
Abstract
Zinc homeostasis plays a critical role in cellular function, yet its dysregulation may lead to cytotoxicity. Based on the unexpected finding that excessive zinc downregulates the transcription of cardiac-related factors in mouse cardiomyocytes, this study reveals a mechanistic pathway wherein cytoplasmic zinc overload reduces histone acetyltransferase activity, subsequently lowering histone acetylation and ultimately decreasing the transcriptional levels of target genes. The universality of this mechanism was further confirmed across multiple cell types. By investigating the phenomenon of zinc-induced autophagy regulated by acetylation, we explored the potential therapeutic implications of zinc as a drug. Through comparative analysis of cells with varying sensitivity to zinc, we identified aberrant expression of zinc transporters under physiological conditions as a primary factor contributing to zinc-induced toxicity. This finding suggests that zinc transporters may serve as potential therapeutic targets. This study is the first to elucidate the molecular link between zinc homeostasis and histone acetylation, providing a novel perspective for understanding zinc metabolism-related diseases and zinc-targeted therapies.
Similar content being viewed by others
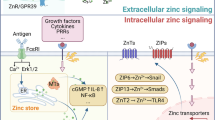


Introduction
As early as 1961, zinc had already been identified as an essential element for humans1. As the second most abundant trace element, zinc is present in approximately 10% of human proteins2,3. Zinc deficiency manifests through diverse pathological consequences including anemia, growth retardation, dermatological abnormalities, immune dysfunction, and neurological disorders2. The maintenance of metal ion homeostasis constitutes a fundamental biological imperative, with precise regulation by specialized transporters ensuring proper cellular function and organismal development. Disruptions in this delicate equilibrium can precipitate systemic dysfunction, as exemplified by copper-induced cuproptosis and calcium-mediated calcicoptosis4,5,6. Zinc toxicity similarly affects multiple organ systems, with documented impacts on gastrointestinal, dermatological, respiratory, and neurological functions7. The zinc pyrithione (ZPT)-induced intracellular zinc overload can trigger apoptosis in lymphocytes, dermal fibroblasts, and Hepatoma cells8,9,10. In the field of tumor immunotherapy, zinc-induced DNA damage and pyroptosis are utilized to enhance tumor immunogenicity11,12.
Protein lysine acetylation represents a ubiquitous post-translational modification with broad regulatory implications. This modification occurs on both histone and non-histone proteins13. Histones are essential components for maintaining chromosome structure. The acetylation of histone lysine residue catalyzed by histone acetyltransferases (HATs) weakens the electrostatic interactions between histones and DNA, and produce a more relaxed chromatin structure, which allows transcription factors to bind to DNA more easily, thereby facilitating the transcription of genes near the histone modification sites14,15. Conversely, reduced histone acetylation caused by histone deacetylases (HDACs) produces the opposite effects16. The proper regulation of gene expression requires the coordinated action of both HATs and HDACs17. Histone acetylation modulates genes implicated in diverse biological processes, including development, cell cycle, metabolism and oncogenesis16,18,19,20. Additionally, proteomic analyses revealed that acetylation modifications of non-histone proteins are functionally associated with essential cellular processes including translation, protein folding, metabolism, RNA processing, and chromatin remodeling21,22,23.
In this study, we investigated the transcriptional consequences of zinc overload in murine and human cardiomyocytes, and further explored its broader cellular implications.
Materials and methods
Cell Lines
Mouse cardiac muscle cell line HL-1 was obtained from Sigma-Aldrich (SCC065). Cells were cultured in a Claycomb medium (51800C, Sigma-Aldrich) supplemented with 10% fetal bovine serum (FBS) (Life Technologies, Inc., Grand Island, NY), 1% penicillin/streptomycin, Norepinephrine 10 mM (A0937, Sigma-Aldrich), and L-Glutamine 2 mM (A7506, Sigma-Aldrich). AC16 human cardiomyocyte cell line was purchased from Sigma-Aldrich (SCC109). HEK-293 human kidney epithelial cell line was purchased from ATCC (CRL-1573). AGS human gastric adenocarcinoma cell line was purchased from ATCC (CRL-1739). Human placental trophoblast cell line TEV-1, human endometrial carcinoma cell line Ishikawa, and human ovarian carcinoma cell line A2780 were all gifted by Prof. Tang. Cells were cultured in high glucose DMEM (Life Technologies, Inc., Grand Island, NY) containing 10% fetal bovine serum (FBS) (FBS, Life Technologies, Inc., Grand Island, NY), 1% antibiotics penicillin–streptomycin. All the cells were maintained at 37 °C with 5% CO2.
Cell viability assay
Cell viability was assayed by Cell Counting Kit-8 (HY-K0301, Med Chem Express) according to the manufacturer’s protocols. Briefly, HL-1 and AC16 cells were seeded into 96-well plates and cultured for 12 h. After treatment with different concentrations of zinc sulfate for 24 h, 10 μL of CCK-8 solution was added to each well and incubated for 2 h at 37 °C. The absorbance was then recorded at 450 nm using a microplate reader (Spark, TECAN). Three independent experiments were conducted.
Primary culture of neonatal mouse cardiomyocytes
C57BL/6 J mice (SLAC Laboratory Animal Co. Ltd, Shanghai, China) were kept under standard animal room conditions (temperature 21 ± 1 °C; humidity 55–60%) with food and water continuously available. Neonatal mice (1–3 days old) were placed in an anesthesia induction chamber containing 3% isoflurane and promptly decapitated with sharp scissors following anesthesia. Cardiomyocytes were isolated as follows. Briefly, after hearts were digested by 0.06 mg/mL trypsin and 1 mg/mL Collagenase II, cells were suspended in DMEM with 15% FBS, and pre-cultured in humidified incubator (5% CO2) for 90 min to obtain cardiac fibroblasts for their selective adhesion. Then the suspended cardiomyocytes were plated in another dish. The purity of cardiomyocytes was increased by supplementing BrdU (5-Bromo-2’-deoxyuridine) to prevent non-cardiomyocytes from developing. Culture medium was renewed after 48 h. This study was reported in accordance with the ARRIVE guidelines.
Antibodies and chemicals
Primary antibodies used to detect proteins were: BMP4 (ab235114, Abcam), KAT2A (ER63516, Huabio, Hangzhou, China), Histone H3 (ET1701-64, Huabio, Hangzhou, China), Histone H3 (acetyl K9) (HA722132, Huabio, Hangzhou, China), LC3B (ET1701-652, Huabio, Hangzhou, China), GAPDH (10,494-1-AP, Proteintech), and β-actin (HA722023, Huabio, Hangzhou, China). secondary antibody: goat anti-rabbit IgG-h + HRP Conjugated (HA1023 Huabio, Hangzhou, China). Zinc sulfate heptahydrate was purchased from Sigma-Aldrich (Z0251). Vorinostat, Entinostat, Romidepsin, Pyrithione, and Zinpyr-1 were purchased from Med Chem Express (HY-10221, HY-12163, HY-15149, HY-B1747, and HY-D0155).
RNA Isolation and quantitative real-time PCR (RT-PCR)
Total RNA was isolated from HL-1 cells, AC16 or neonatal mouse cardiomyocytes by using a TRIzol reagent (Takara Biotechnology, Dalian, China) according to the manufacturer’s instructions. 5 µg total RNA in a volume of 20 µL was reversely transcribed by using Hifair® Ⅲ1st Strand cDNA Synthesis SuperMix (11137ES60, Yeasen, Shanghai, China). After the termination of cDNA synthesis, mRNA levels of target genes were determined by qRT-PCR. Amplification was performed by CFX Opus 96 (Bio-Rad, USA). Real-time PCR was cycled in 95 °C/10 s, 60 °C/20 s and 72 °C/30 s for 40 cycles, after an initial denaturation step at 95 °C for 5 min using Hieff® qPCR SYBR Green Master Mix (11201ES08, Yeasen, Shanghai, China).The relative amounts of the mRNA levels of the target genes were normalized to the β-actin levels, respectively, and the relative difference in mRNA levels was calculated by 2 − ΔΔCt method. The primers for quantitative Real-time PCR (RT-PCR) were listed in the Supplementary Information (Table S1).
Statistical analysis
Experiments were independently triplicated, and results were qualitatively identical. Representative experiments are shown. All data are expressed as mean ± SD. And the data of biological replicates were analyzed by one-way ANOVA and Tukey–Kramer multiple comparison test (GraphPad Prism Version 8.0.2(263), https://www.graphpad.com/features). Pearson test was also performed on GraphPad Prism. Statistical significance was assessed at p < 0.05, p < 0.01, and p < 0.001. All Western blot bands and green fluorescence were quantified using ImageJ (Version 1.53a, https://imagej.net/ij/download.html).
Results
Excess zinc triggers downregulation of cardiac-related genes in mouse myocardial cells
To assess the effects of zinc overload on cardiomyocytes, we performed CCK-8 viability assays on both mouse (HL-1) and human (AC16) cardiomyocyte cell lines across a range of ZnSO4 concentrations (0–538 μM).
(Fig. 1A). Based on dose–response curves, we selected 70 μM ZnSO4 as the experimental concentration, which provided maximal cellular stimulation while maintaining normal growth and metabolic activity. We then examined the transcriptional response of 13 key cardiac developmental genes (Bmp4, Foxa2 Gata4, Hand1, Hand2, Isl1, Mef2c, Nkx2-5, Notch1, Tbx1, Tbx5, Wnt2b, and Wnt3a)24,25 to zinc overload. The results showed that under ZnSO4 treatment, the transcription of Bmp4 and Isl1 in HL-1 were significantly downregulated (Fig. 1B). However, the transcriptional changes of the 13 genes were relatively modest in AC16 cells (Fig. 1C). Furthermore, we ruled out the possibility that the sulfate group influences gene transcription (Fig. S1).

Zinc inhibits the transcription of Bmp4 in mouse cardiomyocytes. (A) CCK-8 viability assay in HL-1 and AC16 cardiomyocytes. N = 4. (B) HL-1 cells, (C) AC16 cells and (D)neonatal mouse cardiomyocytes were treated with 70 μM ZnSO4 for 24 h. mRNA levels of 13 genes were analyzed. N = 3.All data are presented as means ± SD. *p < 0.05; **p < 0.01; ***p < 0.001 versus relevant control.
The downregulation of Bmp4 induced by zinc was also confirmed in neonatal mouse cardiomyocytes (> twofold decrease, p < 0.01) (Fig. 1D). Comparative analysis of basal gene expression profiles in neonatal cardiomyocytes (Fig. S2) and ZnSO4-treated HL-1 cells (Fig. 1B) identified Bmp4 as the most consistently responsive target. Given its pronounced sensitivity to zinc overload, we selected Bmp4 as a target gene to further investigate the mechanism of zinc overload-induced gene downregulation in HL-1 cells.
The reduction in histone acetylation induced by zinc leads to the downregulation of the cardiac-related factor Bmp4
In HL-1 cells, the transcriptional regulation of bmp4 is highly dependent on zinc concentration (Fig. 2A). Time-course analysis revealed this modulation is rapid and reversible (Fig. 2B). In immunoblotting analysis, the inhibition of BMP4 by zinc lagged behind the transcriptional change (4 h), with discernible differences only becoming apparent at 6 h (Fig. 2C).

The inhibition of Bmp4 in HL-1 cardiomyocytes by zinc is both (A) concentration- and (B) time- dependent. 70 μM ZnSO4. N = 3. Concentration-dependent test lasted for 24 h. (C) Immunoblotting analyses of BMP4 at different time points after treatment with ZnSO4. All data are presented as means ± SD. ***p < 0.001 versus relevant control.
Bmp4 is known to promote cardiomyocyte differentiation and is considered as an important cardiomyocyte marker26. Some studies suggest that both cardiac growth and maintenance of homeostasis are associated with covalent modifications of HDACs27,28,29,30. Therefore, we utilized a broad-spectrum.
HDAC inhibitor, Vorinostat, to treat HL-1 in combination with zinc. The result indicates that Vorinostat is able to counteract the effects of zinc and maintain.
Bmp4 mRNA at its original level in HL-1 cells (Fig. 3A). This infer that Bmp4 could be regulated by HDACs. HDACs exert their catalytic function in conjunction with other proteins as part of a complex, and these HDAC complexes exhibit specificity in their transcriptional regulation13. Therefore,in the next step, we need to determine which specific HDACs regulate Bmp4 transcription. The results show that HDAC inhibitors Entinostat (for HDAC1,2,3) and Romidepsin (for HDAC1,2) both inhibited the downregulation of Bmp4. Moreover, when all of the inhibitors acted alone, the expression of Bmp4 were markedly increased (Fig. 3A). Thus, we propose that HDAC1/2 are capable of regulating Bmp4.

Zinc inhibits the transcription of Bmp4 through reducing the acetylation of histones. (A) Vorinostat, Entinostat, and Romidepsin were all capable of reversing the downregulation of Bmp4 induced by ZnSO4, with their working concentrations being 5 μM, 4 μM, and 50 nM, respectively. (B) Zinc significantly reduced H3K9ac. (C) Zinc significantly reversed H3K9ac increased by Romidepsin. (D) Quantification reveals a high correlation between H3K9ac protein and Bmp4 mRNA levels (see integrated data from A and C). All data are presented as means ± SD. **p < 0.01 versus relevant control.
HDAC1 and HDAC2 share a common catalytic site in histones at H3K913. Then the acetylation levels at H3K9 were tested. Markedly, Zinc reduced H3K9ac, even at 50 μM (Fig. 3B). Furthermore, we ruled out the possibility that the sulfate group influences H3K9ac (Fig. S3). Moreover, zinc reversed the elevated H3K9ac caused by Romidepsin (Fig. 3C). Integrated analysis of Fig. 2A and 2C demonstrates that transcriptional changes of Bmp4 correlate strongly with alterations in H3K9 acetylation (Fig. 3D). The deacetylation modulation by zinc operates in direct opposition to HDAC inhibitor-mediated acetylation. Therefore, we conclude that zinc downregulates Bmp4 by reducing acetylation levels.
Disruption of cytoplasmic zinc homeostasis leads to decreased histone acetylation levels in mouse myocardial cells
Using zinc ion probe Zinpyr-131, we detected significantly higher concentrations of free zinc ions in the cytoplasm of HL-1 cells treated with 70 μM ZnSO4 compared to untreated cells (Fig. 4A). This prompted us to investigate whether the observed reduction in acetylation levels was triggered by the zinc ions with high concentration in culture medium or by the intracellular zinc ions within the cytoplasm. As a zinc ionophore, pyrithione allows zinc ions to bypass zinc transporters, resulting in a substantial influx into the cytoplasm. Through experimental condition optimization, we found that treatment with 70 μM ZnSO4 alone generated intracellular free zinc ion concentrations equivalent to those produced by 0.7–1.4 μM zinc in the presence of pyrithione (Fig. 4A). Consistently, western blot analysis demonstrated equivalent reductions in acetylation levels under both experimental conditions (Fig. 4B). The results also demonstrate a positive correlation between cytoplasmic zinc ion levels and the transcriptional suppression of Bmp4 (Fig. S4, to Fig. 2A). These results clearly established that the reduction in histone acetylation levels is caused by abnormal intracellular zinc ions in the cytoplasm, rather than extracellular zinc with high concentrations in culture medium through zinc-associated membrane proteins.

Elevated cytoplasmic zinc levels reduce histone acetylation. (A) The distribution of free zinc ions in HL-1 with or without pyrithione after 4 h of treatment with zinc. Pyrithione, zinc ionophores, 20 µM. HL-1 cells labeled with 5 µM Zinpyr-1 for 0.5 h at 37 °C. Scale bar, 50 µm (B) Immunoblotting analyses of H3K9ac with or without pyrithione after 4 h of treatment with zinc. All data are presented as means ± SD. *p < 0.05; **p < 0.01 versus relevant control.
The aberrant response of zinc transporters in mouse myocardial cells increases their susceptibility to zinc toxicity
Unlike HL-1 cells, AC16 cells showed no change in H3K9ac with treatment of zinc (Fig. 5A). We hypothesized that the insensitivity of AC16 cells to zinc might be due to zinc ions’ inability to enter the cytoplasm. The results demonstrated that even when the extracellular zinc concentration reached 70 μM, there was no significant increase in zinc ions within the cytoplasm of AC16 cells (Fig. 5B). However, with zinc ionophores, even low zinc levels significantly decrease H3K9ac in AC16 cells (Fig. 5C). This suggests that zinc-induced reduction in histone acetylation levels may be a widespread phenomenon. For some cell types, zinc fails to exert cytotoxicity simply because it cannot enter the cytoplasm in large quantities. Under normal physiological conditions, the influx and efflux of zinc are tightly regulated by the Zrt- and Irt-like proteins (ZIP) family and the zinc transporter (ZnT) family, respectively2. They work in coordination to maintain cellular zinc homeostasis. We then investigated the changes in transcriptional levels of ZIPs (Fig. 5D) and ZnTs (Fig. 5E) in both HL-1 cells and AC16 cells under high zinc concentration. In human cardiomyocyte AC16 cells, the zinc influx transporter ZIPs were generally downregulated, with the ZIP2 and ZIP4 showing particularly significant decreases. However, in mouse cardiomyocytes, most ZIPs were downregulated, but interestingly, ZIP1, ZIP2, and ZIP5 were upregulated instead. This indicates that the response of zinc transporters in mouse cardiomyocytes to high extracellular zinc concentrations is aberrant, which would lead to increased zinc influx into the cytoplasm and disrupt zinc homeostasis.Regarding zinc efflux proteins (ZNTs), the transcriptional levels of ZnT1, ZnT2, and ZnT10 were upregulated in both cell types. The upregulation of ZnT2 in AC16 cells was especially pronounced, reaching approximately 12-fold compared to the control. Therefore, in human cardiomyocytes, the coordinated regulation of zinc transporters is effective, enabling the maintenance of intracellular zinc homeostasis and protecting cells from the toxicity of zinc overload.

Efficient regulation of zinc transporters protects cells from toxicity. (A) AC16 cells treated with ZnSO4 alone. 4 h. (B) The distribution of free zinc ions in AC16 cells after treatment with ZnSO4. AC16 cells labeled with 5 µM Zinpyr-1 for 0.5 h at 37 °C. Scale bar, 20 µm. (C) AC16 cells treated with ZnSO4 in the presence of pyrithione. Pyrithione, 20 µM.Transcriptional changes of (D) ZIPs and (E)ZnTs in AC16 and HL-1 cells after 4 h treatment with 70 μM ZnCl₂. All data are presented as means ± SD. *p < 0.05; **p < 0.01; ***p < 0.001 versus relevant control.
Zinc reduces histone acetylation levels by downregulating the transcription of acetyltransferases
HATs and HDACs exert opposing effects and work together to maintain dynamic equilibrium in histone acetylation. After treating HL-1 with ZnSO4, we found no significant change in mRNA levels for 11 HDACs (Fig. 6A). However, for acetyltransferases (Kat2a, Kat2b, Kat6a, Kat7, and Kat14) capable of producing H3K9ac, their transcription were all downregulated (Fig. 6B). Furthermore, we ruled out the possibility that the sulfate group influences HATs gene transcription (Fig. S5). Immunoblotting confirmed that Kat2a expression was suppressed by zinc treatment (Fig. 6C). Thus, we proposed that zinc disrupts the previously maintained balance in histone acetylation levels through downregulating HATs.

Zinc reduces histone acetylation by inhibiting the expression of HATs rather than HDACs. mRNA expressions of (A) 11 HDACs and (B) 5 HATs under treatment of ZnSO4. 4 h, N = 3. (C) Immunoblotting analyses of Kat2a under treatment of ZnSO4. 4 h, N = 3. All data are presented as means ± SD. *p < 0.05; **p < 0.01; ***p < 0.001 versus relevant control.
Zinc-induced reduction in histone acetylation is ubiquitous and sufficient to induce autophagy
As mentioned above, the zinc-induced decrease in acetylation may be a widespread phenomenon. This was validated in human placental trophoblast cell line TEV-1 and human kidney epithelial cell line HEK-293 (Fig. 7). We then examined three different cancer cell lines from distinct organs-human gastric adenocarcinoma cells (AGS), human endometrial carcinoma cells (Ishikawa), and human ovarian carcinoma cells (A2780)-and found that all three cell lines exhibited significant H3K9ac reduction (Fig. 7), although lower zinc concentrations failed to induce noticeable decreases. It should also be noted that even at relatively low concentrations (1.4 μM), zinc causes a significant reduction in acetylation levels in tumor cell line AGS compared to normal cells, indicating that zinc exhibits a certain selectivity toward human gastric adenocarcinoma cells.

Zinc homeostasis disruption leads to decreased histone acetylation in both normal and tumor cells. Normal cell lines and tumor cell lines treated with ZnSO4 in the presence of pyrithione. Pyrithione, 20 µM. All data are presented as means ± SD. *p < 0.05; **p < 0.01; ***p < 0.001 versus relevant control.
Some studies indicate that zinc can induce cellular autophagy32,33, while enhanced autophagy has been linked to reduced activity of the acetyltransferase EP30034. Therefore, we hypothesized that the overall decrease in acetylation levels triggered by zinc is responsible for inducing autophagy. In our experiments, we found that 1.4 μM zinc, in combination with a zinc ionophore, induced significantly enhanced autophagy in HL-1 cells (Fig. 8A). However, this autophagic response was markedly alleviated upon the addition of a deacetylase inhibitor. Subsequently, we selected the zinc-sensitive tumor cell line AGS and treated it with a gradient of zinc concentrations to investigate zinc’s role in autophagy. Interestingly, when zinc.

Zinc modulates cellular autophagy via acetylation. (A) Zinc reduces acetylation to regulate autophagy in HL-1 cells. ZnCl2, 4 h. (B) Effect of varying zinc concentrations on autophagy levels in AGS cells. ZnCl2, 4 h. (C) The diagram shows a mechanism for the transcription inhibition by zinc in cardiomyocytes. (+) stimulation; (−) inhibition. All data are presented as means ± SD. *p < 0.05; **p < 0.01; ***p < 0.001 versus relevant control.
exceeded a certain threshold, autophagy became progressively weaker (Fig. 8B). This demonstrates that, in addition to being regulated by acetylation, other zinc-induced alterations can also modulate autophagy.
Discussion
In this study, we discovered that excess zinc downregulates Bmp4 by reducing histone acetylation levels in HL-1 cells. We further demonstrated that this decrease in histone acetylation is directly caused by disrupted cytoplasmic zinc homeostasis. Mechanistically, zinc was found to suppress histone acetyltransferases (HATs), thereby lowering acetylation levels (Fig. 8C). Finally, we validated the broad relevance of this phenomenon by observing similar effects in other cell types, including tumor cells. For the first time, our study uncovering the link between zinc and epigenetic. Our study provides a theoretical framework for understanding diseases associated with zinc dyshomeostasis, while also offering novel therapeutic potential for malignant cellular disorders.
In our study, the transcription of Bmp4 in HL-1 cells initially decreased and then gradually recovered during 36 h of ZnSO4 treatment. This process is highly consistent with the reversibility of histone acetylation. We speculate that this reversibility may be attributed to the coordinated action of zinc transporters. For humans, the concentration of zinc in peripheral blood (ranging from 10.71 to 18.36 μM) shows a significant difference compared to cytoplasmic levels (typically in the range of pM to nM)2,35. Multiple zinc transporters work in concert to maintain zinc homeostasis. Once zinc transport is impaired, elevated cytoplasmic zinc will reduce histone acetylation levels, leading to downregulation of critical proteins such as Bmp4 and even oncogenes36. For example, if the ZnT2 zinc transporter, which was upregulated 11-fold in this experiment, is knocked down or inhibited, zinc-induced cytotoxicity would be significantly enhanced. In addition, our study reveals that overloaded zinc is able to downregulate the protein levels of acetyltransferases. Notably, acetyltransferases target not only histones but also non-histone proteins, which govern critical biological processes21,22,23. Thus, disruption of zinc homeostasis exerts widespread and profound impacts on living organisms. Under normal physiological conditions, the efficient functioning of zinc transporters is essential for maintaining cellular growth. Based on this, we speculate that zinc transporters are promising novel drug targets for treating hyperplastic diseases, with particular efficacy against neoplastic cells.
Moderate induction of autophagy effectively removes aged organelles such as mitochondria and misfolded proteins from cells and plays a crucial role during the progression of cardiac hypertrophy and heart failure37. Furthermore, the suppression of autophagy in tumor cells has been established as an effective therapeutic strategy38. Our study proposes that zinc induces autophagy through acetylation. Moreover, the level of autophagy can be modulated by adjusting the dosage of zinc. This suggests that zinc can target various types of diseases through autophagy, indicating its significant medical implications. While our study is the first to report the phenomenon of zinc-regulated acetylation, its underlying pathways require more detailed investigation. Furthermore, the current research is confined to in vitro experiments. We anticipate that this regulatory mechanism will exhibit more intriguing aspects within the complex multicellular environment of an animal model.
Data availability
The original contributions presented in the study are included in the article/**Supplementary Information**, further inquiries can be directed to the corresponding author.
References
Prasad, A. S., Halsted, J. A. & Nadimi, M. Syndrome of iron deficiency anemia, hepatosplenomegaly, hypogonadism, dwarfism and geophagia. Am J Med 31, 532–546. https://doi.org/10.1016/0002-9343(61)90137-1 (1961).
Kambe, T. et al. The physiological, biochemical, and molecular roles of zinc transporters in zinc homeostasis and metabolism. Physiol. Rev. 95(3), 749–784. https://doi.org/10.1152/physrev.00035.2014 (2015).
Andreini, C. et al. Counting the zinc-proteins encoded in the human genome. J. Proteome. Res. 5(1), 196–201. https://doi.org/10.1021/pr050361j (2006).
Chen, L., Min, J. & Wang, F. Copper homeostasis and cuproptosis in health and disease. Signal Transduct. Target Ther. 7(1), 378. https://doi.org/10.1038/s41392-022-01229-y (2022).
Zhang, M. et al. Calcium-overload-mediated tumor therapy by calcium peroxide nanoparticles. Chem 5(8), 2171–2182. https://doi.org/10.1016/j.chempr.2019.06.003 (2019).
Du, Y. et al. Calcium influx-induced lytic cell death disrupts skin immune homeostasis. Cell Discov. 9(1), 124. https://doi.org/10.1038/s41421-023-00623-2 (2023).
Rahimzadeh, M. R. et al. Zinc Poisoning - symptoms, causes. Treatments. Mini Rev. Med. Chem. 20(15), 1489–1498. https://doi.org/10.2174/1389557520666200414161944 (2020).
Mann, J. J. & Fraker, P. J. Zinc pyrithione induces apoptosis and increases expression of Bim. Apoptosis 10(2), 369–379. https://doi.org/10.1007/s10495-005-0811-9 (2005).
Rudolf, E. & Cervinka, M. Stress responses of human dermal fibroblasts exposed to zinc pyrithione. Toxicol. Lett. 204(2–3), 164–173. https://doi.org/10.1016/j.toxlet.2011.04.028 (2011).
Mo, J. et al. Apoptosis in HepG2 cells induced by zinc pyrithione via mitochondrial dysfunction pathway: Involvement of zinc accumulation and oxidative stress. Ecotoxicol. Environ. Saf. 161, 515–525. https://doi.org/10.1016/j.ecoenv.2018.06.026 (2018).
Su, X. et al. Disruption of zinc homeostasis by a novel platinum(IV)-terthiophene complex for antitumor immunity. Angewandte Chem. Int. Ed. 62(8), e202216917. https://doi.org/10.1002/anie.202216917 (2023).
Zhang, H. et al. Pyrithione zinc alters mismatch repair to trigger tumor immunogenicity. Oncogene 44(14), 983–995. https://doi.org/10.1038/s41388-024-03272-1 (2025).
Shvedunova, M. & Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell. Biol. 23(5), 329–349. https://doi.org/10.1038/s41580-021-00441-y (2022).
Tropberger, P. & Schneider, R. Going global: novel histone modifications in the globular domain of H3. Epigenetics 5(2), 112–117. https://doi.org/10.4161/epi.5.2.11075 (2010).
Tse, C. et al. Disruption of higher-order folding by core histone acetylation dramatically enhances transcription of nucleosomal arrays by RNA polymerase III. Mol. Cell Biol. 18(8), 4629–4638. https://doi.org/10.1128/mcb.18.8.4629 (1998).
Haberland, M., Montgomery, R. L. & Olson, E. N. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet. 10(1), 32–42. https://doi.org/10.1038/nrg2485 (2009).
Struhl, K. Histone acetylation and transcriptional regulatory mechanisms. Genes Dev. 12(5), 599–606. https://doi.org/10.1101/gad.12.5.599 (1998).
Koprinarova, M., Schnekenburger, M. & Diederich, M. Role of histone acetylation in cell cycle regulation. Curr. Top. Med. Chem. 16(7), 732–744. https://doi.org/10.2174/1568026615666150825140822 (2016).
Charidemou, E. & Kirmizis, A. A two-way relationship between histone acetylation and metabolism. Trends Biochem. Sci. 49(12), 1046–1062. https://doi.org/10.1016/j.tibs.2024.10.005 (2024).
Fraga, M. F. et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 37(4), 391–400. https://doi.org/10.1038/ng1531 (2005).
Kim, S. C. et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23(4), 607–618. https://doi.org/10.1016/j.molcel.2006.06.026 (2006).
Choudhary, C. et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 325(5942), 834–840. https://doi.org/10.1126/science.1175371 (2009).
Weinert, B.T. et al. Proteome-wide mapping of the Drosophila acetylome demonstrates a high degree of conservation of lysine acetylation. Sci. Signal. 4(183): ra48.https://doi.org/10.1126/scisignal.2001902 (2011).
Lescroart, F. et al. Defining the earliest step of cardiovascular lineage segregation by single-cell RNA-seq. Science 359(6380), 1177–1181. https://doi.org/10.1126/science.aao4174 (2018).
Ieda, M. et al. Direct reprogramming of fibroblasts into functional cardiomyocytes by defined factors. Cell 142(3), 375–386. https://doi.org/10.1016/j.cell.2010.07.002 (2010).
Schultheiss, T. M., Burch, J. B. & Lassar, A. B. A role for bone morphogenetic proteins in the induction of cardiac myogenesis. Genes Dev. 11(4), 451–462. https://doi.org/10.1101/gad.11.4.451 (1997).
Montgomery, R. L. et al. Histone deacetylases 1 and 2 redundantly regulate cardiac morphogenesis, growth, and contractility. Genes Dev. 21(14), 1790–1802. https://doi.org/10.1101/gad.1563807 (2007).
Trivedi, C. M. et al. Hdac2 regulates the cardiac hypertrophic response by modulating Gsk3 beta activity. Nat. Med. 13(3), 324–331. https://doi.org/10.1038/nm1552 (2007).
Zhang, C. L. et al. Class II histone deacetylases act as signal-responsive repressors of cardiac hypertrophy. Cell 110(4), 479–488. https://doi.org/10.1016/s0092-8674(02)00861-9 (2002).
Montgomery, R. L. et al. Maintenance of cardiac energy metabolism by histone deacetylase 3 in mice. J. Clin. Invest. 118(11), 3588–3597. https://doi.org/10.1172/jci35847 (2008).
Burdette, S. C. et al. Fluorescent sensors for Zn(2+) based on a fluorescein platform: synthesis, properties and intracellular distribution. J. Am. Chem. Soc. 123(32), 7831–7841. https://doi.org/10.1021/ja010059l (2001).
Liuzzi, J. P. & Yoo, C. Role of zinc in the regulation of autophagy during ethanol exposure in human hepatoma cells. Biol. Trace. Elem. Res. 156(1–3), 350–356. https://doi.org/10.1007/s12011-013-9816-3 (2013).
Hung, H. H., Huang, W. P. & Pan, C. Y. Dopamine- and zinc-induced autophagosome formation facilitates PC12 cell survival. Cell Biol. Toxicol. 29(6), 415–429. https://doi.org/10.1007/s10565-013-9261-2 (2013).
Mariño, G. et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol. Cell 53(5), 710–725. https://doi.org/10.1016/j.molcel.2014.01.016 (2014).
Ruktanonchai, D. et al. Zinc deficiency-associated dermatitis in infants during a nationwide shortage of injectable zinc - Washington, DC, and Houston, Texas, 2012–2013. MMWR Morb. Mortal. Wkly. Rep. 63(2), 35–37 (2014).
Li, Y. & E. Seto, HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb Perspect Med 6(10).https://doi.org/10.1101/cshperspect.a026831 (2016).
Sciarretta, S. et al. The role of autophagy in the heart. Annu. Rev. Physiol. 80, 1–26. https://doi.org/10.1146/annurev-physiol-021317-121427 (2018).
Pimentel, J. M., Zhou, J. Y. & Wu, G. S. Autophagy and cancer therapy. Cancer Lett. 605, 17285. https://doi.org/10.1016/j.canlet.2024.217285 (2024).
Acknowledgements
This research was supported by the National Nature Science Foundation of China (NO. 82270309).
Funding
National Nature Science Foundation of China (NO. 82270309).
Author information
Authors and Affiliations
Contributions
Shu Xu, Chao Tang, and Weize Xu designed the experiments. Shu Xu conducted the experiments and wrote the manuscript. Yuzhuang Hu provided discussions for the experiment.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethical approval
Animal experiments were approved (approval no. 21045) by the Animal Ethics Committee of Zhejiang University (Zhejiang, China) and performed according to the Guide for the Care and Use of Laboratory Animals (NIH Publication no. 85‑23, revised 1996).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Xu, S., Hu, Y., Tang, C. et al. Disruption of zinc homeostasis reduces histone acetylation levels in normal and tumor cells. Sci Rep 16, 4983 (2026). https://doi.org/10.1038/s41598-026-35270-6
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-35270-6
