequencing.
We thank the South African National Research Foundation (NRF) for grants 92739 and 110448, and 109350, NIH NIAID R01AI136832, and the Belgian Directorate General for Development Cooperation Framework FA4 (ITM-DGCD) awarded to Marinda Oosthuizen, as well as the Agricultural Sector Education Training Authority (AgriSETA; BU18UP124-18.2) Scholarship and the Meat Industry Trust for the bursary awarded to Rebecca E. Ackermann. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funding bodies.
Author information
Authors and Affiliations
Contributions
The research was conceptualised and designed by K.A.B., C.A.G., N.E.C., M.C.O. and R.E.A. Iv.W., R.E.A., C.A.G. and J.W. did sample acquisition. Data acquisition was done by R.E.A. and A.O.K. R.E.A. performed the analysis. R.E.A., M.C.O., K.A.B., N.E.C. and C.A.G. interpreted the data. The manuscript was drafted by REA and revised and edited by R.A.E., C.A.G., K.A.B., N.E.C., Iv.W., A.O.K. and M.C.O.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
The following committees approved this project: The Faculty of Veterinary Science, University of Pretoria (UP) Research Ethics Committee (REC010-18), the UP Animal Ethics Committee (V012-18 and V064-16), and the UP Faculty of Humanities Research Ethics Committee (GW20180719HS). Permission was obtained to conduct the research, in terms of Sect. 20 of the Animal Diseases Act of 1984, from the Department of Agriculture, Land Reform and Rural Development (DALRRD) South Africa, with reference numbers 12/11/1/1/8 and 12/11/1/1. Animal owners provided informed consent for us to work with their animals, by signing a consent form that was translated into the local language (Shangaan); interpreters were available to translate any questions.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Ackermann, R.E., Gall, C.A., Brayton, K.A. et al. Temporal changes in the bacterial microbiome of the salivary gland and midgut tissues of Rhipicephalus sanguineus (s.l.) ticks in South Africa. Sci Rep 15, 17434 (2025). https://doi.org/10.1038/s41598-025-99189-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-99189-0
