
Abstract
This study investigates the role of extracellular vesicles (EVs) in predicting melanoma patients’ responses to anti-PD1 immunotherapy. Nine patients with advanced melanoma provided blood samples at three stages: before treatment, before the second dose, and either at disease progression or nine months later. EVs were isolated from serum and analyzed using mass-spectrometry proteomics, followed by network and enrichment analyses. Six out of nine patients progressed despite treatment. Before therapy, responders exhibited higher levels of adaptive immune and cell adhesion proteins, while proteins related to UV radiation response were deplected. An eight-protein signature and cellular adhesion markers correlated with longer progression-free survival. After treatment, non-responders had EV proteins enriched in proteasome activity and metabolic pathways, especially glycolysis. Finally, dynamic changes in EV protein over time showed decreased coagulation proteins, along with an increase in MHC proteins in patients with progressive disease. Overall, EV protein profiles differed between responders and non-responders both before and during therapy. These findings suggest that EVs could provide predictive biomarkers and insights into resistance mechanisms, potentially guiding more effective melanoma treatment strategies.
Similar content being viewed by others


Introduction
Melanoma is the most lethal cutaneous cancer. Ten years ago, the prognosis of metastatic melanoma was poor, with a median overall survival of less than 12 months. With the advent of targeted therapies against BRAF mutations and immunotherapy based on immune checkpoint inhibitors (ICIs), prognosis has improved1. The median overall survival of these patients now exceeds two years and around 40% of the patients become long-term responders2,3,4.
The main limitation of immunotherapy treatment is the presence of primary resistance. One third of the patients have a progression to treatment in the first six months, and an additional 20–30% progress thereafter.
Different predictive markers of response to immunotherapy have been proposed. Tumors with BRAF mutations seem to have a better response to immunotherapy. High expression of PD-L1, a high mutational burden, or a high expression of gamma-interferon signature also correlate with response5,6. However, these methods are not routinely used in the clinic. Therefore, it is still necessary the identification of good biomarkers of response to anti-PD1 inhibitors.
Extracellular vesicles (EVs) are blood-circulating, double-membrane vesicles involved in signaling processes and intercellular communications7. EVs implication in carcinogenesis, tumor progression and immunity is being studied8. The presence of EVs with high levels of PD-L1 has been linked to resistance to immunotherapy9,10.
In the present study, the protein content of circulating EVs was characterized in patients with advanced melanoma who received anti-PD1 therapy. The aim was to find predictive biomarkers of response and characterization of the mechanisms of resistance to this treatment.
Results
Patients’ characteristics
Nine patients diagnosed with advanced melanoma were included in this study. All patients were treated with anti-PD1 inhibitors. Three patients had a response to treatment (one partial response and two stable diseases) while six patients had progressive disease. Detailed clinical data of each patient can be found at Sup Table 1.
EV isolation
EV specific markers (CD63, CD9, CD81) were detected by western blot (Fig. 1A). The isolated EVs showed typical morphology and size (< 200 nm) by electron microscopy (Fig. 1B). The size (< 200 nm) was confirmed by NanoSight, and a concentration of 3.01e-08 particles/mL was measured (Fig. 1C). Full uncropped gel images can be found at Sup Fig. 1.

Extracellular vesicles (EVs) characterization. (A) Presence of specific EVs markers (CD9, CD63, and CD81) detected by Western Blot in pooled samples in each time point. Pre-Tx: pre-treated samples. Post-Tx: post-treatment samples. Endpoint: Samples at progression or at nine months. (B) Transmission electron microscopy (TEM) images showing the characteristic size and form of the EVs. (C) NTA analysis report showing the average size and concentration of the EVs in one representative sample.
Proteomics experiments
EV content was characterized using MS proteomics. 1,431 proteins were identified by DIA MS proteomics. After removing duplicates and applying quality filters, a database including 969 proteins was further analyzed.
Identification of innate mechanisms of resistance and prediction of response in pre-treated samples
With the aim of characterizing innate mechanisms of resistance to anti-PD1 therapy, differences in pre-treated samples between responding patients and patients who progressed to anti-PD1 therapy were assessed. A functional network was built: this network had 29 functional nodes (Fig. 2A). Patients who progressed to anti-PD1 inhibitors presented higher functional node activity of node1_ adaptive immune response & immunoglobulin. This node was mostly composed of immunoglobulin chains and complement factors (Fig. 2B, Sup Table 2).

Network analyses to identify biological processes involved in innate mechanisms of resistance to anti-PD1 inhibitors. (A). Network built using the proteins identified in the analysis of extracellular vesicles in pre-treated samples. Each color represents a functional node to which an overrepresented biological function has been asigned. (B). Differential functional node activities according to progression to anti-PD1 inhibitors in pre-treated samples.
Additionally, GSEA was used to identify specific hallmarks of cancer involved in these innate mechanisms of resistance. Samples from responder patients were enriched in proteins related to apical junction, genes downregulated in response to UV radiation, and estrogen early response hallmarks (FDR < 5%) (Sup Table 3). No statistically significant hallmarks were enriched in patients with progressive disease to anti-PD1 inhibitors.
The relation between both functional node activities and individual proteins with PFS has also been studied. Node20 cell-cell adhesion & focal adhesion divided patients into two groups with different PFS (p = 0.05, HR = 3.62, AUC = 0.80) (Fig. 3).

Predictive value of the functional node 20_cell-cell adhesion & focal adhesion. (A). Progression-free survival in low and high-risk groups identified by the node 20. (B). ROC time-dependent curve of the node 20 predictive signature.
On the other hand, forty-nine individual proteins were correlated with PFS (p-value < 0.05) (Sup Table 4). Of these proteins, eight were selected based on their p-value to build a predictive signature (Table 1; Fig. 4). The individual risk associated with this predictive signature was calculated using the following formula: ∑iwi xi + 25.93092, where wi and xi are the weight and logged protein expression for the i-th gene respectively. A sample is classified as high risk if its prognostic index is larger than − 0.58. After 1,000 random permutations, the p-value obtained for this signature was 0.042 and the AUC was 0.86.

Eight protein predictive signature classification. (A). Progression-free survival in low and high-risk groups identified by the eigth protein signature. (B). ROC time-dependent curve of the eigth protein predictive signature.
Identification of acquired mechanisms of resistance in post-treatment samples
With the aim of defining acquired mechanisms of resistance, post-treatment samples were analyzed. Again, a functional network using post-treatment samples was built and functional node activities were used to identify differential biological processes between samples from patients who did or did not progress to anti-PD1 inhibitors. In this case, 30 functional nodes were defined and Node 16_ proteasome complex & metabolism presented differential functional node activity (Fig. 5, Sup Table 5).

Network analysis to identify biological processes involved in acquired mechanisms of resistance to anti-PD1 inhibitors. (A). Network built using the proteins identified in the analysis of extracellular vesicles in post-treatment samples. Each color represents a functional node to which an overrepresented biological function has been asigned. (B). Differential functional node activities according to progression to anti-PD1 inhibitors in post-treatment samples.
Again, a GSEA was used to define hallmarks of cancer involved in acquired mechanisms of resistance to anti-PD1 therapy in advanced melanoma patients. Samples from patients who had a progression were enriched in cholesterol homeostasis, apical, MTORC1 signaling, MYC targets version1, heme metabolism, interferon alpha response and interferon gamma response gene sets (FDR < 5%) (Sup Table 6). The proteins enriched in interferon response and MYC pathway are proteins related to proteasome. MTORC1 signaling highlighted proteins involved in glycolysis like LDHA, GAPDH, G6P, PGK1, TPI, or GPI (Sup Table 7).
Differences across time in EV protein content according to progression to anti-PD1 inhibitors
Finally, the evolution of EV protein content across time in responding vs. progressing patients was studied. A network using the 969 filtered measured proteins in all samples was built (Fig. 6A). Twenty-seven functional nodes with an overrepresented biological function were defined in this network (Sup Table 8). Then, differences due to progression to treatment were evaluated using mixed lineal models. Node 6_platelet activation and cell adhesion showed a decrease whereas node 15_antigen processing and MHC I showed an increase in patients progressing to anti-PD1 therapy (Fig. 6B).

Network analyses of the EV proteins measured in all the samples at the three time points. (A). Network using the proteins measured in extracellular vesicles in all the samples. Each color indicates a functional node to which an overrepresented biological function has been asigned. (B). Evolution of the differential functional node activities at the three time points (pre-treated, post-treatment and progression) according to progression to anti-PD1 inhibitors. P-values were calculated using mixed lineal models with fixed effects.
Discussion
This study explores the role of extracellular vesicles (EVs) as carriers of relevant information in tumor progression and resistance to immunotherapy in patients with advanced melanoma. It focuses on the proteomics analysis of EVs isolated from patient samples before and after treatment with PD-1 inhibitors. EVs isolated from cancer patients transmit information that can be related to tumor progression and metastasis. These particles have been proposed not only as diagnostic and prognostic markers but also as therapeutic targets11,12. The role of EVs in melanoma was first described in 1988 by Taylor et al.13, while Mears et al. demonstrated the presence of proteins in melanoma EVs14. However, the studies of EVs in advanced melanoma are scarce, mostly focused on diagnosis rather than in prediction of response. Some studies describing a differential EV content between melanoma patients and healthy controls have been reported. For instance, exosomes expressing caveolin 1 were significantly increased in plasma from melanoma patients compared to healthy donors15. More recently, high levels of PD-L1 in these EVs have been correlated to response to immune checkpoint inhibitors16,17 whereas high levels of PD-1 have been correlated with resistance10,18. In this work, we used mass-spectrometry proteomics to characterize EV cargo at different time points from nine patients with advanced melanoma who received anti-PD1 therapy.
Pre-treated samples were analyzed to search for innate mechanisms of resistance. Adaptive immune response markers and immunoglobulins were higher in EVs from patients who progressed to anti-PD1 inhibitors. This functional node mostly contained immunoglobulins and complement activator factors but also several pro-inflammatory proteins such as CD36 or CD5L. A pro-inflammatory environment is associated with a poor response to immunotherapy19 and another immunosuppressive proteins have been previously found in melanoma-derived exosomes, supporting the possibility of their implication in immune evasion20.
GSEA also highlighted several processes enriched in pre-treated samples from responder patients: apical junction, genes downregulated in response to UV radiation, and genes defining early response to estrogen. Among the proteins involved in response to UV radiation, KIT was enriched in EVs from patients who did not progress. The presence of KIT, a relevant oncogene in melanoma, has been previously described in melanoma exosomes by Lazar et al.21 and its role in this scenario should be further investigated.
Regarding the functional nodes previously identified in the PGM protein network, node 20_cell-cell adhesion & focal adhesion correlated with PFS. This node included integrins, cofilin, myosin, tropomyosins, and tubulins, among others. EVs have an important role in preparing the metastatic niche22, so it is not surprising that this node was related to PFS. It was not possible to determine if the node had prognostic or predictive value, as all patients were treated. However, it seems that EVs containing proteins related to cell-cell adhesion and focal adhesion influence PFS in advanced melanoma.
In addition, an eight protein-based signature predicted response to anti-PD1 therapy in pre-treatment samples. Protein signatures in serum samples from melanoma patients have been previously established, but studies related to response to anti-PD1 inhibitors are scarce. Most of the studies were focused on finding diagnostic signatures, comparing exosome content between melanoma patients and healthy controls. In one study, exosomes from patients with advanced melanoma contained a protein signature including melanoma-specific protein tyrosinase-related protein-2 (TYRP2), very late antigen 4 (VLA-4), HSP70, and MET oncoprotein23. In another study, circulating exosomes from patients with active melanoma contained more S100B and Melanoma Inhibitory Activity (MIA) protein quantity than healthy controls and patients free of disease, MIA also being related with survival24. Finally, in a study of 15 patients with metastatic melanoma, a set of 8 proteins was differentially expressed between patients with progression and those with a response to treatment. However, the samples were collected after treatment, therefore not being suitable for prediction of response before treatment administration, and no detailed information about the administered treatments was provided25. On the contrary, in our series a predictive signature was built from pre-treatment serum samples.
Then, we explored the biological role of these eigth proteins in melanoma. Corneodesmosin (CDSN) protein express mainly in skin tissue26. It is related to corneodesmosomes, which localize to human epidermis and other cornified squamous epithelia, and has been associated with skin diseases such as psoriasis, hypotrichosis and peeling skin syndrome. CDSN seems to be downregulated in metastatic melanoma tissue in comparison with primary melanoma tissue27. A role of ANKRD13A as a gatekeeper of the early cell-death checkpoint, an escape mechanism from cell death in ovary cancer, has been suggested28. ITGA2B has been included in panel of four proteins predicting early breast cancer in serum29. CES1 has been suggested as a potential therapeutic target in melanoma due to its implication in monoacyglycerols metabolism30. No previous reports related to RSBN1L, GP9, PITPNM2 or RAB10 proteins with melanoma disease exist.
Some patients with advanced melanoma initially respond to anti-PD1 therapy but eventually progress, developing acquired or secondary resistance31. With the aim of characterizing acquired mechanisms of resistance, post-treatment samples were analyzed. In this case, node 16, which contained proteins related to proteasome complex & metabolism, had higher node activity in patients who progressed to anti-PD1 inhibitors. The interaction between proteasome and immune cells and its role in regulating PD-1/PD-L1 expression has been described previously, and proteasome inhibitors are being tested in combination with immunotherapy in several clinical trials32, suggesting that EVs could be used as a source of biomarkers in the management of this proteasome-related drugs.
Additionally, GSEA highlighted other processes in patients who progressed, such as MYC, MTOR, or interferon alpha and gamma pathways. Again, the enriched proteins related to interferon alpha and gamma, and MYC pathway are mostly related to the proteasome complex. Interestingly, the MTOR pathway included several proteins related to glucose metabolism and Warburg effect, like GAPDH, G6P, PGK1, TPI, GPI or LDHA, that were enriched in patients who progressed. Warburg effect consists of an increase of glucose uptake and fermentation into lactate, considered one of the hallmarks of cancer33,34. It was described that elevated LDHA expression in melanoma leads to a lactate accumulation that inhibits T cell activation, leading to tumor immune scape35. Moreover, melanoma patients with high expression of glycolytic genes response poorly to anti-PD1 inhibitors, while it has been shown that diclofenac lowers lactate levels and restore T cell function, improving response to checkpoint therapy in vivo36. Therefore, the use of metabolic inhibitors is a potential treatment to restore response to anti-PD1 inhibitors. Additionally, the presence of proteins related to glycolysis in exosomes has been previously associated with metastatic melanoma21.
Finally, dynamic changes in EV proteins during treatment administration and tumor evolution have been studied using the samples collected in all the time points. A significant decrease in proteins from node 6, involved in platelet activation and coagulation, in patients who progress to treatment during time has been shown. Tumor EVs promote coagulation, and the coagulation process is relevant in cancer progression and metastasis37. This node contained, among others, platelet glycoproteins, with a crucial role in hemostasis38. On the contrary, MHC related proteins increased during time in patients who progressed. In addition, it is described that tumor EVs mediate tumor antigen presentation to MHC-I, leading to the inhibition of CD8 + T cells39 which are necessary for the efficacy of anti-PD1 therapy40. The MHC related proteins identified are proteasome subunits, which are the source of most antigenic peptides presented to the immune system on MHC41. It was previously suggested that functional antigen proteasome presentation machinery is transferred to platelet-derived EVs, but no data about serum-derived EVs is available42. This increase in proteasome subunits and antigen presentation machinery in non-responders does not match with results observed in tumors, where a higher expression of an antigen presentation machinery signature has been associated with better DFS43.
The study has several limitations. First of all, the reduced number of samples analyzed constrains the statistical power of the study. To mitigate this, we used probabilistic graphical models to analyze our proteomics data, reducing the number of variables analyzed and increasing the power of the study. In addition, we limited our study to patients receiving anti-PD1 monotherapy, but it would be interesting to study the differences in EV content according to different immunotherapy schemes, including anti-CTLA4 or anti-LAG3 therapies. Finally, this is a proof-of-concept with the aim of demonstrate the feasibility to use EV from serum samples in prediction of response to anti-PD1 therapy. Our results regarding the implication of these proteins in prediction of treatment response and treatment resistance need to be validated in larger, independent cohorts.
The results support the potential of EVs as biomarkers for predicting immunotherapy response and understanding mechanisms of resistance. These findings pave the way for future research with larger cohorts and different therapeutic combinations.
Methods
Cohort
Samples from patients diagnosed with advanced melanoma who initiated anti-PD1 therapy in a single institution from 2022 to 2024 were prospectively included in this study. Uveal melamoma was excluded. The local ethical committee approved the study (PI-5403) and written informed consent from participants was obtained. All methods were performed in accordance with relevant guidelines.
Computed-tomography scan was used as per standard clinical practice to assess response using iRECIST44. Patients with progressive disease as best response were considered as non-responders, whereas patients with an objective response or stable disease were considered as responders. Progression-free survival (PFS) was calculated from treatment initiation to the time of progression or last follow-up. Overall survival (OS) was calculated from treatment initiation to the date of death or last follow up.
Sample processing
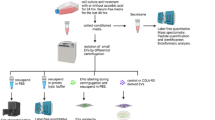
Blood samples were collected prior to treatment, after a first cycle of treatment, and at the time of progression or nine months after treatment initiation for each patient. Blood samples were collected in serum separator tubes and coagulated for 30 min. After that, blood samples were centrifuged at 3,000 g at 4 °C for 15 min to isolate the serum. Serum samples were aliquoted and stored at −80 °C until further use.
Extracellular vesicle isolation
Extracellular vesicles (EV) were isolated from 250 µL of serum using ExoQuick ULTRA EV precipitation solution (SBI, System Biosciences Mountain View, CA, USA), following manufacturer’s instructions. Briefly, ExoQuick solution was added, mixed and incubated for 30 min at 4 °C. The mixture serum/Exoquick was centrifuged at 3,000 g at 4 °C for 10 min. The obtained pellet was resolubilized and albumin and immunoglobulins were largely removed using binding columns, which were centrifuged at 1,000 g for 30 s. The obtained EVs were stored at −80 °C until further use.
Extracellular vesicle characterization
EVs characterization comprised three different techniques: Transmission electron microscopy (TEM), Western Blot, and NanoSight assessment.
The size and shape of the isolated EVs was examined using transmission electron microscopy (TEM). The EVs were fixed (5 min) on a drop of PFA 2%, washed two times with Milli-Q, and negatively stained with 2% aqueous uranyl acetate (1 min). The EVs were observed through a Tecnai 12 FEI electron microscope operated at 120 kV, and the images were recorded with a CCD (charged coupled device) FEI Ceta camera.
Western-Blot using a 4–10% SDS-PAGE electrophoresis gel was performed. 20 µg of total protein concentration were loaded in each western blot line based on Pierce™ BCA Protein Assay Kit (Thermo Scientific, USA) (working range: 20–2000 µg/mL). Specific antibodies against EV tetraspanins were used: anti-CD63 (1:1000, Invitrogen, Carlsbad, USA), anti-CD81 (1:1000, Invitrogen, Carlsbad, USA) and anti-CD9 (1:1000, Invitrogen, Carlsbad, USA), followed by a secondary HRP antibody (1:10000, Abcam, Cambridge, UK). Albumin was included as a negative (co-precipitated) marker to assess sample purity. Image was acquired using Pierce ECL chemoluminescence (Thermo Fisher Scientific, Waltham, USA) on an Uvitec-Cambridge image acquisition system (Uvitec, Cambridge, Reino Unido).
Finally, size and number of EVs were analyzed using a NanoSight LN10 (Malvern Instruments Ltd., Malvern, UK). Nanoparticle tracking analysis (NTA) was performed by applying light scattering and Brownian motion properties to determine the size and number of particles in a liquid suspension. EVs were diluted in 300 µL of sterile Phosphate Buffered Saline (PBS). Particle movement was captured by triplicate in 60-second videos at a detection threshold of 3. Recordings were analyzed using the NTA Software v. 4.1 (Malvern Instruments Ltd., Malvern, UK).
Sample Preparation for mass-spectrometry experiments
Isolated EV-samples were vacuum concentrated to dryness and resuspended in 100 µl of lysis buffer (2% sodium dodecyl sulfate (SDS), 1% sodium deoxycholate, 0,1% dodecyl-D-moltoside in 50 mM triethylammonium bicarbonate buffer pH 8.5 (TEAB)). Protein extraction was performed using a tissue homogenizer (TissueLyser II, QUIAGEN) for 4 min at 30 Hz. After that the samples were processed with high-intensity-focused ultrasound (Hielscher Ultrasonics GmbH) for 2 × 1 min (100% amplitude, 60% cycle time). Finally, the samples were incubated in a thermos shaker at 95 °C and 800 rmp for 10 min. The samples were centrifuged at 20,000 x g for 10 min and the protein concentration was determined using a Lunatic plate reader (Unchained Labs) with a 1:30 dilution of the original sample. For each sample 50 ug of protein were taken and reduced with 2 mM TCEP(tris(2-carboxyethyl)phosphine) and alkylated with 15 mM chloroacetamide at 60 °C for 30 min.
Protein purification, digest and peptide clean-up were performed using the sp3-protocol45 on a KingFisher Flex System (Thermo Fisher Scientific). Magnetic beads (Cytiva) were conditioned following the manufacturer’s instructions, consisting of 3 washes with water at a concentration of 1 ug/ul. Samples were diluted with an equal volume of 100% ethanol (60% ethanol final concentration). The beads, wash solutions and the samples were loaded into 96 deep well- or micro-plates and transferred to the KingFisher. The protocol on the robot carried out the following steps: collecting of beads from the last water wash, binding of the protein to the beads (14 min), washing the beads in wash1 (80% ethanol, 3 min), whash2 (80% ethanol, 3 min), wash3 (80% ethanol, 3 min), digesting the proteins at 37 °C (offline from the KingFisher, overnight at RT with trypsin to protein ratio of 1:50 in 50 mM TEAB pH 8.5) and eluting the remaining peptides from the magnetic beads (deionized water, 6 min). The digest solution and the water elution were combined and dried to completeness. Afterwards, the peptides were re-solubilized with 40 µl of 3% acetonitrile, 0.1% formic acid for MS analysis.
Mass-spectrometry experiments
Mass spectrometry analysis was performed on an Orbitrap Exploris mass spectrometer (Thermo Scientific) equipped with a Digital PicoView source (New Objective) and coupled to a M-Class UPLC (Waters). Solvent composition at the two channels was 0.1% formic acid in water for channel A and 0.1% formic acid in acetonitrile for channel B. For each sample 2 µL of peptides were loaded on a commercial MZ Symmetry C18 Trap Column (100 Å, 5 μm, 180 μm x 20 mm, Waters) followed by nanoEase MZ C18 HSS T3 Column (100 Å, 1.8 μm, 75 μm x 250 mm, Waters). The peptides were eluted at a flow rate of 300 nl/min by a gradient from 5 to 22% B in 80 min, 32% B in 10 min and 95% B in 1 min. Samples were acquired in a randomized order. The mass spectrometer was operated in data-independent mode (DIA), acquiring a full-scan MS (396–1000 m/z) at a resolution of 60’000 at 200 m/z. Followed by HCD (higher-energy collision dissociation) fragmentation on 70 windows with 8 m/z width between 400 and 960 m/z. The MS2 resolution was set to 30’000 and a normalized collision energy of 30% was used. The samples were acquired using internal lock mass calibration on m/z 371.1012 and 445.1200.
The mass spectrometry proteomics data were handled using the local laboratory information management system (LIMS)46 and all relevant data have been deposited to the ProteomeXchange Consortium via the RIDE (https://www.ebi.ac.uk/pride/login) partner repository with the data set identifier PXD062378, Username: reviewer_pxd062378@ebi.ac.uk Password: x4OPUvEoaRma47.
Spectral library generation and protein quantification
The identification and quantification of proteins from MS data was performed using the DIA-NN workflow (version 1.8.248,. The following parameters for the library-free search were used: precursor charge + 2 and + 3, precursor mass range 350 m/z to 1500 m/z, fragment mass range 200 m/z to 1800 m/z, mass accuracy MS1 15 ppm, MS2 20 ppm, enzyme specificity trypsin allowing one missed cleavage, fixed modification carbamidomethylation of cysteine. The in-silico spectra were generated using a canonical protein database for humans, concatenated with common protein contaminants (fgcz_9606_1spg_20230330.fasta, ID9606, release date 20230330) and the maximum false discovery rate (FDR) was set to 0.01.
Statistical modelling of proteomics data
The R package prolfqua49 was used to analyze the differential expression and to determine group differences, confidence intervals, and false discovery rates for all quantifiable proteins. Starting with the report.tsv file generated by DIA-NN, which does report the precursor ion abundances for each raw file, we determined protein abundances by first aggregating the precursor abundances to peptidoform abundances. Then, we employed the Tukeys-Median Polish to estimate protein abundances. Furthermore, before fitting the linear models, we transformed the protein abundances using the variance stabilizing normalization50.
Preprocessing of proteomics data
Perseus software was used for proteomics data preprocessing51. First, data was log2 transformed. Then, two filtering criteria were applied: a percentage of valid values in 100% of the samples from the same patient and at least 50% of valid values in total per protein. Lastly, missing values were imputed using a normal distribution with Perseus default setting.
Protein networks built using probabilistic graphical models
High-dimensional compatible probabilistic graphical models (PGM) were used to build protein networks and define differentially biological processes between responders and non-responders, as previously described52. Briefly, the network was built in two sequential steps: first, the spanning tree with the maximum likelihood was determined and, then, Bayesian Information Criterion (BIC) was used to penalize more complex models53. PGM was built using functions of grapHD54 R package implemented in Python.
The obtained network was split into nodes using Louvain community detection algorithm55. The overrepresented function of each defined node was determined by gene ontology analyses using the GOAtools Python package and DAVID database56. Cellular compartment, molecular function and biological process databases were used for the analysis. Homo sapiens was selected as background. To select an overrepresented function for each node, gene ontology categories enriched in the complete dataset were excluded from the analysis, and then, gene ontology categories significantly overrepresented in each functional node were defined, using an FDR adjusted p-value < 0.05 on a Fisher’s exact test.
To make comparisons between responders and non-responders, the functional node activity, i.e. the mean expression of the proteins that conform this node, was calculated52.
Statistical analyses
Comparisons between groups of samples at a one time-point were done using a non-parametric Mann-Whitney test. P-values were corrected using Benjamin-Hochberg. These comparisons were done using Graph Pad Prism version 6. Comparisons between the three time points were done using mixed lineal models with fixed effects. The time point and progression or not to PD1 inhibitors were set as fixed effects whereas the patient was set as the variable effect. P-values were two-sided and corrected using FDR. Mixed lineal models were calculated using R with glmer and emmeans packages.
Gene Set Enrichment Analysis (GSEA) was used to identify, on the one hand, mechanisms of innate resistance in pre-treated samples, and, on the other hand, acquired mechanisms of resistance in post-treatment samples. Hallmarks of cancer database was used as background and gene sets were considered statistically significant when FDR < 5%.
Construction of predictive signatures
Predictive signatures were built using BRB Array Tools57. First, proteins or node activities related to PFS were selected based on their p-values. Then, a predictive signature was built using the supervised principal component method of E. Bair and R. Tibshirani58, based on a Cox proportional hazards model. The threshold significance level was set to 0.05. Leave-one-out cross validation was used to determine the predictive accuracy of the models. A sample is omitted and the prognostic index was calculated. Having determined a prognostic index function for that training set, it is used to compute a prognostic index for the omitted observation. That value is compared to the prognostic index for the n-1 cases included in that training set. The prognostic index for the omitted sample is ranked relative to the prognostic index for the samples included in the cross-validated training set. The omitted sample is classified into a risk group based on its percentile ranking, the number of risk groups specified, and the cut-off percentiles specified for defining the risk groups. This analysis is repeated from scratch n times, leaving out a different case each time. The cut-off was defined a priori in all cases and the log-rank test p-value was evaluated using 1,000 random permutations. In each permutation, the test is re-calculated in order to determine a p-value.
Data availability
All relevant data have been deposited to the ProteomeXchange Consortium via the PRIDE (https://www.ebi.ac.uk/pride/login) partner repository with the data set identifier PXD062378.
References
Dummer, R., Keilholz, U. & Committee, E. G. Appendix 2: cutaneous melanoma (2): eUpdate. Ann. Oncol. 27, v136–v137. http://www.esmo.org/Guidelines/Melanoma (2016).
Robert, C. et al. Pembrolizumab versus ipilimumab in advanced melanoma (KEYNOTE-006): post-hoc 5-year results from an open-label, multicentre, randomised, controlled, phase 3 study. Lancet Oncol. 20, 1239–1251 (2019).
Robert, C. et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl. J. Med. 372, 2521–2532 (2015).
Robert, C. et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl. J. Med. 372, 320–330 (2015).
Ayers, M. et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 Blockade. J. Clin. Invest. 127, 2930–2940 (2017).
Morrison, C. et al. Predicting response to checkpoint inhibitors in melanoma beyond PD-L1 and mutational burden. J. Immunother Cancer. 6, 32 (2018).
Yáñez-Mó, M. et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles. 4, 27066 (2015).
Becker, A. et al. Extracellular vesicles in cancer: Cell-to-Cell mediators of metastasis. Cancer Cell. 30, 836–848 (2016).
Fan, Y. et al. Exosomal PD-L1 retains immunosuppressive activity and is associated with gastric cancer prognosis. Ann. Surg. Oncol. 26, 3745–3755 (2019).
Chen, G. et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 560, 382–386 (2018).
Lener, T. et al. Applying extracellular vesicles based therapeutics in clinical trials - an ISEV position paper. J. Extracell. Vesicles. 4, 30087 (2015).
Bernardi, S. et al. Extracellular vesicles in the chronic myeloid leukemia scenario: an update about the shuttling of disease markers and therapeutic molecules. Front. Oncol. 13, 1239042 (2023).
Taylor, D. D., Taylor, C. G., Jiang, C. G. & Black, P. H. Characterization of plasma membrane shedding from murine melanoma cells. Int. J. Cancer. 41, 629–635 (1988).
Mears, R. et al. Proteomic analysis of melanoma-derived exosomes by two-dimensional polyacrylamide gel electrophoresis and mass spectrometry. Proteomics 4, 4019–4031 (2004).
Logozzi, M. et al. High levels of exosomes expressing CD63 and caveolin-1 in plasma of melanoma patients. PLoS One. 4, e5219 (2009).
Poggio, M. et al. Suppression of Exosomal PD-L1 induces systemic Anti-tumor immunity and memory. Cell 177, 414–427e413 (2019).
Cordonnier, M. et al. Tracking the evolution of Circulating exosomal-PD-L1 to monitor melanoma patients. J. Extracell. Vesicles. 9, 1710899 (2020).
Serratì, S. et al. Circulating extracellular vesicles expressing PD1 and PD-L1 predict response and mediate resistance to checkpoint inhibitors immunotherapy in metastatic melanoma. Mol. Cancer. 21, 20 (2022).
Macciò, A. & Madeddu, C. Blocking inflammation to improve immunotherapy of advanced cancer. Immunology 159, 357–364 (2020).
Gyukity-Sebestyén, E. et al. Melanoma-Derived exosomes induce PD-1 overexpression and tumor progression via mesenchymal stem cell oncogenic reprogramming. Front. Immunol. 10, 2459 (2019).
Lazar, I. et al. Proteome characterization of melanoma exosomes reveals a specific signature for metastatic cell lines. Pigment Cell. Melanoma Res. 28, 464–475 (2015).
Aguado, B. A. et al. Extracellular matrix mediators of metastatic cell colonization characterized using scaffold mimics of the pre-metastatic niche. Acta Biomater. 33, 13–24 (2016).
Peinado, H. et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 18, 883–891 (2012).
Alegre, E. et al. Circulating melanoma exosomes as diagnostic and prognosis biomarkers. Clin. Chim. Acta. 454, 28–32 (2016).
Pietrowska, M. et al. Proteomic profile of melanoma cell-derived small extracellular vesicles in patients’ plasma: a potential correlate of melanoma progression. J. Extracell. Vesicles. 10, e12063 (2021).
Fagerberg, L. et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol. Cell. Proteom. 13, 397–406 (2014).
Li, K. et al. Identification of keratinocyte Differentiation-Involved genes for metastatic melanoma by gene expression profiles. Comput. Math. Methods Med. 2021, 9652768 (2021).
Won, M. et al. ANKRD13a controls early cell-death checkpoint by interacting with RIP1 independent of NF-κB. Cell. Death Differ. 29, 1152–1163 (2022).
Fredolini, C. et al. Shotgun proteomics coupled to nanoparticle-based biomarker enrichment reveals a novel panel of extracellular matrix proteins as candidate serum protein biomarkers for early-stage breast cancer detection. Breast Cancer Res. 22, 135 (2020).
Morozova, V., Pellegata, D., Charles, R. P. & Gertsch, J. Carboxylesterase 1-mediated endocannabinoid metabolism in skin: role in melanoma progression in BRaf. Cancer Metab. 13, 8 (2025).
Trojaniello, C., Vitale, M. G., Scarpato, L., Esposito, A. & Ascierto, P. A. Melanoma immunotherapy: strategies to overcome Pharmacological resistance. Expert Rev. Anticancer Ther. 20, 289–304 (2020).
Wang, Y., Li, S. & Wang, W. The ubiquitin-proteasome system in the tumor immune microenvironment: a key force in combination therapy. Front. Immunol. 15, 1436174 (2024).
Warburg, O. The metabolism of carcinoma cells. J. Cancer Res. 9, 148–163 (1925).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Brand, A. et al. LDHA-Associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell. Metab. 24, 657–671 (2016).
Renner, K. et al. Restricting Glycolysis preserves T cell effector functions and augments checkpoint therapy. Cell. Rep. 29, 135–150e139 (2019).
Yu, J. L. et al. Oncogenic events regulate tissue factor expression in colorectal cancer cells: implications for tumor progression and angiogenesis. Blood 105, 1734–1741 (2005).
Thomas, S. Platelet membrane glycoproteins in haemostasis. Clin. Lab. 48, 247–262 (2002).
Leary, N. et al. Melanoma-derived extracellular vesicles mediate lymphatic remodelling and impair tumour immunity in draining lymph nodes. J. Extracell. Vesicles. 11, e12197 (2022).
Tumeh, P. C. et al. PD-1 Blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571 (2014).
Gaczynska, M., Rock, K. L. & Goldberg, A. L. Role of proteasomes in antigen presentation. Enzyme Protein. 47, 354–369 (1993).
Marcoux, G. et al. Platelet EVs contain an active proteasome involved in protein processing for antigen presentation via MHC-I molecules. Blood 138, 2607–2620 (2021).
Thompson, J. C. et al. Gene signature of antigen processing and presentation machinery predicts response to checkpoint Blockade in non-small cell lung cancer (NSCLC) and melanoma. J Immunother Cancer 8(2). (2020).
Seymour, L. et al. iRECIST: guidelines for response criteria for use in trials testing immunotherapeutics. Lancet Oncol. 18, e143–e152 (2017).
Hughes, C. S. et al. Single-pot, solid-phase-enhanced sample Preparation for proteomics experiments. Nat. Protoc. 14, 68–85 (2019).
Turker, C. A. et al. R. B-Fabric: the Swiss Army Knife for Life Sciences EDBT 2010 (Lausanne, Switzerland 2010).
Deutsch, E. W. et al. The proteomexchange consortium at 10 years: 2023 update. Nucleic Acids Res. 51, D1539–D1548 (2023).
Demichev, V., Messner, C. B., Vernardis, S. I., Lilley, K. S. & Ralser, M. DIA-NN: neural networks and interference correction enable deep proteome coverage in high throughput. Nat. Methods. 17, 41–44 (2020).
Wolski, W. et al. prolfquapp - A user-friendly command-line tool simplifying differential expression analysis in quantitative proteomics. bioRxiv, (2024).
Huber, W., von Heydebreck, A., Sültmann, H., Poustka, A. & Vingron, M. Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18 (Suppl 1), S96–104 (2002).
Tyanova, S. et al. The perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 13, 731–740 (2016).
Gámez-Pozo, A. et al. Combined label-free quantitative proteomics and MicroRNA expression analysis of breast cancer unravel molecular differences with clinical implications. Cancer Res. 75, 2243–2253 (2015).
Lauritzen, S. Graphical Models (Oxford University Press, 1996).
Abreu, G., Edwards, D. & Labouriau, R. High-Dimensional Graphical Model Search with the gRapHD R Package 37 pp 1–18 (Journal of Statistical Software, 2010).
Blondel, V. D., Guillaume, J-L., Lambiotte, R. & Lefebvre, E. Fast unfolding of communities in large networks, 10. Journal of Statistical Mechanics: Theory & Experiment, p P10008. (2008).
Huang dW, Sherman, B. T. & Lempicki, R. A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4, 44–57 (2009).
Simon, R. Roadmap for developing and validating therapeutically relevant genomic classifiers. J. Clin. Oncol. 23, 7332–7341 (2005).
Bair, E. & Tibshirani, R. Semi-supervised methods to predict patient survival from gene expression data. PLoS Biol. 2, E108 (2004).
Acknowledgements
We would like to thank you to the Hospital La Paz Biobank and Transmission Electronic Microscopy Service of Universidad Autónoma de Madrid.
Funding
This work was supported by a grant from GEM (Spanish Melanoma Group).
Author information
Authors and Affiliations
Contributions
LT-F, AG-P, FL-G, GM, FB-G, LS, AG-C, RL-V, MB, BR, MG-F, and PN contributed to methodology (sample processing and experiments). LT-F, AG-P, FB-G, MD-A, PL-D, and JAFV did the formal analyses. LT-F and AG-P wrote the original draft. EE contributed to data curation, conceptualization, funding acquisition, supervision, and revised the original draft.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics approval statement
The ethical committee of La Paz University Hospital approved the study (PI-5403) and written informed consent from participants was obtained.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Trilla-Fuertes, L., Gámez-Pozo, A., Laso-García, F. et al. Protein content of extracellular vesicles from patients with advanced melanoma changes upon progression to anti-PD1 therapy. Sci Rep 16, 5891 (2026). https://doi.org/10.1038/s41598-026-35848-0
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-35848-0
