
Abstract
Fumarole fields on active volcanoes are habitats that host unique microbial ecosystems. However, DNA extraction from them for further analysis is rather challenging. In this study, we compared two different ways of sample homogenization for DNA extraction to further profile the microbial communities of active fumarolic fields from Elbrus and Ushkovsky volcanoes and the frozen fumarole deposits of Fujiyama. Vertical homogenizer gave significantly higher DNA concentrations for the Elbrus samples, and more archaeal amplicon sequence variants for Elbrus and Ushkovsky samples compared to the horizontal one. This suggests that vertical homogenizer might be preferable for DNA extraction from sandy and rocky soils. Independent of the homogenizer type, the dominant phyla for Elbrus were Acidobacteriota and Pseudomonadota, and Crenarchaeota for Ushkovsky. The bacterial community of Fuji was less diverse, with Actinomycetota, Pseudomonadota and Bacillota being the dominant phyla. Thus, the studied fumaroles showed distinct microbial profiles, revealing unique adaptations to their respective extreme environments. Within the fungal community, Ascomycota, Basidiomycota and Chytridiomycota were the most dominant phyla for all three volcanoes, but their abundance varied. This study offers the first comprehensive analysis of microbial and fungal communities of active and frozen fumarolic fields, and demonstrates that the choice of methodology can significantly influence the understanding of microbial diversity in extreme environments.
Similar content being viewed by others


Introduction
Environments once thought to be lifeless actually host diverse forms of microorganisms. From polar ice caps to hydrothermal vents, from the deep ocean and terrestrial subsurface to the upper stratosphere, these are some of Earth’s harshest environments that have been found to support life1. However, only a few studies analyzed the microbial diversity of extreme environments associated with volcanic activity2,3. Due to high temperatures, the volcanic environments, especially fumaroles and thermal springs, are typically considered to be inhabited solely by thermophiles and hyperthermophiles. Fumaroles are areas at the surface where volcanic gasses and vapors are emitted. These are common features on active volcanoes, and are an important sign that a volcano is active in that fumaroles indicate the presence of heat from volcanic sources and often lead to formation of mineral deposits on cooler surfaces nearby4. Typically, these environments with steam deposits, rich in metals, exposed to gasses and temperatures ranging from mild to extremely high, harbor low-biomass communities5 and are challenging to study. Understanding of biosignatures within systems generated by volcano-ice interaction is insufficient, and this represents a significant research interest.
Preservation of microorganisms and their DNA in extreme environments, including fumaroles and permafrost, may be achieved by encapsulating microbes in cryogenic opal-a, which is formed when silica-rich hot fluids freeze. This process could preserve cellular structures and genetic material, although research into its effectiveness of this preservation is limited6. At that, identification and functional analysis of permafrost microbial communities is strongly dependent on the effectiveness of DNA extraction.
While extensive approaches to DNA extraction have been evaluated and developed for a variety of environmental samples7,8,9,10, there is no universal method suitable for all sample types. Proper lysis of microbial cells from heterogeneous communities, especially unculturable microbial species, without damaging their genomes is a major challenge. The main aim of lysis is to disrupt cells and release nucleic acids from bacteria into the solution for subsequent extraction. Although the mechanical lysis has been shown to be effective for both bacteria and archaea11,12, its impact on the DNA yield, resulting sequencing coverage of the microbiome composition is poorly understood, particularly for low biomass samples from harsh environments13,14,15. It is still uncertain whether the extracted DNA accurately reflects the true composition of the microbial community, as the efficiency of lysis may preferentially impact certain microbial cells over others.
Communities of different fumarolic fields have a distinct microbial composition. A study of the geothermal region of Favara Grande (Pantelleria, Italy)16 showed that the dominant phyla were Actinobacteria (Actinomycetota), Chloroflexi (Chloroflexota ), Firmicutes (Bacillota), and Verrucomicrobia (Verrucomicrobiota). Members of the Firmicutes (Bacillota) and Proteobacteria (Pseudomonadota) phyla, including sequences associated with thermophilic and sulfate-reducing bacteria, were the most abundant bacterial populations in fumarole niches of the Paricutín volcano in Michoacán (Mexico)3. Ascomycota and Basidiomycota are the most abundant members of fungal communities in hydrothermal environments17. Geothermal environments with temperatures > 80 °C tend to be dominated by archaea over bacteria18. The microbial composition of extreme environments may be affected by geochemical characteristics, such as pH, temperature, pressure, radiation, and high concentrations of salt or heavy metals.
Here, we explore the microbial composition of underexplored high mountain fumarole fields of the Elbrus, Ushkovsky, and Fuji volcanoes. The composition of the prokaryotic and fungal communities of the fumarole fields was assessed by the 16S rRNA and ITS gene analysis, respectively. Due to the challenges of extracting DNA from low-biomass samples19, we used two different homogenizers to compare the effect of vertical and horizontal homogenization on DNA yield. Our results show that the use of a vertical homogenizer TissueLyser LT (QIAGEN) in DNA extraction of low biomass samples yields higher average concentration of DNA and more archaeal ASVs (amplicon sequence variants). The microbial community composition was described for each site.
Methods and materials
Site description and sampling
Soil samples were collected from three different locations: Elbrus (Caucasus, Russia), Fuji (Japan) and Ushkovsky (Kamchatka, Russia) active stratovolcanoes. Topsoil samples from Elbrus and Ushkovsky were taken at natural humidity by a sterile spoon. The Fuji samples (cores with diameter 6–8 cm and length 15–30 cm) were collected using a dry drilling technique, without any fluid or compressed air. All samples were stored at − 20 °C upon collection to preserve the composition of the microbial communities.
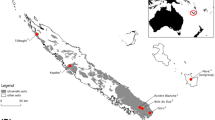
Three Elbrus samples were collected in August 2020 from fumarole fields on the eastern slope of the volcano (43° 20.979′ N; 42° 27.241′ E; 5590 m above sea level) (Fig. 1a). Sample FM5 was collected from a point under the snow in the air cavity of an active fumarole. The sample is a fine soil from the surface (0 cm) containing fragments of moss, both living and dead. Samples FP1 and FP2 were collected from a point located in the same fumarole cavity under the snow. The samples are weakly cemented fine soil with inclusions of clastic material and were taken from the wall of the pits from the surface (FP1 at 0–5 cm) and subsurface layers (FP2 at 5–10 cm). The soil surface temperature at the time of sampling was + 22.5 °C for all samples.

Location of the sample collection sites. (a) Fumarole fields on the eastern slope of the Elbrus volcano (43° 20.979′ N; 42° 27.241′ E; 5590 m above sea level). (b) Fumarole field on the western rim of the Hertz crater on the Ushkovsky volcano (56.07° N; 160.47° E; 3943 m a.s.l.). (c) Recent Activity Point Fuji (RAPFuji) drill hole, Mountain Fuji (35° 21.776′ N; 138° 44.056′ E; 3734 m a.s.l.).
In the Summer of 2022, subsurface (0–5 cm depth) samples were collected at the Ushkovsky volcano, specifically from an snow-free area at a fumarole field located on the western rim of the Hertz Crater (56.07°N; 160.47°E; 3943 m above sea level) (Fig. 1b).The sediments are fine soils of hydrothermally altered basalts and are characterized by a rich ochre color. The surface temperature at the time of sampling was + 68.4 °C. The sampling site is under the influence of the neighboring Kliuchevskoi volcano, so the sampled material is likely to contain an admixture of ash of calcareous-alkaline high-aluminous subaphyric basalts from the Kliuchevskoi ash.
The Fujiyama samples were collected frozen in 2012 from extinct fumarole fields located at the rim of the Fujiyama volcano crater. The samples are core fragments from the Recent Activity Point Fuji (RAPFuji) drill hole (35° 21.776′ N; 138° 44.056′ E; 3734 m above sea level) (Fig. 1c). These samples were collected from two depths of 60–67 cm (Fj60) and 75–80 cm (Fj75). They represent frozen scoria, ochre-rust colored with inclusions of clastic material. According to historical records, the surface temperature was + 80 °C in 1897, + 50 °C in 1957 and became ambient in 198220.
DNA isolation and sequencing
DNA was isolated using the DNeasy PowerSoil Pro kit (Qiagen, Germany) from 800 mg of each sample, in triplicate. Samples were taken from the middle of the frozen kern, and put directly to the PowerBead Pro tube with 800 ul of CD1, without prior defrosting. Then they were homogenized for 10 min using two different homogenizers at the same frequency of 50 Hz: the horizontal homogenizer Vortex-Genie 2 (MoBio, USA) and the vertical homogenizer TissueLyser LT (Qiagen, Germany). Following sample disruption, DNA extraction was made using the DNeasy PowerSoil Pro kit (Qiagen, Germany) according to the manufacturer’s instructions. DNA concentrations were measured using the Qubit fluorometer (Thermo Fisher Scientific, USA) with the dsDNA HS assay kit (Thermo Fisher Scientific, USA). The isolated DNA was stored at − 20 °C until the start of the amplicon library preparation.
The V4 variable region of the 16S rRNA gene was amplified using the universal primers 515F 5′-GTGCCAGCMGCCGCGGTAA-3′ and 806R 5′-GGACTACHVGGGTWTCTAAT-3′21. To avoid the adaptor ligation step, Illumina adaptors 1 and 2 were added to the primer sequences. The fungal ITS region was amplified with ITS3_KYO2–F 5′- GATGAAGAACGYAGYRAA-3′ and ITS4-R 5′-TCCTCCGCTTATTGATATGC-3′22. Phusion 2X Master Mix with the HS buffer (New England Biolabs) was used for amplification. The DNA was denatured (95 °C; 2 min), followed by 27 cycles (30 for ITS) of denaturation (95 °C; 25 s), annealing (58 °C; 30 s), extension (72 °C; 40 s), final extension (72 °C; 3 min), and holding (12 °C). The quality of amplicons was initially tested using electrophoresis in 1.5% agarose in the 1xTAE buffer. Amplicons were then purified with the AMPure XP beads (1:0.9 V:V, Beckman Coulter), and their concentration was measured on the Qubit fluorometer. To control possible contamination, two negative PCR controls with sterile water were made for each primer pair. The Nextera XT index kits (Illumina, USA) were then used to index the amplicons. The resulting libraries were cleaned using the AMPure XP beads, quantified, and sequenced in the Skoltech Core Genomics Facility on Illumina MiSeq (Illumina, USA) as 250 PE using the MiSeq reagent kit V2 (500 cycles).
qPCR
The relative bacterial load in the samples was assessed by qPCR of the 16S rRNA genes23. qPCR was made using the DT-lite machine (DNA-Technology LLC, Russia) and SYBR Green 5 × qPCRMix-HS (Evrogen, Russia). For all qPCR assays, the following cycling conditions were used: denaturation (95 °C; 2 min) followed by 35 cycles of denaturation (95 °C; 25 s), annealing (58 °C; 30 s), extension (72 °C; 40 s), final extension (72 °C; 3 min), and holding (12 °C). All reactions were performed in triplicate, Comparative Ct quantification (2-ΔCt method) was used to obtain qPCR results, with horizontal homogenization samples used as a control. Real-time PCR results represent the average concentration of three independent DNA extractions.
Data analysis
Raw read quality was evaluated using FASTQC24. Trimmomatic-0.3925 was used to trim adapter sequences and low-quality reads with the following parameters: ILLUMINACLIP:NexteraPE-PE.fa:2:30:10:2 LEADING:20 TRAILING:20 SLIDINGWINDOW:20:20 MINLEN:40. Trimmed reads from all samples were analyzed using QIIME2, version 2023.526. Reads were denoised, filtered, chimera checked, and paired-end read joined using DADA227. Amplicon sequence variants ASV were generated. Multiple sequence alignment was performed using MAFFT28 and phylogenetic trees were generated with FastTree29. Bacterial and archeal taxonomy was assigned to ASVs using the q2-feature-classifier plugin30 with the classifier pre-trained for the V4 region, silva-138-99-515-806-nb-classifier.qza, available from the QIIME2 data resources site. For fungal reads we trained the feature classifier with UNITE QIIME release for Fungi version 18.07.202331 with the QIIME feature-classifier fit-classifier-naive-bayes plugin32. The final ASV count table, taxonomy, tree, and sample metadata were merged into a phyloseq object in R version 4.3.3 (2024-02-29) for further analysis. Contaminants were removed using the Decontam (version 1.20.0) package33. All samples were rarefied to the lowest observed number of reads. The Phyloseq package (version 1.44.0)34 was used to normalize the ASVs into percentages as relative abundance. Alpha diversity indices were plotted using the plot_richness function in Phyloseq.
Results
Effect of homogenisation on the yield of bacterial and fungal DNA
We first compared the concentration of total DNA after sample homogenization with the vertical Qiagen LT or horizontal MoBio Vortex-Genie 2 homogenizers (Table 1). In five out of six samples, the overall DNA concentration was higher after sample disruption with Qiagen TissueLyser LT, with the highest effect for the Elbrus FM5 sample (for the second Fuji sample the concentration was low in both cases). Gels run after PCR with the 505F-806R 16S rRNA primers were in line with this observation, showing higher amounts of bacterial DNA with the use of vertical homogenizer (Supplementary Figure S1a). To quantify the results, qPCR had been set up that further confirmed that this difference in concentrations indeed reflected the amount of bacterial DNA in the samples (Fig. 2; gel is shown in Supplementary Fig. S2). Again, the largest difference was detected for Elbrus FM5 (Fig. 2). The same observation was made for fungal DNA (gel after amplification with the ITS3-ITS4 primers is shown in Supplementary Fig. S1b). Though the overall fungal load in all studied samples was predictably low, for the FM5 DNA the ITS3-4 product was observed. This might be partially explained with the fragments of living and dead moss contained in the sample.

Relative qPCR yield of total DNA after sample disruption with vertical Qiagen TissueLyser LT (red) and MoBio Vortex-Genie 2 horizontal (green) homogenizers. Error bars were calculated based on two replicates, with three technical replicates for each.
Thus, sample disruption with the vertical Qiagen TissueLyser LT homogenizer yielded higher overall DNA concentration and higher amount of bacterial DNA. In addition, this technique allowed for recovery of a larger number of archaeal ASVs (Fig. 3a, Supplementary Table S1).

Homogenization effects. (a) Microbial community compositions at the domain level, determined by 16S rRNA amplicon sequencing. Samples are divided by location. (b) Alpha diversity indices (Chao1, ACE, Shannon, Fisher). In the SampleID, letter “h” means horizontal homogenizer, letter “v” means vertical homogenizer.
When vertical homogenization was used on samples from the Elbrus volcano, archaea constituted from 1.12 to 2.13%, while when the horizontal homogenizer was used on the same samples, the archaeal domain was completely absent or present in much smaller quantities (Table S1). For Ushkovsky’s sample the vertical homogenizer yielded up to 8% more archaea compared to the horizontal one. While bacteria were present in all the Fuji samples, no evidence for the presence of archaea was found (Fig. 3a). Alpha diversity indices (Shannon, Chao1, ACE, and Fisher indices; Fig. 3b, Table S2, Table S3) were not significantly different when two homogenizers were used.
Microbial composition of fumaroles
A total of 194,806 reads were acquired for 12 libraries of 16S rRNA genes; after removing adapters, 99,759 reads remained; after denoising in QIIME2, 83,596 reads were merged and used for downstream analysis. Alpha diversity indices Chao1, ACE, and Fisher were lower in all Fuji, Elbrus FP2, and Ushkovsky Ushk_v bacterial communities compared to other samples (Fig. 3b, Table S2, S3). Beta diversity estimated using the Bray–Curtis distance demonstrated significant differences between bacterial samples (Fig. 4a), with microbiomes from different sites clearly separated.

Beta diversity of bacterial (a) and fungal (b) communities assessed by the Bray–Curtis index (PCoA).
The composition at the phylum level is presented in Fig. 5a and Table S4. The featured proportion represents the percentage of 30 most common genera, including “Other” genera (those not in top 30) and unclassified bacteria (Fig. 5b, Table S5). Based on the relative abundance of 16S rRNA genes, phyla Acidobacteria (Acidobacteriota), Proteobacteria (Pseudomonadota), Actinobacteriota(Actinomycetota), Chloroflexi (Chloroflexota) were the most abundant in almost all samples, followed by other, relatively weakly represented phyla (Fig. 5a).

(a) Taxonomic composition at the phylum level of fumarole microbial communities. (b) Taxonomic composition at the genus level of fumarole microbial communities, including “other” genera (those not in the top 30) and unclassified bacteria. Samples are split by site. In the SampleID, letter “h” means horizontal homogenizer, letter “v” means vertical homogenizer.
The microbial composition of the Elbrus fumarole field is composed of diverse phyla and genera of bacteria and archaea. Bacteria from phyla Acidobacteria (Acidobacteriota), Proteobacteria (Pseudomonadota), and Chloroflexi (Chloroflexota) (Fig. 5a, Table S4) predominate in all samples. Archaea from phylum Crenarchaeota (Thermoproteota ) were also observed, as well as various soil bacteria, including Verrucomicrobium (Verrucomicrobiota) and Planctomycetes (Planctomycetota), are also prominent. Minor differences in microbial communities between topsoil layers were observed in the Elbrus samples; in particular, Candidatus_Koribacter was identified only in FP1 (0–5 cm) and FP2 (5–10 cm depth) samples, and Acinetobacter was found only in FP2_v (5–10 cm depth). The following genera were abundant in the Elbrus samples: Subgroup_2, WPS-2, Candidatus_Udaeobacter, HSB_OF53-F07, AD3 and Rokubacteriales were found in FM5. Subgroup_7, Ellin6067, Candidatus_Koribacter, AD3, WPS-2, Subgroup_13, Rokubacteriales were abundant in the FP1 and FP2 samples (Fig. 5b, Table S5).
The microbial communities of the Ushkovsky fumarole are dominantly composed of Crenarcheota (Thermoproteota) phylum, including mainly genus Group_1.1c and Thaumarchaeota archaeon SCGC AB-179-E04. Furthermore, thermophilic bacteria such as Thermus were also identified. The presence of bacteria from genera Subgroup_2 and 11–24 from phylum Acidobacteria (Acidobacteriota), was also notable (Fig. 5a, b, Table S4, S5).
The microbial composition of the Fuji frozen fumarole included bacterial phyla such as Proteobacteria (Pseudomonadota), Actinobacteriota (Actinomycetota), Firmicutes (Bacillota) (Fig. 5a, Table S4). Genera Cutibacterium, Staphylococcus, Acinetobacter, Schlegelella, Corynebacterium were predominant. Thermus was also found (Fig. 5b, Table S5).
Fungal community of fumaroles
For the ITS region, a total of 211,818 reads were acquired for 12 libraries; after removing adapters, 47,260 reads remained; after denoising in QIIME2, 35,768 reads were merged and used for further analysis. Beta diversity assessed by the Bray–Curtis distance showed significant differences in fungal communities across the samples (Fig. 4b). The proportion of fungal genera represents the percentage of 30 most common genera (Fig. 6b, Table S6). The most abundant phyla were Ascomycota, Basidiomycota and Chytridiomycota (Fig. 6a, Table S7).

Taxonomic composition at the phylum (a) and genus (b) level of fumarole fungal communities determined ITS region amplicon sequencing. The proportion of all fungal genera represents the percentage of the 30 most common genera, including “other” genera (those not in the top 30) and unclassified fungi. Samples were split by site. In the SampleID, letter “h” means horizontal homogenizer, letter “v” means vertical homogenizer.
The fungal composition of the Elbrus fumarole included phyla such as Ascomycota and Basidiomycota and less abundant Chytridiomycota, Mortierellomycota, Rozellomycota and others. Samples FM5_h, FP1_h and FP1_v were also dominated by unclassified fungi, while others were dominated by members of Ascomycota (FP2_h and FP2_v). The genera Alternaria and Helotiales_gen_Incertae_sedis were present only in the FM5 samples.
The dominant fungal phyla in the Ushkovsky fumaroles were Ascomycota and Basidiomycota. Fomes and Botryobasidium dominated in the Ushk_h sample; Helotiales_gen_Incertae_sedis and Rozellomycota_gen_Incertae_sedis, in the Ushk_v samples.
The most abundant phyla in the Fuji fumaroles were Ascomycota and Basidiomycota, dependent on depths. Samples from the Fuji depths of 75–80 cm, Fj75 contained mainly Ascomycota. Samples from the 60–67 cm depth, Fj60 contained Basidiomycota outnumbering Ascomycota. The following genera were dominant in the Fuji samples: in the 75–80 cm depth samples (Fj75_h), genera Alternaria and Cladosporium were noted. In contrast, the same sample but with vertical homogenization (Fj75_v) contained mainly Cladosporium. In the 60–67 cm depth samples, Fj60_v contained Hyphodontia, Trametes, and Cladosporium, while Fj60_h contained Antrodiella and Cladosporium (Fig. 6b, Table S6).
Discussion
Fumarole sites have been studied worldwide and have been found to contain unique microbial communities2,3,16. The problem with rocky fumarole samples may be related to the specific composition of the soil35, in particular the limited surface area on which microorganisms and DNA molecules can be absorbed, and also the lower amount of unfrozen water, which favors the preservation of microbial cells in frozen sediments. We studied the microbial communities of fumarole fields from active volcanoes, Elbrus (Caucasus, Russia), Ushkovsky (Kamchatka, Russia), and Mount Fuji (Japan), and the effects of homogenization techniques on the DNA yield and microbial abundance. We found that the vertical homogenizer yielded higher DNA concentrations in most samples, compared to the horizontal homogenizer. The Elbrus FP2 sample was the only exception that might reflect soil features in this horizon.
In particular, in the Elbrus samples, the vertical homogenizer improved the recovery of archaeal ASVs compared to the horizontal method. Sample FM5, which contained living and dead moss fragments, showed particularly high DNA yields, suggesting that vertical homogenization can effectively disrupt complex samples containing microbial communities associated with soil organic matter. We did not separate the moss fragments from the soil particles so as not to disturb the microbial profile. However, their impact was minimal, since amplification was performed with bacterial primers, the contribution of the chloroplasts was less than 3% in the FM5 samples, while mitochondrial reads were almost absent.
In contrast, the horizontal homogenizer yielded lower concentrations of DNA and in some cases failed to detect the archaeal diversity. This may indicate that a horizontal MoBio homogenizer might be not suitable for all types of samples, particularly those containing tough or fibrous materials such as plant matter. Our results may suggest that the limitation of the DNA extraction methodology from fumarole samples is due to the first step of the process, homogenization.
Of all studied samples, the most unfavorable soil for DNA extraction was the volcanic scoria of the Fuji due to the high content of rock fragments. Interestingly, this was the only volcano where we did not find any archaeal ASVs. This might be either due to natural reasons such as the absence of archaeal communities in this type of frozen slag, or due to the DNA degradation. This can also be due to previously reported technical reasons2,3.
Overall, the microbial communities observed in the fumarole fields of the Elbrus, Ushkovsky, and Fuji volcanoes are consistent with previous studies in similar environments. Acidobacteria (Acidobacteriota), Actinobacteria (Actinomycetota), Chloroflexi (Chloroflexota), and Proteobacteria (Pseudomonadota) are commonly found in volcanic ecosystems such as the active Santorini-Kolumbo volcanic field36, the Antarctic Peninsula37. Specifically Chloroflexi (Chloroflexota) were found in every fumarole sample in the Deception Island Glaciers vs. Fumaroles study38. In fumaroles on the island of Hawai’i39, ASVs belonging to Chloroflexi (Chloroflexota) and Proteobacteria (Pseudomonadota) were the most abundant consortia. Gemmatimonadetes (Gemmatimonadota), found in the Fuji and Elbrus samples, was one of the most abundant phyla in the bacterial communities associated with speleothems from two lava tubes on Santa Cruz Island40. In addition, Planctomycetes (Planctomycetota) were the most abundant in the Elbrus fumarole. They are usually considered mesophilic and are recognised as common soil inhabitants41. Among the fungi, Ascomycota and Basidiomycetes, which occupy different niches42, were the largest and most diverse groups in the studied fumaroles.
Temperature appeared to be a critical factor influencing the microbial community structure at the fumarole sites. Higher temperatures of the Ushkovsky fumaroles (+ 68.4 °C) favored the presence of thermophilic microorganisms, predominantly Crenarchaeota (Thermoproteota). This is consistent with observations of thermophilic prokaryotes typical of such geothermal environments. In contrast, milder temperatures of the Elbrus fumaroles (+ 22.5 °C) favored a more diverse range of mesophilic or psychrotolerant microorganisms. The presence of apparently thermophilic Thermus in the frozen environment of Mount Fuji can be explained by remaining DNA from dead cells.
In the Elbrus fumaroles, small differences in microbial communities were found between different soil layers, probably due to the heterogeneity of samples and their respective communities. This illustrates a challenge in metagenomic studies. For example, unique bacterial members such as Candidatus_Koribacter which were found at topsoil and Acinetobacter were uniquely present in the FP2 sample at the 5–10 cm depth, and fungal communities in the samples also showed notable differences depending on depth. Conversely, in the Fuji fumaroles, while there was a clear difference in microbial profiles, with the presence of phylum Planctomycetes (Planctomycetota) at the 75 cm depth, genera Acinetobacter, Cutibacterium, and Thermus were present in both layers. This suggests that even within a single fumarole, abiotic variables such as depth can significantly influence the microbial community structure.
Several studies describe bacterial and archaeal communities from Fuji43,44, but not the fumaroles. Specifically, one study described changes in bacterial diversity with elevation43. Briefly, the soil samples taken at elevation intervals between the base of the mountain at 1000 m and its summit at 3700 m showed that the most abundant phyla in the entire sample set were Proteobacteria (Pseudomonadota), followed by Actinobacteriota (Actinomycetota), Chloroflexi (Chloroflexota) and Bacteroidetes (Bacteroidota), which partially correlated with our results. Another study showed elevation patterns for archaeal phyla Thaumarchaeota and Euryarchaeota44. At that, no archaea were found in our samples.
While our findings align with those of similar studies38,39, they underscore the necessity of considering individual fumarole characteristics such as the thermal regime and geological context. The diversity of microbial communities found in different fumaroles are unique and fumarole-specific, providing valuable information about the ecology of these extreme environments.
Conclusion
Our findings indicate that the vertical homogenization method may have advantages in terms of DNA yield and the detection of archaeal members compared to horizontal homogenization. This suggests that vertical homogenization might be a more effective approach for studying microbial communities in extreme environments such as fumaroles.To validate these findings, further confirmation of the homogenization effect and a more detailed study with a larger sample size for statistical significance are needed. The observed results on the dynamics of microbial community composition in fumarole fields suggest a critical role of environmental characteristics in shaping the microbial diversity. Our central hypothesis is that the microbial diversity and community structure in fumarole environments are significantly influenced by the unique geochemical and thermal characteristics of each fumarole, as well as the physical properties of the surrounding substrate.
Data availability
Raw sequencing reads are deposited to the GenBank SRA database with under the Bioproject number PRJNA1149287.
References
Rampelotto, P. Extremophiles and extreme environments. Life 3, 482–485 (2013).
Mayhew, L. E., Geist, D. J., Childers, S. E. & Pierson, J. D. Microbial community comparisons as a function of the physical and geochemical conditions of Galápagos Island fumaroles. Geomicrobiol. J. 24, 615–625 (2007).
Medrano-Santillana, M., Souza-Brito, E. M., Duran, R., Gutierrez-Corona, F. & Reyna-López, G. E. Bacterial diversity in fumarole environments of the Paricutín volcano, Michoacán (Mexico). Extremophiles 21, 499–511 (2017).
Gresse, M. et al. Anatomy of a fumarolic system inferred from a multiphysics approach. Sci. Rep. 8, 7580 (2018).
Westmeijer, G. et al. Continental scientific drilling and microbiology: (Extremely) low biomass in crystalline bedrock of central Sweden. Preprint at https://doi.org/10.5194/bg-2023-147 (2023).
Channing, A. & Butler, I. B. Cryogenic opal-A deposition from Yellowstone hot springs. Earth Planet. Sci. Lett. 257, 121–131 (2007).
Miller, D. N., Bryant, J. E., Madsen, E. L. & Ghiorse, W. C. Evaluation and optimization of DNA extraction and purification procedures for soil and sediment samples. Appl. Environ. Microbiol. 65, 4715–4724 (1999).
Bürgmann, H., Pesaro, M., Widmer, F. & Zeyer, J. A strategy for optimizing quality and quantity of DNA extracted from soil. J. Microbiol. Methods 45, 7–20 (2001).
Zhou, J., Bruns, M. A. & Tiedje, J. M. DNA recovery from soils of diverse composition. Appl. Environ. Microbiol. 62, 316–322 (1996).
Elbrecht, V., Peinert, B. & Leese, F. Sorting things out: Assessing effects of unequal specimen biomass on DNA metabarcoding. Ecol. Evol. 7, 6918–6926 (2017).
Salonen, A. et al. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: Effective recovery of bacterial and archaeal DNA using mechanical cell lysis. J. Microbiol. Methods 81, 127–134 (2010).
Wang, H. & Edwards, K. J. Bacterial and archaeal DNA extracted from inoculated experiments: Implication for the optimization of DNA extraction from deep-sea basalts. Geomicrobiol. J. 26, 463–469 (2009).
Henneberger, R. M., Walter, M. R. & Anitori, R. P. Extraction of DNA from acidic, hydrothermally modified volcanic soils. Environ. Chem. 3, 100 (2006).
Hestetun, J. T., Lanzén, A., Skaar, K. S. & Dahlgren, T. G. The impact of DNA extract homogenization and replication on marine sediment metabarcoding diversity and heterogeneity. Environ. DNA 3, 997–1006 (2021).
Yu, V. M. Bead beating offers high-performance homogenization for molecular biology downstream processing of tough and difficult samples. In Sample Preparation Techniques for Soil, Plant, and Animal Samples (ed. Micic, M.) 85–97 (Springer, 2016). https://doi.org/10.1007/978-1-4939-3185-9_7.
Picone, N. et al. Geothermal gases shape the microbial community of the volcanic soil of pantelleria, Italy. mSystems 5, e00517-e520 (2020).
Odilia, A. S., Huxley, M. M., Remmy, W. K. & Hamadi, I. B. Isolation and characterization of fungi from a hot-spring on the shores of Lake Bogoria. Kenya. J. Yeast Fungal Res. 9, 1–13 (2018).
Meyer-Dombard, D. R., Shock, E. L. & Amend, J. P. Archaeal and bacterial communities in geochemically diverse hot springs of Yellowstone National Park, USA. Geobiology 3, 211–227 (2005).
Barton, H. A., Taylor, N. M., Lubbers, B. R. & Pemberton, A. C. DNA extraction from low-biomass carbonate rock: An improved method with reduced contamination and the low-biomass contaminant database. J. Microbiol. Methods 66, 21–31 (2006).
Global Volcanism Program. Report on Fujisan (Japan). Bull. Glob. Volcanism Netw. 38 (2013).
Caporaso, J. G. et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624 (2012).
Toju, H., Tanabe, A. S., Yamamoto, S. & Sato, H. High-coverage ITS primers for the DNA-based identification of ascomycetes and basidiomycetes in environmental samples. PLoS ONE 7, e40863 (2012).
Zaytsev, V. et al. Monitoring of meat quality and change-point detection by a sensor array and profiling of bacterial communities. Anal. Chim. Acta 1320, 343022 (2024).
Andrews S. FastQC: A quality control tool for high throughput sequence data. (2010).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Bolyen, E. et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857 (2019).
Callahan, B. J. et al. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583 (2016).
Katoh, K. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066 (2002).
Price, M. N., Dehal, P. S. & Arkin, A. P. FastTree 2—Approximately maximum-likelihood trees for large alignments. PLoS ONE 5, e9490 (2010).
Bokulich, N. A. et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6, 90 (2018).
Abarenkov, K. et al. The UNITE database for molecular identification and taxonomic communication of fungi and other eukaryotes: sequences, taxa and classifications reconsidered. Nucleic Acids Res. 52, D791–D797 (2024).
Pedregosa, F. et al. Scikit-learn: machine learning in python. J. Mach. Learn. Res. 12, 2825–2830 (2011).
Davis, N. M., Proctor, D. M., Holmes, S. P., Relman, D. A. & Callahan, B. J. Simple statistical identification and removal of contaminant sequences in marker-gene and metagenomics data. Preprint at https://doi.org/10.1101/221499 (2017).
McMurdie, P. J. & Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 (2013).
Lombard, N., Prestat, E., Van Elsas, J. D. & Simonet, P. Soil-specific limitations for access and analysis of soil microbial communities by metagenomics: Limitations in soil metagenomics. FEMS Microbiol. Ecol. 78, 31–49 (2011).
Polymenakou, P. N. et al. Taxonomic diversity of microbial communities in sub-seafloor hydrothermal sediments of the active Santorini-Kolumbo volcanic field. Front. Microbiol. 14, 1188544 (2023).
Bottos, E. M., Scarrow, J. W., Archer, S. D. J., McDonald, I. R. & Cary, S. C. Bacterial community structures of antarctic soils. In Antarctic Terrestrial Microbiology (ed. Cowan, D. A.) 9–33 (Springer, 2014). https://doi.org/10.1007/978-3-642-45213-0_2.
Bendia, A. G. et al. A mosaic of geothermal and marine features shapes microbial community structure on deception Island Volcano, Antarctica. Front. Microbiol. 9, 899 (2018).
Prescott, R. D. et al. Islands within Islands: Bacterial phylogenetic structure and consortia in Hawaiian lava caves and fumaroles. Front. Microbiol. 13, 934708 (2022).
Miller, A. Z. et al. Colored microbial coatings in show caves from the Galapagos Islands (Ecuador): First microbiological approach. Coatings 10, 1134 (2020).
Fuerst, J. A. & Sagulenko, E. Beyond the bacterium: planctomycetes challenge our concepts of microbial structure and function. Nat. Rev. Microbiol. 9, 403–413 (2011).
Jones, E. B. G. et al. Classification of marine Ascomycota, Basidiomycota, Blastocladiomycota and Chytridiomycota. Fungal Divers. 73, 1–72 (2015).
Singh, D., Takahashi, K., Kim, M., Chun, J. & Adams, J. M. A hump-backed trend in bacterial diversity with elevation on mount Fuji, Japan. Microb. Ecol. 63, 429–437 (2012).
Singh, D., Takahashi, K. & Adams, J. M. Elevational patterns in archaeal diversity on Mt. Fuji. PLoS ONE 7, e44494 (2012).
Acknowledgements
This study was supported by the state assignment № FFRW-2024-0004 "Comparative genomics analysis of regulatory systems in pro- and eukaryotes". We thank Go Iwahana, Vasily Mironov for the Fujiyama sampling, and the Hokkaido University for support of drilling operations. Sampling at the Elbrus and Ushkovsky summits was supported by state assignment FMRM-2022-0009 and the Institute of Geography, Russian Academy of Science, in collaboration with Vladimir Mikhalenko, Ivan Lavrentiev, and Stas Kutuzov. Sequencing was partially supported by RSF 24-14-00276 (MSG). We also thank Tatiana Bessonova and Anastasiya Shatilovich for technical assistance, Margarita Ezhova and the Skoltech Genomics Core Facility for sequencing.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study. A.A. conducted fieldwork. A.A. and E.R. developed research concepts. N.E., G.U. extracted DNA and prepared amplicons. M.T. prepared sequencing libraries. G.U. and M.T. performed qPCR, comparative Ct quantification and visualization. A.S. and P.S. analyzed data. A.S. wrote the original manuscript. M.G. and T.V. gained funding. M.G., T.V., M.T. and A.A. revised the manuscript. All authors have read and agreed to the submitted version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Shevchenko, A.Y., Ursalov, G.I., Eromasova, N.I. et al. Microbial diversity of high-elevated fumarole fields, low-biomass communities on the boundary between ice and fire. Sci Rep 15, 16600 (2025). https://doi.org/10.1038/s41598-025-99782-3
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-025-99782-3
