
Abstract
Multiplexed immunofluorescence (mIF) imaging has revolutionized the study of cellular interactions within tissue microenvironments, enabling complex pattern analysis critical to understanding disease biology. However, current analytical methods assume uniform cellular patterns across tissues, overlooking the spatial heterogeneity that characterizes tumor microenvironments. Here, we introduce ISPat (Informed Spatially aware Patterns), a fully Bayesian framework that identifies both shared and region-specific interaction patterns while integrating domain knowledge to enhance spatial pattern estimation. ISPat models spatial cellular densities through kernel density estimation, then constructs interaction networks from precision matrices that capture conditional dependencies between cell types while controlling for confounding effects. The resulting networks reveal direct cellular relationships, with non-zero precision matrix entries indicating significant interactions. We applied ISPat to analyze 119 pancreatic ductal adenocarcinoma (PDAC) and 53 intraductal papillary mucinous neoplasm (IPMN) patients, partitioning tissues into five regions based on tumor intensity gradients. Our analysis revealed fundamentally distinct immune architectures: PDAC maintains a rigid, stable immunosuppressive microenvironment across tumor heterogeneity gradients, whereas IPMN exhibits dynamic spatial remodeling with marked regional variability. Critically, we identified multiple ligand-receptor (LR) interactions that consistently differ between disease conditions specifically in intermediate tumor intensity regions, while extreme conditions showed no significant differences. These include interactions spanning multiple functional axes of anti-tumor immunity: antigen presentation and T cell activation (APC\(\leftrightarrow\)CTL, THelper\(\leftrightarrow\)APC, Epithelial\(\leftrightarrow\)APC), effector function and tumor cell killing (Epithelial\(\leftrightarrow\)CTL, CTL\(\leftrightarrow\)Treg), and immune regulation (Epithelial\(\leftrightarrow\)Treg, Treg\(\leftrightarrow\)APC, THelper\(\leftrightarrow\)Treg). Notably, the APC\(\leftrightarrow\)CTL interaction, fundamental for adaptive immunity activation, differs significantly in high tumor density regions, alongside Epithelial\(\leftrightarrow\)CTL interactions critical for direct tumor elimination. These spatially resolved signatures provide quantitative evidence for distinct immune evasion mechanisms and represent promising biomarker candidates for disease classification and risk stratification. Through simulation studies, we demonstrated ISPat’s accuracy in pattern recovery and its computational efficiency, achieving 8-10 fold speedup over comparable methods through variational Bayesian inference. The framework exhibits robust scalability and handles naturally occurring partition size imbalances, making it well-suited for analyzing heterogeneous tissues. Our findings demonstrate that spatial context fundamentally shapes cellular interactions in pancreatic cancer, with critical implications for understanding immune evasion mechanisms and developing spatially informed therapeutic strategies. The identification of differential interactions across antigen presentation, effector function, and immune regulation pathways suggests that therapeutic interventions must address multiple axes of immune dysfunction rather than single targets. ISPat provides a generalizable framework for spatial analysis applicable to emerging technologies, enabling precision oncology approaches guided by spatially resolved biomarkers.
Similar content being viewed by others

Introduction
Pancreatic cancer is among the most prevalent gastrointestinal (GI) malignancies in the United States, with the lowest five-year relative survival rate among major cancers1. Among the most lethal malignancies, pancreatic ductal adenocarcinoma (PDAC) accounts for majority of all pancreatic cancers2. This aggressive tumor type arises from the pancreatic ductal epithelium and remains difficult to treat3,4,5. PDAC treatment remains particularly challenging due to three key factors: the pancreas’s inaccessible anatomical position, the tumor’s rapid metastatic potential, and its typically silent early progression6. Despite medical advances across oncology, PDAC prognosis remains poor as incidence rates continue climbing7. However, not all pancreatic lesions share same prognosis for example, intraductal papillary mucinous neoplasms (IPMNs) accounting for smaller number of all pancreatic cancers, demonstrate substantially better outcomes when appropriately managed8. Therefore comparing these two entities, invasive PDAC and precancerous IPMN can characterize immune dysfunction differences across the pancreatic cancer progression spectrum.
PDAC exhibits profound immune dysfunction through coordinated suppression across multiple cellular compartments. Regulatory T cells (Tregs) are central to this immunosuppressive network, accumulating within the TME to foster immune tolerance9 and counteract the protective effects of cytotoxic CD8+ T lymphocytes (CTLs). PDAC’s characteristic desmoplastic stroma compounds this dysfunction by creating physical barriers to CTL infiltration while establishing inhibitory signaling pathways that promote T-cell exhaustion10,11. Consequently, PDAC tumors exhibit sparse or functionally exhausted CTL populations. Antigen presenting cells (APCs) are similarly compromised, with dendritic cells and tumor-associated macrophages frequently depleted or adopting tolerogenic, M2-polarized phenotypes that undermine effective T-cell priming12. CD4+ helper T-cell responses are also skewed, favoring Th2 over Th1 polarization, a shift associated with disease progression and poor prognosis13,14. This multilayered immunosuppressive architecture surrounding suppressive Tregs, constrained CTLs, dysfunctional APCs, and skewed CD4+ responses establishes PDAC as a highly immunoresistant malignancy. This dysfunction underlies the limited efficacy of current immunotherapies and necessitates combinatorial strategies to restore antigen presentation, promote Th1 polarization, and relieve CD8+ suppression11. However, IPMNs initially exhibit enhanced immune surveillance, but as they progress toward malignancy they converge on the same immunosuppressive features seen in PDAC, including Treg accumulation, CTL dysfunction, and impaired antigen presentation, thereby providing a natural point of comparison between precancerous and malignant states15,16.
Recent advances in Multiplexed Immunofluorescence (mIF), enhanced our understanding of cellular patterns in tissues under both healthy and diseased conditions. mIF allow simultaneous visualization of multiple cell types within their native spatial context17. mIF provided us with a premise for detailed study of the Tumor Microenvironment (TME). A TME is an elaborate ecosystem composed of various cells and vascular connected systems such as stromal cells, blood vessels, and extracellular components18,19,20,21. TME poses significant importance in cancer research due to its major influence on tumorigenesis22. Tumor heterogeneity is an area of cancer research that specifically investigates the inter- and intra-tumor differences for assessment and monitor disease development and progression. Rapid increase of imaging technologies, allowed us to evaluate tumor heterogeneity on multiple levels. It is among key drivers for development, progression and therapy responses23,24,25.
Spatially enabled methods in cancer biology helps in understanding heterogeneity across intratumoral, cancerous sites and interplay between different type of cells in the ecosystem under consideration26. Understanding how cells communicate across space has become paramount in this field, as these intercellular signaling networks hold the keys to unraveling disease mechanisms27,28. Networks or patterns are utilized in various fields as a medium for better understanding of inter-dependencies among variables29. Studying cell-networks or patterns can help researchers interpret cellular function, regulation, and organization within different tissue zones30,31. Current methods mostly focus on cell co-localization or nearest-neighbor analysis32,33,34. Though informative, majority of these approaches lack the ability to simultaneously capture complex, multivariate dependencies among cells \((>1)\). While modeling direct relationships among variables controlling for confounding effects, Network-based methods from systems biology offer an alternating route31.
Studying cellular co-localization patterns can identify novel ligand receptor (LR) interactions35. Spatial co-expression studies must overcome the computational challenge of partitioning cellular interactions into those with broad tissue relevance and those with region-specific significance. Current computational approaches often reduce data dimensions to manage complexity, but may overlook crucial spatial dependencies36,37. Previous computational methods in this domain focused on network visualization and ligand-receptor interactions38. However, existing methods are computationally challenging and typically assume spatial homogeneity, limiting their scope in capturing region-specific interaction patterns that may be critical for understanding tumor biology39,40. Studies found that the extraction of characterizing features from biomedical imaging often captures physiological and morphological heterogeneity of tumors41,42. Such studies are pivotal in designing precision medicine i.e., construction of personalized therapeutic strategies for cancer, enabling guided monitoring of disease development or progression. Spatial arrangement of cells within cancerous tissues are increasingly acknowledged as a key prognostic element in physiologically different tumor types43,44,45. Similarly gene networks facilitate the identification of gene regulatory communities and pathways associated with disease46,47.
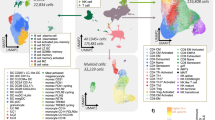
To address these limitations, and our interest in studying tumor heterogeneity, we developed ISPat (Informed Spatially Aware Patterns), a Bayesian statistical framework designed to (i) construct patterns that incorporate both shared and region-specific components across spatial domains consolidating tumor heterogeneity and (ii) additionally incorporate domain knowledge into pattern construction if available. ISPat incorporates spatial dependencies, allowing for a more nuanced exploration of cellular localization, variation across different spatial regions in a tissue. Our method first estimates continuous intensity functions for each cell type for overall tissue, then partitions tissue into biologically relevant areas and constructs shared and region specific interaction networks. For each image, we use spatial locations of each cell phenotype to understand their individual distribution in the image by generating intensity maps. Factors such as the choice of bandwidth in kernel methods significantly impact accuracy of density estimation, thus a cross-validated bandwidth selection was applied. We examined conditional dependencies across cell types and scrutinize their relationships accounting for all cells in the TME. We generate intensity maps of tumorous areas in all images for each group of patients (PDAC and IPMN) for further analyses. ISPat’s performance was evaluated through rigorous simulation studies designed to test pattern accuracy. To identify hub-genes or cells that are consistently conserved, spatially distinct, potentially offering new insights into differential gene or cellular activities and therapeutic pathways across various tissue sub-regions. The capacity to integrate domain knowledge with spatially concurrent data makes ISPat a valuable tool for advancing our understanding of complex biological patterns in health and diseased tissues.
Methods
Study design and patient samples
We conducted a retrospective analysis of tissue samples from patients treated at the University of Michigan Pancreatic Cancer Clinic under institutional review board approval (HUM00045286). We included 119 PDAC patients and 53 IPMN patients who underwent surgical resection between 2015 and 2020. Sample size was determined based on tissue availability and previous studies demonstrating adequate power for spatial analysis with similar cohort sizes48,49. Normal pancreatic tissues were excluded due to lack of clinical indication for biopsy. All samples were formalin-fixed, paraffin-embedded (FFPE) and processed into tissue microarrays (TMAs).
Multiplexed immunofluorescence imaging
Tissue preparation and multiplexed immunofluorescence
We analyzed surgically resected PDAC and IPMN tissue samples following pathological review. Three 0.6 mm diameter cores were extracted from each tissue block and assembled into TMAs. Five-micrometer sections were cut onto charged slides and processed using an Opal 7 manual kit (Akoya Biosciences) following established multiplexed immunofluorescence protocols50. Tissue sections were sequentially stained with primary antibodies against CD3 (Agilent Dako AO452), CD8 (M5390), FOXP3 (Cell Signaling Technology 12653), CD163 (NCL-L-CD163), PD-L1 (Cell Signaling Technology 13684), and pancytokeratin (M3515), followed by Opal Polymer secondary antibodies and Tyramide Signal Amplification. DAPI was used for nuclear counterstaining.
Image acquisition and analysis
Multispectral images were captured using the Vectra Polaris system (Akoya Biosciences) at 20x magnification with standardized 250 ms exposure times across all spectral channels (DAPI, CY3, CY5, CY7, Texas Red, Qdot). Automated cell segmentation and phenotyping were performed using Vectra InForm software version 2.4.8 with standardized protocols. Five primary cell populations were identified based on marker expression: epithelial cells (\(cytokeratin^+\)), helper T cells (helperTs; \(CD3^+CD8^-FOXP3^-\)), cytotoxic T lymphocytes (CTLs; \(CD3^+CD8^+\)), regulatory T cells (Tregs; \(CD3^+FOXP3^+\)), and antigen-presenting cells (APCs; \(CD163^+\)). For PDAC samples, cytokeratin-positive cells represented tumorous epithelium, while in IPMN samples, they indicated neoplastic epithelial cells. All multiplex images underwent validation review by an experienced gastrointestinal pathologist to confirm staining accuracy and phenotyping reliability.
In the analysis, spatial cell distributions on pathology slides are represented as point patterns on a 2-dimensional grid derived from staining. To achieve a smoother representation, spatial intensity functions for each cellular phenotype were estimated using a two-dimensional isotropic Gaussian kernel density estimator, providing a continuous probabilistic representation of cell density across the tissue microenvironment. Cellular spatial coordinates were utilized as input point patterns for the kernel density estimation, with computational grid resolution calibrated to match the original image pixel spacing. The derived marginal intensity functions provided continuous representations of cell density for subsequent statistical analyses. Implementation of our image processing step required partitioning individual intensities into blocks and a pixel grid with pixels of size \(70\times 70\, \upmu\) m was chosen. We finally furnish (i) most abundant LR interactions across tissue images, keeping in mind that metabolic differences across patients can impart an impact in PDAC or IPMN growth and (ii) significantly different LR interactions observed in different areas of tissue between patient groups.

Spatial organization of a PDAC tissue sample. (a) Point pattern representation of different cell types within the tumor microenvironment. (b) Identification of distinct tumor subregions based on underlying cellular heterogeneity, where each colored region represents a different cellular composition profile.
Statistical methodology
Spatial density estimation
For each tissue image \(\mathcal {I}\), we represented cell locations as point patterns in 2-D space. The marginal intensity functions computed from pixel values within image grids of a particular patient’s image was estimated through marginal intensity functions for cell c. We select grid dimensions to ensure that they are proportional to image dimensions in pixels. Let, for cell type c, \(s_j^c\) be the j’th data point, \(\mathcal {K}_{h}\) as Gaussian smoothing kernel with bandwidth h and \(e(s_j^c)\) as edge correction factor accounting boundary effects. The intensity estimate at a point \(\varvec{s}_j\) on the grid was therefore given by, \(\hat{f^c}(s) = \sum _{s_j} \mathcal {K}_{h}(s-s_j^c) e(s_j^c)\). Each computed intensities are in need of correction for edge effect bias by dividing it with correction term \(\frac{1}{e(s_j^c)} = \int _{W} k(s_j^c-v)dv\) i.e, the convolution of Gaussian kernel with the observation window W. We optimized bandwidth h separately for each cell type using leave-one-out cross-validation to account for differences in spatial clustering behavior between cell types. Once we individually obtained these marginal intensities, they are partitioned into non overlapping regions \(\mathcal {I}_q\)’s, spanned over q tumor regions, i.e we have c many cell types for a total of \(N = \sum _{q=1}^Q N_{q}\) many spatial locations, partitioned into Q spatially contiguous but heterogeneous partitions \(\mathcal {I}_q\). These partitions for used Pancreatic Cancer-mIF data are predominantly tumor specific. We define these partitions specific functional estimands as -
The first part of Eq. (1) states that for a cell type c, overall intensity \(f^{c}\), the union of partitioned intensities in each of regions \(\mathcal {I}_q\) in the image \(\mathcal {I}\) are non-overlapping, thus each point s in the space belongs to exactly one region \(\mathcal {I}_q\), ensuring that the indicator function \(\varsigma ^{\mathcal {I}_q}\) properly represents partitioning. However, second part of Eq. (1) deals with these marginal intensity functions belonging in the overall non-linear functional space \(\mathcal {F}\), defined as a comprehensive functional space of valid intensities, each intensity was subject to the condition that these are realizations of the same variable and remain non-negative over spatial domain.
Tissue region definition
Different tumor areas were obtained using quantiles from tumor intensity as shown in Fig. 1b. We partitioned each tissue image into five regions based on tumor cell density quantiles, representing distributional heterogeneity : Very High (80–100th percentile: Area 1), High (60–80th percentile: Area 2), Medium (40–60th percentile: Area 3), Low (20–40th percentile: Area 4) and Very Low (0–20th percentile: Area 5). We name these regions as Area 1, \(\ldots\) , Area 5 respectively. This partitioning captures the gradient of tumor cellularity commonly observed in pancreatic cancer specimens. Each region was defined as spatially contiguous areas with similar tumor density, creating biologically interpretable spatial domains for analysis. Subsequently we focus ourselves with (i) the shared cell-patterns and (ii) individual sub-region specific cell-patterns, (iii) dependent on the prior knowledge of cell-patterns, we could seamlessly incorporate a domain informed pattern into our sub-region specific cell-patterns. In the next few Sections, we explain how we dig deeper into these individual patterns pertaining to such image partitions as well as estimating a shared pattern.
Network construction using precision matrices
We characterize the spatial co-localization patterns of multiple cell types by modeling their joint intensity distribution across q regions of interest (ROIs) using a multivariate Gaussian framework. For each ROI, the joint probability distribution of cell-type densities are as follows:
where j being the number of spatial locations; \(j=1,\ldots , N_q\), i.e individually each of these regions contain \(N_q\) many spatial locations. The marginal intensity function is given by \(f^{c}_{q}(\varvec{s}_j)\mid \ldots\) for cth cell type in the qth partition and the precision matrix is \(\varvec{\Omega }_q = {\bf \Sigma }^{-1}_{q}\). Here non-zero elements of the precision matrix \(\varvec{\Omega }_q^{[i,j]}\) indicate the relationship between cell types i and j. The next part of this mechanism, i.e., the gaussian regression part, where for an image \(\mathcal {I}\) we assume the following model, the standardized marginal intensity functionals \(f^{c}_{q}(\varvec{s}_j) \mid \ldots\) are modeled by a Gaussian process prior (\(\mathcal{G}\mathcal{P}\)). The \(\mathcal{G}\mathcal{P}\) provides flexibility in modeling the spatial structure. \(\mathcal{G}\mathcal{P}\) offers a flexible nonlinear model; works for large-dimensional data as well; but we must select a parametric covariance kernel, i.e., \(\mathcal {K}_{\mathcal {I}}(\varvec{s}_{j},\varvec{s}_{j'})\) which represents a kernel function. Some examples of such kernels are Radial basis function kernel or a Matèrn kernel. We formulate a hierarchical Gaussian process model where the spatial field comprises both fixed effects, represented by the intercept term, and random effects that capture spatial correlation structure. This hierarchical \(\mathcal{G}\mathcal{P}\) framework allows us to model fixed effects for global intensity trends across regions and random effects as spatially correlated \(\mathcal{G}\mathcal{P}\)s, capturing local heterogeneity. The model is:
Prior distributions for the model parameters are specified as follows: the fixed effect intercept parameter across all spatial locations is \(\beta \sim \mathcal {N}(0,1)\), the random effect \(\xi ^{c}_{q}(\varvec{s})\) is a spatially correlated Gaussian process with a predefined covariance structure specific to the cth cell type in the qth region. The \(\xi ^{c}_{q}(\varvec{s})\) captures the spatially correlated structures. The covariance structure of this random effect is specified by a chosen kernel function (RBF or Matérn), which defines how spatial intensities are correlated across space. For example, if we use the RBF kernel, we would have \(\mathcal {K}_{\mathcal {I}_q}(\varvec{s}_{j},\varvec{s}_{j'}) = \sigma _{\mathcal {I}_q}^2 exp\{{ \Vert s_{j} - s_{j'}\Vert ^2}/{2l_{\mathcal {I}_q}^2} \}\). Here we assume that \(l_{\mathcal {I}_q}\) as the 2D isotropic length scale parameter of the spatial kernel. To select a valid length scale, we first compute the pairwise Euclidean distances between the spatial locations for each ROI. These distances are then transformed to the logarithmic scale to accommodate the wide range of values in the image. We choose the middle value from this logarithmic sequence as the length scale parameter. This choice is reasonable because it balances both short-range and long-range spatial dependencies, providing a representative scale for the spatial correlation. The spatial process scale \(\sigma _{\mathcal {I}_q} \sim \mathcal {C}^{+}(0,1)\) and the nuggets, encoding noise \(\sigma _{\varepsilon } \sim \mathcal {C}^{+}(0,1)\).
To this end, we perform these spatial \(\mathcal{G}\mathcal{P}\)-regressions parallelly for individual cell types with a fixed effect for cells and a random effect pertaining to individual regions such a way that incorporates conditional independence structure. This conditional independence drives this paragraph of discussion about Gaussian Markov random fields (\(\mathcal {GMRF}\))51,52. The spatial coordinates were discrete valued. \(\mathcal{G}\mathcal{P}\)-assumes that spatially nearby points have similar observation values. \(\mathcal{G}\mathcal{P}\)’s are seen as an apparatus that generalizes \(\mathcal {GMRF}\), has (i.) a better capacity to represent smoother functions (ii.) offer better performance. The \(f_q^c\) functional intensities are assumed as an \(\mathcal {GMRF}\) and together they are modeled through hierarchical decomposition approach53,54. This allows us to separate interactions that are consistent across all regions from those that are specific to particular spatial domains. The graph be denoted by \(\mathcal {G}\) where \(\mathcal {G}= (V,E)\) where \(V = \{1,\ldots , C\}\) and the set of edges \(E\in V \times V\). For \(\mathcal {G}\), the nodes represent two types of cells (different types) and the edge would denote the functional overlap between intensities of these two cell types. For each of these image partitions, there exist a graph \(\mathcal {G}_q = (V,E)\) where the V corresponds to the set of vertices and the edges are defined through conditional independence. Formally for \(q^{th}\) partitions, \((c',c'') \notin E \iff f_q^{c'} \perp \!\!\! \perp f_q^{c''} \mid f_q^{\setminus (c',c'')}\) for all \(c' \ne cx2019;'\) and \(c', c'' \in (1, \ldots , C)\). Where \(f_q^{\setminus (c',c'')}\) denotes all cell intensities except that of \(c'\) and \(c''\), moreover \((c',c'') \in E \iff \varvec{\Sigma }_q^{(c'c'')} \ne 0\) for all \(c \ne cx2019;'\), thus we can assume the following multi-study factor analytic model to identify shared and study-specific latent factors driving observed variations:
This decomposes the covariance across cell types (dimension C), incorporating shared and partition-specific patterns, which then yields the precision matrix for graph construction. We therefore discuss the interpretations of the terms we require from model (4). Here, \({\bf \Phi }{\bf F}\) represents shared pattern consisting loading matrix \({\bf \Phi }\). The shared patterns are obtained from \({\bf \Phi }{\bf \Phi }^T\). [\({{\bf F}} \sim \mathcal {N}_{K}({\bf 0}, {\bf I}_{k})\) where K being the number of shared latent factor components, \(D_{qj} \sim \mathcal {N}_{Qs}({\bf 0}, {\bf I}_{Qs})\) are the scores with study specific loading matrices \({\bf \Lambda }_{q}\) and the errors \(\mathbb {E}_{qj} \sim \mathcal {N}({\bf 0}, \varvec{\Psi }_q)\) with \(\varvec{\Psi }_q = diag(\psi _{q1}^2, \ldots , \psi _{qC}^2)\) ]. The covariance matrices of each study section \({\bf \Sigma }_q\) for \(q = 1, \cdots , Q\), are decomposed as \({\bf \Sigma }_q = {\bf \Phi }{\bf \Phi }^T + {\bf \Lambda }_{q} {\bf \Lambda }_{q}^T + \varvec{\Psi }_{q}\). The terms that represent the portion of the covariance matrix, referred to common factors and study-specific factors, respectively are \({\bf \Phi }{\bf \Phi }^T\) and \({\bf \Lambda }_{q} {\bf \Lambda }_{q}^T\).
While adjacency matrices cannot be directly utilized within this methodological framework, we circumvented this limitation through covariance matrix transformations. To estimate an overall covariance pattern, one can utilize the following approach to obtain valid covariance matrix from the adjacency matrix55. A function g(x) of a variable x can be expressed as \((g(x)=c+\sum _{i} g_i(x_i))\) in an additive way56, we thus estimate the covariance utilizing the additive decomposition of additive function of Gaussian processes defined over a particular domain57. To this end, depending on a reference covariance matrix (\({\bf \Sigma }^{*}\)), a reference based \(\mathcal {I}_{q}\) specific pattern of cells can be obtained using with \([{\bf \Sigma }^{*}]^{1/2} {\bf \Lambda }_{q}{{\bf \Lambda }_{q}}^T [{\bf \Sigma }^{*}]^{1/2}\).
Model fitting and statistical inference
ISPat exploits conditional independence for cell type marginal intensities where intensities were partitioned over different domains \(\mathcal {I}_q\), for same image \(\mathcal {I}\). Bayesian model fitting with \(\mathcal{G}\mathcal{P}\) was therefore implemented as these functionals were assumed to be Gaussian. We incorporate spatial information through region specific loadings. For precision matrix estimation, variational bayes algorithms provide computational efficiency while maintaining uncertainty quantification. To this end, we note that the factor loading matrices \([{\bf \Phi }, {\bf \Lambda }_{q}]\) are not unique since there exist rotation problem58. But our object of interest \([{\bf \Phi }{\bf \Phi }^T, {\bf \Lambda }_{q}{{\bf \Lambda }_{q}}^T]\) is always unique. Hence fourth, we could set \(\tilde{{\bf \Lambda }_{q}} = [{\bf \Sigma }^{*}_{q}]^{1/2} {\bf \Lambda }_{q}\) incorporating any partition q specific domain knowledge. ISPat represents a scalable, computationally efficient framework that precisely identifies spatially heterogeneous cellular interaction patterns while maintaining uncertainty quantification.
Simulation study

Simulation study.
To assess whether our method can flexibly learn shared and area-specific patterns across diverse scenarios, we conducted comprehensive simulation studies with strategic parameter choices designed to test performance under balanced and unbalanced designs. However, as the partitions have unequal sizes, we systematically varied the number of spatial locations within each partition to assess the impact on pattern recovery performance. We fixed the number of nodes at 5 (motivated by our empirical data) and varied the number of areas from 3 to 5, systematically testing different spatial location distributions per area. For \(Q=3\), we examined eight configurations \((c1-c8)\) varying number of spatial locations under each area or partition \(N_q\) ; \(c1 = (150,150,150)\), \(c2=(150,250,150)\), \(c3= (150,150,250)\), \(c4=(150,250,250)\), \(c5=(250,150,150)\), \(c6 = (250,250,150)\), \(c7=(250,150,250)\) and \(c8=(250,250,250)\). For \(Q=5\) and we tested seven configurations of \(N_q\) choices \((c9-c15)\) ; \(c9=(300,300,300,300,300)\), \(c10=(250,300,250,350,350)\), \(c11=(350,300,250,250,350)\), \(c12=(350,250,300,250,350)\), \(c13=(250,300,300,300,350)\), \(c14=(300,300,250,350,300)\), \(c15=(250,250,300,350,350)\). We first generate \({\bf \Phi }\), \({\bf \Lambda }_q\)’s, and other necessary quantities for multi-study factor analysis, always keeping the number of factors used for the shared loading matrix as 7. We assessed network recovery with RV coefficient59. RV coefficient captures the similarity between two matrices with 1 being the highest value indicating perfect match between true and estimated values. We apply Gaussian process on spatial locations, using two different kernels: RBF and Matérn. All simulations were replicated over 10 datasets, using the same kernel function and Gaussian likelihood. Results showed (Fig. 2a), in most instances mean RV of the estimated covariances while comparing to the true covariance matrices were \(>0.86\), while standard deviations of RV’s were consistently low. Balanced configurations (c1, c8, c9) achieved higher estimation accuracy than unbalanced cases, with particularly strong recovery of shared patterns and area-specific covariances. Fig. 2bshowed computational time for our simulation study as we vary \(N_q\) choices as well as covariance structures. The number of partitions and number of spatial locations in each area impact time consumption. Time increases if we assume a Matérn covariance compared to RBF and from balanced to unbalanced \(N_q\) choice. Cases where \(Q=5\) showed less variability in terms of time than the \(Q=3\). We achieve significant computational speedup (average 15 minutes vs 2-4 hours for comparable methods). All \(\mathcal{G}\mathcal{P}\)-regressions were performed in an Intel(R) Core(TM) \(i7-14700-2.10\) GHz with 32 GB RAM using 25 cores in parallel. We fixed the number of cell types at 5 based on our data and computational considerations. We expect the method to perform similarly for 8-20 cell types, provided partitions contain sufficient spatial observations (\(N_q \ge 150\)) and adequate cell type abundance.
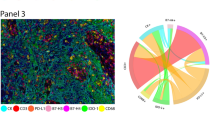
Domain informed knowledge for PDAC
Consistent with our methodology, previous analysis of PDAC datasets derived disease-specific adjacency matrices representing the comprehensive spatial and molecular landscape of PDAC60. We utilize this as our domain knowledge showcasing in Fig. 3(3). Once the covariance matrix was estimated through matrix exponential kernel, obtaining a graph as shown in Fig. 3(3) was immediate. This PDAC specific pattern preserves all ligand receptor interactions being preserved though the strength of the connection varies. It should be noted that these partial correlations were very low but all positive. The ligand receptor interactions that we found interesting were the following, in decreasing order of strength, Epithelial \(\leftrightarrow\) Treg, Epithelial \(\leftrightarrow\) THelper, THelper \(\leftrightarrow\) Treg, Epithelial \(\leftrightarrow\) APC, THelper \(\leftrightarrow\) APC, Treg \(\leftrightarrow\) CTL, Treg \(\leftrightarrow\) APC, THelper \(\leftrightarrow\) CTL, Epithelial \(\leftrightarrow\) CTL and APC \(\leftrightarrow\)CTL. Most interestingly, we note from this that the association of CTL’s and Treg’s, given the effects of rest of cells were much stronger than association of CTL’s with other cell types accounting for the effects of rest. The frequent appearance of Treg-centered interactions in PDAC networks aligns with known biology, as these cells exert broad immunosuppressive effects across diverse immune populations61,62,63..

Here we represent the schematic overview of ISPat framework for analyzing spatial heterogeneity in PDAC and IPMN pancreatic cancer tissues. The pipeline first transforms histopathological mIF images in (1) into point pattern representations of cellular distributions in (2), obtains optional domain-informed networks from spatial and omics modalities in (3), delineates Tumor subregions based on heterogeneity of tumor cells in (4), obtains marginal spatial intensity estimates in (5) and finally identifies tumor subregion based cellular heterogeneity patterns as well as shared cellular patterns in (6) through optional integration of the domain knowledge from step (3). This pipeline enables area-specific spatial characterization of cell patterns while maintaining a shared spatial pattern across the tissue.
Results
Spatial organization of patterns in PDAC
We analyzed 119 PDAC patients focusing on five cell types: Antigen-presenting cells (APC), Cytotoxic T cells (CTL), Epithelial Cells (Epi), Helper T cells (THelper), and Regulatory T cells (Treg). Cells expressing cytokeratin were classified as tumorous. To capture spatial heterogeneity within tumors, we partitioned each tissue into five distinct areas based on tumor intensity quantiles, as shown in Fig. 1b. These areas represent a spectrum from Area 1 (very high tumor intensity, highly heterogeneous) to Area 5 (very low tumor intensity, least heterogeneous in tumor cells). Areas 2, 3, and 4 correspond to high, medium, and low tumor intensity, respectively, exhibiting continuously decreasing tumor heterogeneity gradients and proving most informative for understanding spatial immune organization. We utilized partial correlations to identify ligand-receptor (LR) interaction patterns, as this approach better captures the interdependence between cell types while accounting for confounding effects from other variables. Statistically, partial correlation quantifies the linear association between two variables after controlling for all other variables in the system. The sparsity pattern of the partial correlation matrix directly corresponds to the underlying interaction graph. We applied a threshold of 0.1 to identify significant interactions and constructed the corresponding graphs. Subsequently, we calculated the proportions of positive and negative interactions across all images, confirming the presence of specific LR interactions shown in Fig. 4.
Focusing on shared patterns across all 119 patients, we observed highly consistent cellular organization. Figure 4 revealed that Epithelial\(\leftrightarrow\)THelper, Epithelial\(\leftrightarrow\)APC, and Epithelial\(\leftrightarrow\)CTL interactions were positively present in 97.5% of images, while Epithelial\(\leftrightarrow\)Treg interactions were positively present in 92.4% of images. THelper\(\leftrightarrow\)Treg and Treg\(\leftrightarrow\)CTL interactions were positively present in 89.9% of images. Notably, THelper\(\leftrightarrow\)APC, THelper\(\leftrightarrow\)CTL, and APC\(\leftrightarrow\)CTL interactions showed the highest consistency, appearing positively in 98.3% of images, while Treg\(\leftrightarrow\)APC interactions were positively present in 91.6% of images. Examining area-specific patterns along the tumor intensity gradient revealed remarkable consistency. While Area 1 (very high tumor intensity) and Area 5 (very low tumor intensity) showed expected extreme behaviors, Areas 2, 3, and 4 (high, medium, and low tumor intensity) demonstrated the most informative patterns with continuously decreasing heterogeneity gradients. These intermediate areas maintained similar interaction profiles to the shared patterns, suggesting that PDAC establishes a stable immunosuppressive architecture that persists across varying tumor densities.
Distinct patterns in IPMN
We conducted parallel analyses for 53 IPMN patients using identical methodology. Using the same threshold of 0.1 for partial correlations, we obtained interaction graphs and calculated proportions of positive and negative interactions across all images, as shown in Fig. 4. The shared patterns across all IPMN patients revealed notable differences from PDAC. Epithelial\(\leftrightarrow\)THelper and THelper\(\leftrightarrow\)Treg interactions were positively present in 94.2% of images, while Epithelial\(\leftrightarrow\)APC and Epithelial\(\leftrightarrow\)CTL interactions were positively present in 96.2% of images. THelper\(\leftrightarrow\)APC and THelper\(\leftrightarrow\)CTL interactions showed the highest consistency at 98.1%, and Treg\(\leftrightarrow\)APC and Treg\(\leftrightarrow\)CTL interactions were positively present in 92.3% of images. Notably, Epithelial\(\leftrightarrow\)Treg interactions were positively present in only 88.5% of images (compared to 92.4% in PDAC), while APC\(\leftrightarrow\)CTL interactions appeared positively in all IPMN images. Critically, the area-specific patterns in IPMN diverged substantially from the shared patterns, particularly in Areas 2, 3, and 4. Unlike PDAC, where these intermediate tumor intensity areas maintained consistent interaction profiles, IPMN exhibited marked variability across these regions. This spatial heterogeneity was especially pronounced for Treg-related interactions, which fluctuated notably across the tumor intensity gradient, suggesting dynamic and regionally distinct immune regulatory mechanisms in IPMN.
Comparative analysis: PDAC versus IPMN
Comparing the spatial patterns between PDAC and IPMN revealed several key differences in tumor microenvironment organization. First, IPMN exhibited substantially greater spatial heterogeneity across tissue areas compared to PDAC, as evidenced by more variable interaction patterns between shared and area-specific analyses. This contrast was most striking in Areas 2, 3, and 4 (high, medium, and low tumor intensity), where PDAC maintained remarkable uniformity while IPMN showed considerable divergence from shared patterns. Second, four Treg-related LR interactions showed markedly different abundance patterns between the two patient groups: Epithelial\(\leftrightarrow\)Treg, THelper\(\leftrightarrow\)Treg, Treg\(\leftrightarrow\)APC, and Treg\(\leftrightarrow\)CTL.
The intermediate tumor intensity areas (Areas 2–4) proved particularly informative for distinguishing disease-specific immune architectures. In PDAC, these areas demonstrated a stable immunosuppressive microenvironment with minimal deviation from shared patterns, suggesting that once established, the PDAC immune landscape remains rigid across the tumor heterogeneity gradient. Conversely, IPMN showed dynamic spatial remodeling across these same areas, with Treg-mediated interactions exhibiting area-specific variations that may reflect different stages of immune evasion or context-dependent immunoregulation. The presence of negative interactions was also more pronounced in specific areas of IPMN compared to PDAC, indicating potential spatial compartmentalization of inhibitory cellular relationships. These distinctive patterns motivated further statistical investigation to identify LR interactions that significantly differ between PDAC and IPMN patient groups, as discussed in the subsequent analysis.

Comparison of ligand-receptor interaction patterns between PDAC (right panel, \(n=119\)) and IPMN (left panel, \(n=53\)) across tissue areas. Bars represent the proportion of samples showing positive (dark blue) or negative (yellow) interactions for each cell type pair. Interactions are shown for the shared pattern across all tissue areas and five area-specific patterns, with Areas 2–4 exhibiting continuously decreasing heterogeneity gradients.
Area-specific interaction signatures
Having identified shared and area-specific patterns for both patient groups, we next investigated which ligand-receptor (LR) interactions significantly differ between PDAC and IPMN. For each tissue area, we obtained the total counts of positive and negative interactions across both patient groups and performed statistical tests to identify LR interactions that differ significantly between disease conditions. Specifically, we fit a sequence of generalized linear models for the binomial family with logit link, which is analogous to performing McNemar’s test for comparing two dichotomous variables. To account for the sequential nature of testing multiple LR pairs across disease conditions, we applied Bonferroni correction to control the family-wise error rate. At a significance level of \(\alpha = 0.005\), we observed no significant differences in LR interaction patterns for the shared patterns, Area 1 (very high tumor intensity, highly heterogeneous), or Area 5 (very low tumor intensity, least heterogeneous). In contrast, the intermediate tumor intensity areas (Areas 2–4), where tumor heterogeneity gradients continuously decrease, revealed the most striking differences between PDAC and IPMN, as shown in Figure 5.
Conserved differential interactions across intermediate areas
Four Treg-related LR interactions showed significant differences between PDAC and IPMN consistently across multiple intermediate areas. Epithelial\(\leftrightarrow\)Treg interactions differed significantly across all three intermediate areas (Areas 2, 3, and 4). Similarly, Treg\(\leftrightarrow\)APC and Treg\(\leftrightarrow\)CTL interactions were significantly different across all three intermediate areas (Areas 2, 3, and 4), highlighting the central role of regulatory T cells in distinguishing the immune microenvironments of these two disease conditions. THelper\(\leftrightarrow\)Treg interactions showed significant differences in Areas 3 and 4 (medium and low tumor intensity). The consistent differential patterns of these Treg-mediated interactions across the tumor heterogeneity gradient suggest fundamentally different mechanisms of immune regulation between PDAC and IPMN.
Area-specific differential interactions
Beyond the consistently differential interactions, we identified several area-specific patterns that provide insights into the spatial organization of immune responses. In Area 2 (high tumor intensity), seven LR interactions differed significantly: Epithelial\(\leftrightarrow\)THelper, Epithelial\(\leftrightarrow\)Treg, Epithelial\(\leftrightarrow\)CTL, THelper\(\leftrightarrow\)APC, Treg\(\leftrightarrow\)APC, Treg\(\leftrightarrow\)CTL, and APC\(\leftrightarrow\)CTL. Notably, three interactions were unique to Area 2: Epithelial\(\leftrightarrow\)THelper, THelper\(\leftrightarrow\)APC, and APC\(\leftrightarrow\)CTL, suggesting specific immune dynamics in high tumor density regions. In Area 3 (medium tumor intensity), six interactions were significantly different: Epithelial\(\leftrightarrow\)Treg, Epithelial\(\leftrightarrow\)APC, Epithelial\(\leftrightarrow\)CTL, THelper\(\leftrightarrow\)Treg, Treg\(\leftrightarrow\)APC, and Treg\(\leftrightarrow\)CTL. The Epithelial\(\leftrightarrow\)APC and Epithelial\(\leftrightarrow\)CTL interactions appeared significantly different in both Areas 2 and 3, indicating their importance in regions with higher tumor density. In Area 4 (low tumor intensity), five interactions differed significantly: Epithelial\(\leftrightarrow\)Treg, Epithelial\(\leftrightarrow\)APC, THelper\(\leftrightarrow\)Treg, Treg\(\leftrightarrow\)APC, and Treg\(\leftrightarrow\)CTL. The persistence of Treg-related interactions even in low tumor density regions underscores their critical role in shaping the distinct immune landscapes of PDAC versus IPMN.
Biological interpretation
The spatial pattern of differential interactions reveals key insights into immune microenvironment organization. The absence of significant differences in extreme conditions (Areas 1 and 5) and shared patterns suggests that the fundamental divergence between PDAC and IPMN immune architectures emerges in the intermediate tumor density regions where immune-tumor dynamics are most active. The predominance of Treg-mediated interactions among the consistently differential patterns aligns with our earlier observations of distinct Treg infiltration patterns between the two diseases. These findings indicate that T cell behaviors, particularly those involving regulatory T cells, are fundamentally altered across the tumor heterogeneity gradient in PDAC compared to IPMN. Many of these LR interactions have been documented in the literature as critical mediators of immune regulation in pancreatic cancer9,64,65,66.

Heatmap of significant differences in ligand-receptor interaction frequencies between PDAC and IPMN across tissue areas. Each cell indicates whether the corresponding LR interaction differs significantly between disease conditions at \(\alpha = 0.005\) (Bonferroni-corrected).
Discussion
Principal findings and spatial context
Our spatial analysis of PDAC and IPMN revealed multiple ligand-receptor interactions that consistently differ between these disease conditions across intermediate tumor intensity areas (Areas 2–4), while extreme conditions (Area 1: very high tumor intensity and Area 5: very low tumor intensity) show no significant differences. These differential interactions span multiple functional axes of anti-tumor immunity, revealing comprehensive differences in immune architecture rather than isolated defects. Four regulatory T cell-mediated interactions (Epithelial\(\leftrightarrow\)Treg, Treg\(\leftrightarrow\)APC, Treg\(\leftrightarrow\)CTL, and THelper\(\leftrightarrow\)Treg) differ consistently across Areas 2–4, reflecting fundamental differences in immunosuppressive mechanisms. However, equally critical are interactions involving antigen presentation and effector function. Epithelial\(\leftrightarrow\)APC and Epithelial\(\leftrightarrow\)CTL interactions, which differ significantly in Areas 2 and 3, represent the foundation and execution of anti-tumor immunity, respectively. The APC\(\leftrightarrow\)CTL interaction, fundamental for adaptive immunity activation where dendritic cells present tumor antigens to cytotoxic T cells, differs significantly in Area 2 (high tumor intensity). Additionally, THelper\(\leftrightarrow\)APC interactions in Area 2 reflect the capacity for T cell help in dendritic cell maturation, while Epithelial\(\leftrightarrow\)THelper interactions indicate tumor-helper T cell communication patterns. The spatial distribution of these differential interactions provides critical insights into disease-specific immune architectures. PDAC maintains a rigid, stable immunosuppressive microenvironment across the tumor heterogeneity gradient, with consistent suppression of both antigen presentation and effector function alongside elevated immunoregulation. In contrast, IPMN exhibits dynamic spatial remodeling with marked variability across intermediate tumor intensity regions, suggesting context-dependent immune dynamics that may reflect different stages of immune evasion or malignant transformation. The gradient of decreasing immune cell interactions in high tumor density regions provides quantitative evidence for spatial immune exclusion, a key mechanism of tumor immune evasion with important implications for immunotherapy approaches. The concentration of differential interactions specifically in intermediate tumor density regions (Areas 2–4), rather than in areas of extreme tumor burden or tumor paucity, indicates that these transitional zones represent critical sites where immune architecture diverges between disease subtypes. The absence of significant differences in extreme tumor density regions suggests that fundamental disease-specific immune patterns emerge and are maintained primarily in zones where tumor-immune cell interactions are most dynamic and potentially most therapeutically accessible.
Therapeutic and clinical implications
The spatial gradient of immune interactions revealed by ISPat carries significant therapeutic implications for pancreatic cancer, demonstrating that effective strategies must address defects across antigen presentation, effector function, and immune regulation simultaneously rather than targeting isolated mechanisms. The concentration of differential interactions in intermediate tumor density regions (Areas 2–4) indicates that these transitional zones represent critical therapeutic windows where immune architecture diverges between disease subtypes and may be most amenable to intervention. First, the APC\(\leftrightarrow\)CTL interaction, differing significantly in high tumor density regions, represents a foundational target where dendritic cells present tumor antigens to activate cytotoxic T cells, initiating the adaptive immune response. The differential Epithelial\(\leftrightarrow\)APC patterns suggest that IPMN, with its abundant APC infiltration and active (though inhibited) antigen presentation machinery, may be particularly responsive to therapies enhancing dendritic cell function such as CD40 agonists or therapeutic vaccines71,72, while PDAC’s rigid immunosuppressive architecture with potential defects in antigen processing machinery may require aggressive combination strategies to restore antigen presentation capacity before T cell-directed therapies can succeed. Second, the Epithelial\(\leftrightarrow\)CTL interaction, representing the effector phase where activated cytotoxic T cells directly eliminate tumor cells and differing significantly in Areas 2 and 3, suggests that strategies improving CTL trafficking to tumor sites, increasing activation status, and overcoming local immunosuppressive barriers may need to be disease-specific given distinct tumor microenvironment architectures. The THelper\(\leftrightarrow\)APC interaction in high tumor density regions indicates that helper T cell-mediated enhancement of dendritic cell function may also differ between disease conditions, suggesting potential roles for therapies augmenting CD4+ T cell help. Third, the Treg-mediated interactions (Treg\(\leftrightarrow\)CTL, Treg\(\leftrightarrow\)APC, THelper\(\leftrightarrow\)Treg, Epithelial\(\leftrightarrow\)Treg) concentrated in intermediate tumor density regions represent complementary therapeutic targets focused on relieving immunosuppression through Treg depletion approaches (anti-CD25, anti-CTLA-4), checkpoint inhibition (PD-1/PD-L1, TIGIT, LAG-3 blockade)67,68,69, or combination strategies addressing both Treg-mediated impairment of dendritic cell maturation70 and the complex THelper\(\leftrightarrow\)Treg dynamics potentially involving Th17\(\rightarrow\)Treg plasticity73. However, given concurrent defects across all three functional axes, single-agent approaches are unlikely to achieve clinical success, providing a mechanistic explanation for the limited efficacy of checkpoint inhibitors as monotherapy in pancreatic cancer74 and suggesting that spatially informed combination regimens addressing multiple functional axes simultaneously may be required75. These spatially resolved interaction signatures represent promising biomarker candidates that could distinguish disease subtypes, predict therapeutic response, and inform risk stratification. Future studies should examine whether these interaction patterns predict treatment response to specific therapeutic modalities, correlate with clinical outcomes including progression-free and overall survival, and whether spatial biomarkers can guide personalized therapeutic decision-making. Prospective clinical trials testing spatially informed therapeutic strategies, where treatment selection and intensity are guided by the constellation of interaction defects in individual patients’ intermediate tumor density regions, will be essential to translate these findings into improved outcomes. However, these findings represent correlative associations requiring extensive functional validation to establish causality and therapeutic potential, along with validation in independent cohorts with correlation to clinical outcomes, disease progression rates, and treatment responses before clinical application. Ultimately, the spatial heterogeneity revealed by ISPat suggests that precision oncology in pancreatic cancer must consider not only which cellular interactions are disrupted, but where these disruptions occur within the tumor architecture.
Methodological contributions and ISPat framework
Our ISPat (Informed Spatially aware Patterns) framework addresses several limitations of existing spatial analysis approaches through three key innovations.
First, by modeling conditional dependencies through precision matrices derived from partial correlations, we identified direct relationships between cell types while controlling for confounding effects from other cell populations. This approach provides interpretable results where positive partial correlations indicate coordinated spatial co-localization and negative partial correlations suggest spatial exclusion or inhibitory relationships. The identification of regions showing predominantly negative versus positive interactions revealed spatial compartmentalization of immune regulatory mechanisms, particularly in IPMN where negative interactions in intermediate tumor intensity zones indicate active immune remodeling distinct from PDAC’s uniform immunosuppressive architecture. Second, explicit modeling of spatial heterogeneity through tumor intensity-based area partitioning captured biological patterns that would be missed by methods assuming uniform interactions across tissues. Our decomposition approach, predicated on the assumption of decomposable overall covariance structure, enabled efficient estimation of multiple robust patterns for different tissue regions. From a pattern recognition perspective for functional data, this demonstrates that when overall covariance decomposability holds, we can efficiently obtain spatially resolved estimates that reveal region-specific interaction signatures across different areas of tissue images. Third, the integration of domain knowledge through informed prior covariance matrices in the Gaussian process framework produced substantially improved spatial pattern estimates compared to uninformed approaches. This multi-modal data integration capability allows the method to utilize domain knowledge to produce better estimates in spatial context. From a Bayesian modeling perspective, ISPat implements a two-stage procedure with Gaussian process modeling in R and Stan, which, despite introducing some computational complexity and compilation delays, enables rigorous uncertainty quantification validated through comprehensive simulation experiments.
While many of the ligand-receptor interactions we identified have been previously documented in the pancreatic cancer literature, our primary contribution centers on understanding how spatial context modulates these interactions and how tumor heterogeneity gradients shape distinct immune architectures in PDAC versus IPMN. This spatial perspective provides a framework for understanding why uniform therapeutic approaches may fail to account for the regional complexity of tumor immune microenvironments. Our focus on cancerous tissue regions enabled systematic investigation of intra-tumoral heterogeneity patterns across patients, which is fundamental to precision oncology approaches influencing diagnostic strategies, therapeutic interventions, and treatment response prediction.
Methodological advantages
From a computational perspective, ISPat provided substantial advantages over existing methods.
First, by employing variational Bayesian inference instead of typical MCMC sampling, we achieve significant computational speedup (average 15 min vs 2–4 h for comparable methods) while maintaining uncertainty quantification. The method scales well to high-dimensional data and can incorporate diverse forms of prior biological knowledge. Second, ISPat demonstrates robust scalability across multiple dimensions. Though motivated by our empirical study, we concentrated on a fixed number of cell types in our simulation experiments, but this method can handle 8–20 cell types, provided there are adequate number of spatial locations present in the image partitions. Though computational complexity scales quadratically with the number of cell types C, resulting in \(C(C-1)/2\) pairwise interactions, it remains tractable due to parallelizable Gaussian process regressions across cell types. For datasets with 10–15 cell types, computation times remain acceptable (30–45 min per image on standard computational infrastructure). Third, the framework exhibits robustness to increased spatial partitioning. Our primary analyses focused on \(Q=3/5\) tumor density regions, but can handle more partitions provided each partition contains sufficient spatial locations (\(N_q \ge 150\)). The region-specific patterns \({\bf \Lambda }_q{\bf \Lambda }_q^T\) benefit from increased number of locations in each partition through greater statistical power, while shared patterns \({\bf \Phi }{\bf \Phi }^T\) remain stable when the image has adequate spatial locations. This flexibility allows researchers to define biologically meaningful spatial domains without compromising statistical rigor. Fourth, ISPat handles naturally occurring partition size imbalances through its hierarchical structure. In real multiplexed imaging data, partitions often vary substantially in size due to biological heterogeneity. Our empirical PDAC-IPMN data exhibited imbalance ratios up to 4:1 between largest and smallest partitions, yet recovered consistent biological patterns. The partition-specific estimation of \({\bf \Lambda }_q{\bf \Lambda }_q^T\) ensures that small regions do not bias large regions, while the shared component \({\bf \Phi }{\bf \Phi }^T\) borrows strength across all partitions to stabilize estimation. This design makes ISPat particularly well-suited for analyzing heterogeneous tissues where uniform spatial sampling is impractical.
Generalizability and future applications
Though we focused on highly multiplexed immunofluorescence imaging in this study, ISPat is readily amenable to emerging spatial transcriptomics technologies, where domain knowledge can be derived from single-cell RNA sequencing data to inform spatial pattern estimation. The method’s versatility was confirmed through comprehensive testing across multiple scenarios, demonstrating applicability to both low- and high-dimensional spatial datasets. While we concentrated on pancreatic cancer and tumor regions in this study, the framework can be applied to other cancer types and tissues. Targeting particular regions as tumors develop and progress could help study patterns informed by inter- and intra-tumor heterogeneity across diverse disease contexts.
Our findings offer insights into the role of specific LR interactions that differ between PDAC and IPMN patient groups. The concentration of differential interactions in intermediate tumor intensity regions (Areas 2-4) rather than extreme conditions suggests that these transitional zones play crucial roles in shaping disease-specific immune architectures. This spatial perspective emphasizes that cellular interactions are not uniformly distributed but rather exhibit region-specific organization that reflects underlying tumor biology and immune dynamics.
Limitations
Several important limitations warrant consideration. First, our statistical networks represent conditional dependencies rather than direct physical or functional interactions, and the correlative associations we identified require extensive experimental validation to establish causality and therapeutic potential. Second, our region definitions based on tumor density capture only one aspect of tissue heterogeneity; future work should incorporate additional spatial features such as vascular patterns, stromal characteristics, and proximity to lymphoid structures. Third, the assumption of multivariate normality for transformed intensity values may not always hold for all cell types, though our simulation studies suggest the method is reasonably robust to model misspecification. Fourth, while we accounted for different tissue subregions through area-based stratification, potential uncontrolled confounding factors may exist that influence spatial interaction patterns. Fifth, our analysis focused exclusively on five cell types (APC, CTL, Epithelial cells, THelper, and Treg), while the tumor microenvironment contains additional populations (including tumor-associated macrophages, cancer-associated fibroblasts, and neutrophils) that likely contribute to immune regulation. Sixth, our cohort sizes, while substantial for multiplexed imaging studies (119 PDAC and 53 IPMN patients), may not capture the full spectrum of intra-disease heterogeneity. Finally, the absence of longitudinal sampling prevents determination of how spatial interaction patterns evolve during disease progression or treatment response.
Conclusion
We developed and validated ISPat, a Bayesian statistical framework for identifying spatially heterogeneous cell interaction patterns in multiplexed tissue imaging data. Applied to pancreatic cancer patients, our method revealed distinct immune architectures between PDAC and IPMN spanning multiple functional axes of anti-tumor immunity. The differential interactions encompass antigen presentation (APC\(\leftrightarrow\)CTL, Epithelial\(\leftrightarrow\)APC), effector function (Epithelial\(\leftrightarrow\)CTL, THelper\(\leftrightarrow\)APC), and immune regulation (Epithelial\(\leftrightarrow\)Treg, Treg\(\leftrightarrow\)APC, Treg\(\leftrightarrow\)CTL, THelper\(\leftrightarrow\)Treg), with these differences concentrated in intermediate tumor intensity regions (Areas 2-4) rather than extreme tumor density conditions. PDAC exhibits a rigid, stable immunosuppressive microenvironment with consistent suppression across all three functional axes, whereas IPMN displays dynamic spatial remodeling with marked variability across intermediate tumor intensity regions, suggesting context-dependent immune dynamics that may reflect different stages of immune evasion or malignant transformation. The spatial gradient of interactions provides quantitative evidence for immune exclusion mechanisms and demonstrates that successful therapeutic strategies must address concurrent defects in antigen presentation, effector function, and immune regulation simultaneously rather than targeting isolated mechanisms. The concentration of differential interactions in transitional tumor density zones indicates that these regions represent critical therapeutic windows where immune architecture diverges between disease subtypes and may be most amenable to intervention. ISPat provides a generalizable framework for analyzing spatial cellular interactions across diverse tissue types and disease contexts, with particular utility for precision oncology applications where spatially resolved interaction signatures could serve as biomarkers to distinguish disease subtypes, predict therapeutic response, inform risk stratification, and guide personalized treatment decisions that account for regional heterogeneity within tumors.
Data availability
The multiplexed immunofluorescence imaging dataset is available from the University of Michigan School of Medicine upon reasonable request and with institutional approval, subject to patient privacy protections (contact: M.P.D.M., mpasca@umich.edu).
References
Siegel, R. L., Miller, K. D., Wagle, N. S. & Jemal, A. Cancer statistics, 2023. CA Cancer J. Clin.73, 17–48. https://doi.org/10.3322/caac.21763 (2023). _eprint: https://onlinelibrary.wiley.com/doi/pdf/10.3322/caac.21763.
Adamska, A., Domenichini, A. & Falasca, M. Pancreatic ductal adenocarcinoma: Current and evolving therapies. Int. J. Mol. Sci.18, 1338 (2017).
Singhi, A. D., Koay, E. J., Chari, S. T. & Maitra, A. Early detection of pancreatic cancer: Opportunities and challenges. Gastroenterology156, 2024–2040. https://doi.org/10.1053/j.gastro.2019.01.259 (2019).
Sarantis, P., Koustas, E., Papadimitropoulou, A., Papavassiliou, A. G. & Karamouzis, M. V. Pancreatic ductal adenocarcinoma: Treatment hurdles, tumor microenvironment and immunotherapy. World J. Gastrointest. Oncol.12, 173–181. https://doi.org/10.4251/wjgo.v12.i2.173 (2020).
Mizrahi, J. D., Surana, R., Valle, J. W. & Shroff, R. T. Pancreatic cancer. Lancet (London, England)395, 2008–2020. https://doi.org/10.1016/S0140-6736(20)30974-0 (2020).
Takikawa, T. et al. Clinical features and prognostic impact of asymptomatic pancreatic cancer. Sci. Rep.12, 4262. https://doi.org/10.1038/s41598-022-08083-6 (2022) (publisher: Nature Publishing Group).
Schepis, T. et al. State-of-the-art and upcoming innovations in pancreatic cancer care: A step forward to precision medicine. Cancers15, 3423. https://doi.org/10.3390/cancers15133423 (2023) (number: 13 publisher: Multidisciplinary Digital Publishing Institute).
Tanaka, M. et al. International consensus guidelines 2012 for the management of IPMN and MCN of the pancreas. Pancreatology12, 183–197. https://doi.org/10.1016/j.pan.2012.04.004 (2012).
Shi, X., Liu, H. & Liang, Z. Cellular crosstalk of regulatory T cells in pancreatic ductal adenocarcinoma. J. Pancreatol.07, 131–140. https://doi.org/10.1097/JP9.0000000000000135 (2024) (publisher: Chinese Medical Association Publishing House).
Persky, J. et al. Characterization of natural killer and cytotoxic t-cell immune infiltrates in pancreatic ductal adenocarcinoma. J. Surg. Oncol.129, 885–892 (2024).
Ju, Y. et al. Barriers and opportunities in pancreatic cancer immunotherapy. NPJ Precis. Oncol.8, 199 (2024).
Bräutigam, K., Skok, K., Szymonski, K., Rift, C. V. & Karamitopoulou, E. Tumor immune microenvironment in pancreatic ductal adenocarcinoma revisited-exploring the space. Cancer Lett.622, 217699 (2025).
Fan, J.-Q. et al. Current advances and outlooks in immunotherapy for pancreatic ductal adenocarcinoma. Mol. Cancer19, 32 (2020).
Poyia, F., Neophytou, C. M., Christodoulou, M.-I. & Papageorgis, P. The role of tumor microenvironment in pancreatic cancer immunotherapy: current status and future perspectives. Int. J. Mol. Sci.25, 9555 (2024).
Roth, S. et al. Evolution of the immune landscape during progression of pancreatic intraductal papillary mucinous neoplasms to invasive cancer. EBioMedicine54 (2020).
Pedro, B. A., Wood, L. D. et al. Early neoplastic lesions of the pancreas: Initiation, progression, and opportunities for precancer interception. J. Clin. Invest.135 (2025).
Rao, A., Barkley, D., França, G. S. & Yanai, I. Exploring tissue architecture using spatial transcriptomics. Nature596, 211–220. https://doi.org/10.1038/s41586-021-03634-9 (2021) (publisher: Nature Publishing Group).
Labani-Motlagh, A., Ashja-Mahdavi, M. & Loskog, A. The tumor microenvironment: A milieu hindering and obstructing antitumor immune responses. Front. Immunol.11, 940 (2020).
Wei, R., Liu, S., Zhang, S., Min, L. & Zhu, S. Cellular and extracellular components in tumor microenvironment and their application in early diagnosis of cancers. Anal. Cell. Pathol.2020, 6283796 (2020).
Toninelli, M., Rossetti, G. & Pagani, M. Charting the tumor microenvironment with spatial profiling technologies. Trends Cancer9, 1085–1096 (2023).
Wang, Q. et al. Role of tumor microenvironment in cancer progression and therapeutic strategy. Cancer Med.12, 11149–11165 (2023).
Zhang, L. et al. Targets of tumor microenvironment for potential drug development. MedComm-Oncol.3, e68 (2024).
Juiz, N. A., Iovanna, J. & Dusetti, N. Pancreatic cancer heterogeneity can be explained beyond the genome. Front. Oncol.9. https://doi.org/10.3389/fonc.2019.00246 (2019) (publisher: Frontiers).
Wandmacher, A. M., Mehdorn, A.-S. & Sebens, S. The heterogeneity of the tumor microenvironment as essential determinant of development, progression and therapy response of pancreatic cancer. Cancers13, 4932. https://doi.org/10.3390/cancers13194932 (2021).
Evan, T., Wang, V. M.-Y. & Behrens, A. The roles of intratumour heterogeneity in the biology and treatment of pancreatic ductal adenocarcinoma. Oncogene41, 4686–4695. https://doi.org/10.1038/s41388-022-02448-x (2022) (publisher: Nature Publishing Group).
Hu, A. et al. Harnessing innate immune pathways for therapeutic advancement in cancer. Signal Transduct. Target. Ther.9, 68 (2024).
Boisset, J.-C. et al. Mapping the physical network of cellular interactions. Nat. Methods15, 547–553. https://doi.org/10.1038/s41592-018-0009-z (2018) (publisher: Nature Publishing Group).
Navarro, J. et al. Spatial transcriptomics reveals genes associated with dysregulated mitochondrial functions and stress signaling in Alzheimer disease. iScience23, 101556 (2020).
Borsboom, D. et al. Network analysis of multivariate data in psychological science. Nat. Rev. Methods Primers1, 58 (2021).
Mason, M. et al. Signed weighted gene co-expression network analysis of transcriptional regulation in murine embryonic stem cells. BMC Genomics10, 327 (2009).
Barabási, A.-L. et al. Network medicine: A network-based approach to human disease. Nat. Rev. Genet.12, 56–68 (2011).
Keren, L. et al. A structured tumor-immune microenvironment in triple negative breast cancer revealed by multiplexed ion beam imaging. Cell174, 1373-1387.e19. https://doi.org/10.1016/j.cell.2018.08.039 (2018).
Schurch, C. M. et al. Coordinated cellular neighborhoods orchestrate antitumoral immunity at the colorectal cancer invasive front. Cell182, 1341–1359 (2020).
Lacinski, R. A. et al. Spatial multiplexed immunofluorescence analysis reveals coordinated cellular networks associated with overall survival in metastatic osteosarcoma. Bone Res.12, 55 (2024) (number: 1 publisher: Nature Publishing Group).
Marchetti, L., Porciani, D., Mitola, S. & Giacomelli, C. Editorial: Molecular insights into ligand-receptor interactions on the cell surface. Front. Mol. Biosci.9, 921677. https://doi.org/10.3389/fmolb (2022).
Crow, M., Paul, A., Ballouz, S., Huang, Z. J. & Gillis, J. Exploiting single-cell expression to characterize co-expression replicability. Genome Biol.17, 1–19 (2016).
García-Ruiz, S. et al. Coexp: A web tool for the exploitation of co-expression networks. Front. Genet.12, 630187 (2021).
Salamon, J., Qian, X., Nilsson, M. & Lynn, D. J. Network visualization and analysis of spatially aware gene expression data with Insitunet. Cell Syst.6, 626–630 (2018).
Sun, S., Zhu, J. & Zhou, X. Statistical analysis of spatial expression patterns for spatially resolved transcriptomic studies. Nat. Methods17, 193–200 (2020).
Acharyya, S., Zhou, X. & Baladandayuthapani, V. SpaceX: gene co-expression network estimation for spatial transcriptomics. Bioinformatics38, 5033–5041. https://doi.org/10.1093/bioinformatics/btac645 (2022). https://academic.oup.com/bioinformatics/article-pdf/38/22/5033/47057652/btac645.pdf.
Sengupta, D. & Pratx, G. Imaging metabolic heterogeneity in cancer. Mol. Cancer15, 4. https://doi.org/10.1186/s12943-015-0481-3 (2016).
Becker, A. P., Sells, B. E., Haque, S. J. & Chakravarti, A. Tumor heterogeneity in glioblastomas: From light microscopy to molecular pathology. Cancers13, 761. https://doi.org/10.3390/cancers13040761 (2021) (number: 4 publisher: Multidisciplinary Digital Publishing Institute).
Elyada, E. et al. Cross-species single-cell analysis of pancreatic ductal adenocarcinoma reveals antigen-presenting cancer-associated fibroblasts. Cancer Discov.9, 1102–1123 (2019).
Gruosso, T. et al. Spatially distinct tumor immune microenvironments stratify triple-negative breast cancers. J. Clin. Invest.129, 1785–1800 (2019).
Tsujikawa, T. et al. Prognostic significance of spatial immune profiles in human solid cancers. Cancer Sci.111, 3426–3434 (2020).
Platig, J., Castaldi, P. J., DeMeo, D. & Quackenbush, J. Bipartite community structure of EQTLS. PLoS Comput. Biol.12, e1005033 (2016).
Shang, L., Smith, J. A. & Zhou, X. Leveraging gene co-expression patterns to infer trait-relevant tissues in genome-wide association studies. PLoS Genet.16, e1008734 (2020).
Danenberg, E. et al. Breast tumor microenvironment structures are associated with genomic features and clinical outcome. Nat. Genet.54, 660–669 (2022).
Krishnan, S. N. et al. Proximogram-A multi-omics network-based framework to capture tissue heterogeneity integrating single-cell omics and spatial profiling. Comput. Biol. Med.182, 109082. https://doi.org/10.1016/j.compbiomed.2024.109082 (2024).
Lazarus, J. et al. Spatial and phenotypic immune profiling of metastatic colon cancer. JCI Insight3, e121932 (2018).
Li, X., Guindani, M., Ng, C. S. & Hobbs, B. P. Spatial Bayesian modeling of GLCM with application to malignant lesion characterization. J. Appl. Stat.46, 230–246. https://doi.org/10.1080/02664763.2018.1473348 (2018).
Li, X., Guindani, M., Ng, C. S. & Hobbs, B. P. A Bayesian nonparametric model for textural pattern heterogeneity. J. R. Stat. Soc. Ser. C Appl. Stat.70, 459–480. https://doi.org/10.1111/rssc.12469 (2021).
De Vito, R., Bellio, R., Trippa, L. & Parmigiani, G. Bayesian multistudy factor analysis for high-throughput biological data. Ann. Appl. Stat.15, 1723–1741 (2021).
Hansen, B., Avalos-Pacheco, A., Russo, M. & De Vito, R. Fast variational inference for Bayesian factor analysis in single and multi-study settings. J. Comput. Graph. Stat.34, 96–108 (2025).
Rasmussen, C. E. & Williams, C. K. I. Gaussian processes for machine learning. In Adaptive Computation and Machine Learning. 3 Ed. (MIT Press, 2008).
Hastie, T. & Tibshirani, R. Exploring the nature of covariate effects in the proportional hazards model. Biometrics46, 1005–1016. https://doi.org/10.2307/2532444 (1990) (publisher: Wiley, International Biometric Society).
Bishop, C. M. Pattern Recognition and Machine Learning. Vol. 4 (Springer, 2006).
Sass, D. A. & Schmitt, T. A. A comparative investigation of rotation criteria within exploratory factor analysis. Multivar. Behav. Res.45, 73–103. https://doi.org/10.1080/00273170903504810 (2010).
Robert, P. & Escoufier, Y. A unifying tool for linear multivariate statistical methods: The rv-coefficient. J. R. Stat. Soc. Ser. C Appl. Stat.25, 257–265 (1976).
Krishnan, S. N. et al. Proximogram-A multi-omics network-based framework to capture tissue heterogeneity integrating single-cell omics and spatial profiling. Comput. Biol. Med.182, 109082 (2024).
Jang, J.-E. et al. Crosstalk between regulatory T cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic cancer. Cell Rep.20, 558–571. https://doi.org/10.1016/j.celrep.2017.06.062 (2017) (publisher: Elsevier).
Brouwer, T. et al. A paradoxical role for regulatory T cells in the tumor microenvironment of pancreatic cancer. Cancers14, 3862. https://doi.org/10.3390/cancers14163862 (2022).
Smith, H., Arbe-Barnes, E., Shah, E. A. & Sivakumar, S. Manipulating regulatory T cells: Is it the key to unlocking effective immunotherapy for pancreatic ductal adenocarcinoma? Front. Immunol.15. https://doi.org/10.3389/fimmu.2024.1406250 (2024) (publisher: Frontiers).
Song, X., Lu, Z. & Xu, J. Targeting cluster of differentiation 47 improves the efficacy of anti-cytotoxic T-lymphocyte associated protein 4 treatment via antigen presentation enhancement in pancreatic ductal adenocarcinoma. Exp. Ther. Med.20, 3301–3309. https://doi.org/10.3892/etm.2020.9054 (2020).
Lee, J. J. et al. Elucidation of tumor-stromal heterogeneity and the ligand-receptor interactome by single cell transcriptomics in real-world pancreatic cancer biopsies. Clin. Cancer Res.27, 5912–5921. https://doi.org/10.1158/1078-0432.CCR-20-3925 (2021).
Debesset, A. et al. TNFR2 blockade promotes antitumoral immune response in PDAC by targeting activated Treg and reducing T cell exhaustion. J. Immunother. Cancer12, e008898. https://doi.org/10.1136/jitc-2024-008898 (2024).
Mota Reyes, C. et al. Regulatory t cells in pancreatic cancer: Of mice and men. Cancers14, 4582 (2022).
Zhang, A. et al. Regulatory t cells in immune checkpoint blockade antitumor therapy. Mol. Cancer23, 251 (2024).
Tanaka, A. & Sakaguchi, S. Regulatory t cells in cancer immunotherapy. Cell Res.27, 109–118 (2017).
Jang, J.-E. et al. Crosstalk between regulatory t cells and tumor-associated dendritic cells negates anti-tumor immunity in pancreatic cancer. Cell Rep.20, 558–571 (2017).
Lau, S. P. et al. Dendritic cell vaccination and cd40-agonist combination therapy licenses t cell-dependent antitumor immunity in a pancreatic carcinoma murine model. J. Immunother. Cancer8, e000772 (2020).
Vonderheide, R. H. Cd40 agonist antibodies in cancer immunotherapy. Annu. Rev. Med.71, 47–58 (2020).
Wang, X. et al. Changes of th17/treg cell and related cytokines in pancreatic cancer patients. Int. J. Clin. Exp. Pathol.8, 5702 (2015).
Farhangnia, P., Khorramdelazad, H., Nickho, H. & Delbandi, A.-A. Current and future immunotherapeutic approaches in pancreatic cancer treatment. J. Hematol. Oncol.17, 40 (2024).
Byrne, K. T. et al. Neoadjuvant selicrelumab, an agonist cd40 antibody, induces changes in the tumor microenvironment in patients with resectable pancreatic cancer. Clin. Cancer Res.27, 4574–4586 (2021).
Acknowledgements
We thank the University of Michigan Pancreatic Cancer Clinic for providing tissue samples and clinical expertise. We acknowledge computational support from the University of Michigan Advanced Research Computing cluster. We appreciate the services of the Molecular Pathology Research Laboratory, University of Michigan. We thank the anonymous reviewers for their comments.
Funding
This work was supported by NIH grants R37CA214955 (A.R.).
Author information
Authors and Affiliations
Contributions
S.B. and A.R. conceived the study and developed the statistical methodology. S.B. and S.A. implemented the computational framework. S.B. contributed to data processing. S.B. and S.K. contributed to validation. M.P.D.M. and T.F. provided clinical samples and biological expertise. S.B. was solely responsible for all aspects of manuscript preparation, including conceptualization, writing, data analysis, interpretation of results, figure generation, literature review, and all revisions presented in this manuscript. S.B. conducted all simulation experiments, statistical analyses, and biological interpretation of spatial patterns. All authors reviewed and approved the final version.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Approval and informed consent
This study involved human subjects and was approved by University of Michigan’s Institutional Review Board and conducted in accordance with the Declaration of Helsinki. Patients considered in this study signed an informed consent form prior to enrollment. Deidentification of subjects were performed following HIPAA.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Bhadury, S., Peruzzi, M., Acharyya, S. et al. Informed spatially aware patterns for multiplexed immunofluorescence data. Sci Rep 16, 5015 (2026). https://doi.org/10.1038/s41598-026-35341-8
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-35341-8
