
Abstract
The composition and metabolic characteristics of gut microbiota in heart transplant patients with and without preoperative and postoperative infection were investigated to elucidate the impact of antibiotics and immunosuppressants. Twenty patients undergoing allogeneic heart transplantation were enrolled. Gut microbiota profiles were analyzed via 16 S rRNA gene sequencing and metabolic pathway prediction (PICRUSt2) at 5–9 days preoperatively and 30 days postoperatively. Patients were categorized into preoperative non-infected (Pre-NI), preoperative infected (Pre-I), postoperative non-infected (Post-NI) and postoperative infected (Post-I) groups for comparative analysis. Alpha/beta diversity, Maaslin and functional analyses were performed. Preoperative microbiota had similar α-diversity indices and community structures among groups. Pre-NI microbiota were dominated by Bacteroides and Pre-I samples were enriched with Enterococcus casseliflavus、Limosilactobacillus and Weissella cibaria. Post-NI patients showed higher microbial diversity (Shannon/Simpson indices) and Blautia (butyrate-producer) whereas Post-I patients formed a distinct cluster with Enterococcus faecium dominance. Metabolic analysis highlighted Blautia-associated pathways, e.g., L-1,2-propanediol degradation. Dynamic preoperative shifts in the Bacteroides-Enterococcus-Limosilactobacillus axis and postoperative shifts toward Blautia-Enterococcus dominance were found in heart transplant patients and were associated with infection status and antibiotic exposure. Dual-target intervention with an emphasis on restoring butyrate-producing microbiota and monitoring Enterococcus dynamics is recommended to optimize personalized infection control systems in transplant patients.
Similar content being viewed by others
Introduction
Heart transplantation is the only option to prolong survival in patients with end-stage heart failure. However, postoperative infection is the foremost risk factor, causing early mortality and long-term complications1,2. Postoperative infection rates exceed 40% within the first 30 days, often leading to multi-organ dysfunction and secondary infection associated with immunosuppression1,3. The dynamic equilibrium of gut microbiota may influence the maintenance of postoperative immune homeostasis and metabolic repair. Gut microbiota dysbiosis may promote inflammatory cascades and pathogen colonization through microbiota-host metabolic interactions4. For instance, antibiotic exposure in the early postoperative stage reduces beneficial microbes, enriches conditionally pathogenic species and elevates infection susceptibility5,6. The composition of gut microbiota in heart transplant recipients is affected by preoperative comorbidities, surgical trauma, immunosuppressive regimens and postoperative anti-infective therapies7. Pathophysiological processes associated with heart transplantation include extracorporeal circulation-induced systemic inflammatory storm, broad immunosuppression from potent induction regimens, and postoperative infections and associated antibiotic use. Such conditions may have different impacts on gut microbiota dynamics compared with other solid-organ transplants8. However, previous work has usually focused on static microbiota analyses at single time points, so that perioperative changes in microbiota and causal relationships with infections remain poorly understood.
The current investigation arose from the hypothesis that perioperative gut microbiota undergo compositional and functional shifts in heart transplant recipients and that microbiota-immune-metabolic interactions are imbalanced by dysbiosis with an impact on infection risk. The current longitudinal study integrated high-throughput 16 S rRNA gene sequencing with differential taxa identification using Maaslin29 and functional prediction analysis with PICRUSt10. The following were the three core objectives: (1) to characterize the dynamic evolution of gut microbiota structure during the perioperative period of heart transplantation via 16 S rRNA gene sequencing; (2) to investigate the impact of clinical interventions, including antibiotics and immunosuppressants, on gut microbial composition and (3) to conduct a preliminary exploration of the association between perioperative structural shifts in gut microbiota and predicted metabolic potential and to analyze links with infection risk.
Methods
General patient characteristics
Recipients of allogeneic heart transplants at our center were enrolled between June 2023 and October 2024. Inclusion criteria were: patients with end-stage heart failure due to dilated cardiomyopathy unresponsive to medical therapy or conventional surgical intervention, according to the Technical Guidelines for Preoperative Assessment and Preparation of Allogeneic Heart Transplant Recipients in China (2019)11, age between 18 and 60 years. Infection diagnoses were based on the Hospital-Acquired Infection Diagnostic Standards published by the National Health Commission of China in 200112, adhering to clinical diagnostic criteria with validation by pathogen-specific evidence. Exclusion criteria were: patients with multi-organ transplantation, prior heart re-transplantation, severe comorbidities (e.g., malignant neoplasms, hepatic or renal failure), a history of inflammatory bowel disease or intestinal resection or postoperative severe graft rejection. In addition, insufficient fecal sample quantity, contamination, improper storage, patient refusal to participate or loss to follow-up also resulted in exclusion.
Preoperative fecal samples were collected at 5–9 days before heart transplantation and postoperatively at 30 days post-transplantation.
Subject selection and sample exclusion
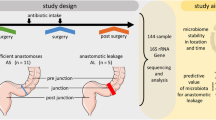
Fifty patients with end-stage dilated cardiomyopathy were initially enrolled from the heart transplant waiting list who had a stable clinical condition for ≥ 2 weeks, with no acute decompensation events, and consecutive three-day stool sampling was performed (For subsequent analyses, only the quality-controlled sample collected closest to the date of heart transplantation was selected from each patient). 25 patients were excluded during the study due to transplantation delays (20 patients failed to find compatible blood/tissue-matched donors and 5 had unsatisfactory preoperative evaluations). An additional 7 preoperative stool specimens were excluded due to DNA concentration < 5 ng/µL or PCR amplification failure. Cohort homogeneity of antibiotic exposure and standardized sampling in the limited postoperative sample were maintained by excluding 6 patients who had received non-β-lactam antibiotics. A further five patients who did not provide samples at the designated postoperative time points were also excluded. 20 subjects were finally enrolled, 14 males and 6 females, yielding 32 qualified stool samples (Fig. 1).

Study flow diagram. n: Number of enrolled patients; s: Fecal samples.
Group definitions
Preoperative Infection (Pre-I, s = 7): patients had confirmed pre-transplant infection based on pathogen-positive results and clinical diagnosis. Preoperative Non-infection (Pre-NI, s = 11): patients showed no evidence of pre-transplant infection and had pathogen-negative results with an absence of clinical symptoms. Postoperative Infection (Post-I, s = 7): patients had confirmed infection within 30 days post-transplantation. Postoperative Non-infection (Post-NI, s = 7): patients showed no evidence of infection within 30 days post-transplantation (see Tables S1 and S2 for clinical characteristics).
Ethical compliance and patient consent
This study is a retrospective data analysis conducted in accordance with the ethical guidelines of the Declaration of Helsinki (2000) and the Istanbul Declaration (2008). It was approved by the Ethics Committee of the Seventh People’s Hospital of Zhengzhou (Approval No.: 2024[ky-018]). We hereby declare that: all transplant organs/tissues originated from voluntary and unpaid donations by Chinese citizens, NO organs/tissues were procured from prisoners. Organ allocation was conducted through the “China Organ Transplant Response System” (COTRS). Written informed consent was obtained from all patients and their immediate family members, and all patient data were de-identified to ensure privacy protection.
Perioperative therapy and antibiotic prophylaxis
Patient demographic and clinical data were accessed from electronic medical records (EMR) and antibiotic use within the two weeks preceding stool sample collection was documented. No adjustment was made for antibiotic exposure in the Pre-I group due to short treatment duration. No adjustment was made for antibiotic exposure in the Post-I group due to minimal variability in antibiotic type and administration timing. All patients received standard perioperative prophylactic regimens of no more than 2 weeks’ duration, consisting of intravenous piperacillin-tazobactam to prevent bacterial infection, ganciclovir to prevent viral infection and trimethoprim-sulfamethoxazole tablets to prevent Pneumocystis jirovecii pneumonia. The post-transplant standard protocol was followed by a triple immunosuppressive regimen of mycophenolate mofetil, prednisone and tacrolimus (see Tables 1, 2 for pathogens and antibiotics used).
Sample collection and DNA extraction
Fecal samples were collected into sterile containers and immediately stored at -80 °C. Genomic DNA was extracted by SDS lysis buffer and freeze-thaw cycles and assessed for quality control (QC) prior to amplification by PCR.
PCR amplification and sequencing of 16 S rRNA gene
Full-length bacterial 16 S rRNA gene sequences were amplified using the forward primer, 27 F, and reverse primer, 1429R. A unique 7-bp barcode sequence was incorporated into the PCR products to enable sample tracking. PCR amplicons were purified using AMPure XP Beads (Beckman Coulter, Indianapolis, IN, USA) and quantified with the Qubit™ dsDNA Assay Kit (Invitrogen, Carlsbad, CA, USA). DNA libraries were sequenced using the Oxford Nanopore Technologies GridION nanopore sequencing platform by DiPu Co., Ltd.
Bioinformatics and statistical analyses
Raw sequencing data were subjected to quality filtering and de novo chimera detection, followed by analysis using QIIME2 and R software (version 3.2.0).
Alpha Diversity Analysis: Alpha diversity indices at the operational taxonomic unit (OTU) level were calculated using QIIME2, including Chao1, ACE, Faith’s phylogenetic diversity (PD_whole_tree), Shannon and Simpson indices. A ranked abundance curve and rarefaction curve were generated to assess species richness and evenness. Intergroup differences in alpha diversity indices were analyzed to compare OTU abundance and evenness among samples.
Beta Diversity Analysis and Inter-Group Difference Testing: Beta diversity was analyzed using QIIME2 to calculate UniFrac distance metrics (unweighted and weighted UniFrac). Principal component analysis (PCA) plots were generated to visualize differences in microbial community structures. Inter-group differences in UniFrac distances were compared by t-test and Monte Carlo permutation test and are visualized as boxplots. ANalysis Of Similarity (ANOSIM) was conducted to compare intergroup differences in community composition with intragroup variation.
Differential Taxonomic and Functional Analyses: Taxonomic groups with significant inter-group abundance differences at phylum-to-genus classification levels were identified by Kruskal-Wallis test using the stats package in R. Differential abundance analysis between groups was performed using Maaslin2 with default parameter settings. Potential functional differences were explored by prediction of microbial community functional profile via PICRUSt2 and pathway annotations were generated. Microbial community functional annotation was conducted using FAPROTAX13, based on taxonomic classification results from 16 S rRNA gene sequences.
All statistical analyses were conducted using python 3.13. Continuous variables are presented as mean ± SD deviation, if normally distributed, or as median with interquartile range [Median (Q1-Q3)], if non-normally distributed. Categorical variables are presented as frequencies and percentages [n (%)]. Intergroup comparisons of continuous variables were made by Student’s t-test for normally distributed data (assessed via the Shapiro-Wilk test) or Wilcoxon rank-sum test for non-normal distributions. Categorical variables were compared using Fisher’s exact test and a p value < 0.05 was considered to indicate a significant difference.
Results
Analysis of gut microbiota diversity
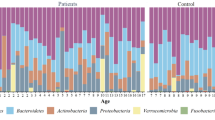
After quality-filtering, denoising and operational taxonomic unit (OTU) clustering of sequencing data using the QIIME 2 pipeline, alpha-diversity metrics including the Chao1 and Shannon indices were calculated by vegan package in R to assess intra-community species richness and evenness. No significant differences in gut microbiota alpha diversity indices, Ace, Chao, Shannon and Simpson, were observed between Pre-NI and Pre-I groups (p > 0.05, Table 3), suggesting comparable species richness. By contrast, no differences in the OTU, Ace and Chao indices were seen for Post-NI and Post-I groups, but Shannon and Simpson indices were significantly higher for Post-NI than for Post-I, indicating differences in species composition (Table 4). The dilution curve plateaued at higher sequencing depths, indicating the reflection of total microbial community information by the sequencing data (Fig. S1). A subset of Amplicon Sequence Variants (ASVs) identified after taxonomic annotation could be annotated at the species level with the majority classified at the genus or species level (Table 5). Subsequent analyses therefore focused primarily on genus and species levels. In addition, the number of effective sequencing reads per sample (Fig. S2) met the analytical requirements, ensuring that the current sequencing depth was sufficient to uncover biologically significant findings with statistical rigor. Genus-level abundance analysis showed great similarity between Pre-I and Pre-NI community structures (Fig. 2a) but Post-NI samples had higher microbial diversity and a more balanced community structure compared with Post-I in which increased pathogenic genera, such as Enterococcus, could be seen (Fig. 2b). Overall, Post-I patients had reduced alpha diversity and abnormally enriched Enterococcus, indicating potential microbial dysbiosis.

Genus-level taxonomic composition: stacked bar plots. a Pre-I vs. Pre-NI and b Post-NI vs. Post-I. The horizontal axis lists sample identifiers; the vertical axis denotes the relative abundance percentage of taxonomic units. Individual samples are represented by stacked bars with different colors assigned to different genera (taxonomic units). The length of each colored segment within a bar reflects the proportional contribution of a specific genus to the sample’s microbial composition with longer segments showing higher relative abundance.
Inter-group comparison of gut microbiota composition
Beta-diversity analysis was conducted using principal component analysis (PCA) based on Bray-Curtis dissimilarity matrices and the significance of inter-group community structure differences tested via permutational multivariate analysis of variance (PERMANOVA). Grouping validity was assessed by non-parametric analysis of similarities (ANOSIM), comparing inter-group versus intra-group differences to assess the statistical meaningfulness of grouping. No statistically significant difference was observed between the Pre-NI and Pre-I groups (R > 0, p > 0.05; Fig. 3a). A significant distinction between Post-NI and Post-I groups was found (R > 0, p < 0.05; Fig. 3b). PCA allowed inter-sample species diversity differences to be visualized. Pre-NI, Pre-I and Post-NI groups had clustered distributions primarily along the positive direction of the PC1 axis with overlapping clusters and minimal intra-group sample dispersion. By contrast, Post-I group samples were significantly enriched along the negative PC1 axis, showing clear segregation from other groups (Fig. 4). In addition, the hierarchical clustering shown in Fig. 3c which was based on the similarity of taxonomic units across samples, vertically presents clustering outcomes to illustrate associations between sample similarities and genus-level community composition. Consistent with the PCoA results, the Post-I group formed an independent cluster, reinforcing observed separation trends.

Comparative analysis of gut microbiota group variability. a Pre-NI vs. Pre-I: ANOSIM-Based boxplot of bray-curtis dissimilarity. b Post-NI vs. Post-I: ANOSIM-based boxplot of bray-curtis dissimilarity. The x-axis shows group labels. “Between” represents the rank of pairwise sample distances sorted by Bray-Curtis dissimilarity. The y-axis indicates the numerical distribution of these ranks. Statistical significance is denoted by p values (p < 0.05 indicates significance). c Hierarchical clustering based on genus-level relative abundance. Genus-level relative abundance data were hierarchically clustered and ordered by sample similarity. Colored labels distinguish groups; closer clustering indicates greater microbial composition similarity.

Principal component analysis (PCA) Plot. The horizontal axis corresponds to the first principal component (PC1) and its contribution rate; the vertical axis represents the second principal component (PC2) and its contribution rate. Individual samples are represented by data points, colored by group origin. Proximity between points reflects sample similarity with samples from the same group circled for emphasis.
Maaslin identification of differentially abundant taxa as potential biomarkers
Maaslin analysis illustrated individual microbial signatures for the different groupings (Fig. 5a). Bacteroides dominated the Pre-NI samples, exhibiting hierarchical dominance across multiple taxonomic levels, including class, order, family, and genus. By contrast, the dominant bacterial taxa in the Pre-I group included Enterococcus casseliflavus, Limosilactobacillus, Clostridium phoceensis and Weissella cibaria.

Identification of significant taxonomic biomarkers via inter-group Maaslin analysis. The x-axis represents the log-fold change (logFC) of the correlation coefficients(coef), indicating the magnitude of change in feature abundance per unit change in the target variable after adjusting for covariates. The y-axis lists the significantly associated omics features. Feature selection was based on a false discovery rate (FDR)-corrected q-value < 0.05 and an absolute correlation coefficient > 0.5. A positive coefficient indicates a positive correlation between the target variable and feature abundance, while a negative coefficient indicates a negative correlation. a Pre-NI vs. Pre-I. b Post-NI vs. Post-I.
Post-I samples were enriched in the class of Bacilli, order Lactobacillales, genus Enterococcus and family Enterococcaceae with Enterococcus faecium being the dominant species. Post-NI samples were dominated by genera within the Lachnospiraceae family, including Fusicatenibacter, Dorea, Lachnospira and Blautia (Fig. 5b). Dominant Post-NI species were Fusicatenibacter saccharivorans, Ruminococcus torques, Blautia provencensis, Blautia luti, Blautia wexlerae and Blautia obeum.
Microbiome and metabolome correlation
Microbial metabolic functional potentials based on 16 S rRNA gene sequencing data were predicted by PICRUSt2 algorithm and inter-group differential analysis performed via STAMP software. Significant inter-group differences in predicted microbial metabolic functional profiles were found, accompanied by distinct correlation patterns. For instance, the predicted functional potentials of pathways including formaldehyde assimilation II (RuMP cycle), uridine degradation to glyoxylate III and D-galacturonic acid degradation I were significantly different between Pre-NI and Pre-I groups (Fig. 6a). Correlation analysis showed a positive correlation between Enterococcus casseliflavus abundance and predicted potential for acetone fermentation (p < 0.05, Fig. 6b), suggesting a potential role of this bacterium in acetone metabolism.

Association analysis of the functional potential of microbial metabolic pathways predicted by PICRUSt2 and FAPROTAX. a Pre-NI vs. Pre-I and c Post-NI vs. Post-I. The x-axis shows functional gene abundance (log2 CPM); the y-axis lists metabolic pathways. Bubble size reflects pathway abundance magnitude. Color saturation indicates confidence (greener shades denote higher confidence). Pathways with p-values < 0.05 are highlighted. b and d Microbial-Metabolic Pathway Correlation Heatmaps. Rows represent microbial taxa; columns represent metabolic pathways. Color gradients from blue (negative correlation) to red (positive correlation) depict correlation strength. Asterisks (*) denote significant correlations (p-value < 0.05).
Differences in gut microbiota structure between Post-NI and Post-I groups extended to predicted metabolic functional potentials. Analysis identified significant differences in the predicted potentials of multiple pathways, including L-1,2-propanediol degradation which showed the most pronounced disparity (Fig. 6c). Further association analysis indicated that the abundance of the genus Blautia was positively correlated with predicted potentials of pathways including L-1,2-propanediol degradation, arginine, ornithine and proline interconversion (p < 0.05, Fig. 6d). These findings imply that the observed differences in Blautia abundance between Post-NI and Post-I groups may reflect functional gene variations in microbial communities associated with specific substrates, such as propanediol and amino acids. Against the backdrop of limited microbiome studies post-cardiac transplantation infection, this work delineates specific correlation patterns between enriched gut microbial taxa in heart transplant recipients and their predicted metabolic functions. These findings offer novel insights that bridge descriptive microbial profiling and functional inference, illuminating how specific microbes may participate in host physiological processes through distinct metabolic pathways in this clinical setting. Although derived from exploratory correlation analyses, our results provide preliminary evidence and generate testable hypotheses for future investigations aimed at elucidating the functional roles and potential impacts of key intestinal microorganisms following heart transplantation.
Discussion
Investigations of bidirectional modulation between immunosuppressive therapy and gut microbiota in solid organ transplantation (SOT) have given insights into the impact on transplantation outcomes but few studies have focused on cardiac transplantation14. The current study examined dynamic changes in gut microbiota composition and metabolic profiles in cardiac transplant recipients in pre- and postoperative non-infected and infected states. Post-NI patients had greater gut microbiota diversity and a more stable community structure at the genus level at 30 days post-surgery. Non-infected patients were dominated by Bacteroides at baseline but composition shifted toward Blautia enrichment at the post-operative stage. Enterococcus is the predominant pathogen in patients with postoperative infections. These findings suggest that surgical intervention and infection status influence the nature of dominant strains in the microbiota. The shift toward Enterococcus for Post-I patients indicates dysbiosis of microbiota diversity due to infection and antibiotic use.
The healthy gut has microbiota primarily belonging to Bacteroidetes and Firmicutes with minor contributions from other phyla, such as Proteobacteria, Actinobacteria and Verrucomicrobia15. However, stressors, such as infection, disrupt this balance16. Maaslin analysis revealed that the dominant Bacteroides of the Pre-NI microbiota maintained microecological homeostasis via polysaccharide degradation and immune regulation17,18,19,20. By contrast, the enrichment of the Post-NI microbiota with butyrate-producing Blautia indicates the production of short-chain fatty acids (SCFAs), which promote immunostability and infection resistance21,22,23. The coexistence of Blautia with Fusicatenibacter and Roseburia (Lachnospiraceae family) stabilized SCFA production and suppressed proliferation of pathogens. The dominant Enterococcus faecium of Post-I patients has been associated with antibiotic resistance, poor prognosis and elevated mortality in critical care settings24,25,26. Previous studies have shown increased Enterococcus in liver transplant recipients within one year post-surgery27, consistent with the changes presented for Post-I cardiac transplant recipients in the present cohort. A greater abundance of Enterococcus, especially in lower-diversity communities, characterizes gut microbiota diversity of critically ill ICU patients and elevated Enterococcus at hospital admission is considered a significant predictor of mortality or infection in ICU patients28,29. The current Pre-I patients had a transient “beneficial-pathogenic” gut microbial composition with pathogenic E. casseliflavus and probiotic Limosilactobacillus and Weissella cibaria co-enriched, possibly reflecting compensatory adaptation to recent infection or antibiotic exposure30. Competitive inhibition within this dynamic equilibrium may regulate infection risk. A strong association between post-cardiac transplant infections and Enterococcus dominance was seen, and restoration of butyrate-producing bacteria by probiotic supplementation may be a potential therapy to reverse the “infection-antibiotic-dysbiosis” vicious cycle. Dual-target intervention focusing on promoting the restoration of butyrate-producing microbiota and enhancing dynamic monitoring of Enterococcus is a theoretical basis for the optimization of personalized infection prevention for transplant patients. The understanding of post-transplant infection mechanisms and development of targeted intervention strategies may be advanced by these findings.
Samples from the Post-I group exhibited distinct clustering (Fig. 2), indicating a marked deviation from the preoperative baseline, with the degree of alteration surpassing that caused by surgical trauma alone. Considering that all patients in this group received antibiotic therapy, this study posits that infection and antibiotic exposure collectively represent the key drivers behind the profound alterations in the Post-I group’s microbiota structure, findings consistent with prior studies on antibiotic effects on gut microbiota31. Notably, despite all postoperative patients undergoing immunosuppressive therapy, the Post-NI group maintained a relatively stable gut microbiota structure within 30 days post-surgery. This suggests that during the early postoperative phase, the direct perturbation of the gut microbiota by immunosuppression may be less pronounced compared to infection/antibiotic events. This aligns with the understanding that microbiota changes following solid organ transplantation (SOT) are primarily concentrated in the early postoperative period and eventually stabilize8, exemplified by hepatic and renal transplant recipients whose microbiota diversity undergoes significant fluctuations within weeks to months post-transplantation before showing recovery trajectories32,33. However, due to the limited sample size and short-term follow-up (30 days) of this study, the long-term effects of immunosuppressive regimens remain unclarified, necessitating future research with expanded cohorts and prolonged follow-up periods for further validation.
16 S rRNA gene sequencing was correlated with PICRUSt2 functional prediction in the current work to achieve a preliminary association between perioperative gut microbiota structural changes and predicted metabolic functional potentials. Comparison of Pre-NI with Pre-I groups showed Enterococcus casseliflavus to be positively correlated with “pyruvate fermentation to acetone”, suggesting that this bacterium may participate in anaerobic gut energy metabolism through atypical metabolic pathways. Pyruvate may be enzymatically fermented into acetate and acetone in strict anaerobes34,35,36. No complete acetone fermentation gene cluster has been reported in the genus Enterococcus, but this finding implies potential metabolic plasticity with mechanisms requiring further validation through genomic and proteomic analyses. Post-I vs. Post-NI comparisons showed the predicted functional potential of the L-1,2-propanediol degradation pathway to have the most significant difference and this difference was positively correlated with the abundance of the Blautia genus. There are clear biological implications underlying this functional prediction. L-1,2-propanediol is a key intermediate in dietary fiber fermentation with the final metabolic product being propionate, a short-chain fatty acid critical for gut barrier function, immune regulation and energy metabolism37,38. Previous studies have demonstrated that intestinal symbiotic Blautia species maintain colonic mucosal function under low-fiber consumption by secreting short-chain fatty acids. The current findings suggest that the Blautia-dominated microbiota of the Post-NI group may maintain higher L-1,2-propanediol metabolic potential. By contrast, reduced Blautia abundance and decreased predicted L-1,2-propanediol metabolic potential in postoperative infection patients may indicate impaired capacity of gut microbiota to synthesize beneficial metabolites, explaining gut dysbiosis in this group. It must be emphasized that the conclusions of this study are based on functional potential predictions with the primary limitation being the inability to confirm actual metabolic activities. However, these findings clarify directions for future validation: (1) shotgun metagenomics should be used to verify whether Blautia and related strains carry a complete L-1,2-propanediol degradation gene cluster; (2) targeted metabolomics should quantify levels of key metabolites, such as propionate, in patient gut samples and (3) functional experiments in animal models, such as fecal microbiota transplantation, are required to establish causality. Such studies will facilitate the transformation of current associative findings into a mechanistic understanding of the influence of microbial function on host health.
We acknowledge some limitations to the current work. Single-center data may limit generalizability; small sample sizes may reduce the robustness of subgroup analysis and long-term microbiota dynamics post-30 days remain unexplored. In addition, metabolic pathway predictions by PICRUSt2/FAPROTAX require validation via metagenomic or metabolomic profiling. Future studies should integrate host immune markers, such as cytokine profiles, to analyze microbiota-metabolism-immunity interactions and guide personalized interventions.
Conclusion
The current study investigated perioperative intestinal microbiota dynamics in cardiac transplant patients, revealing stage-specific evolution characterized by transition from Bacteroides dominance pre-operatively to Blautia-mediated post-operative protective colonization in the non-infected group versus Enterococcus pathogenic proliferation in the infected group. Infection-related stress and antibiotic exposure were the primary drivers of this microbiota shift. A dual-strategy intervention is proposed to restore butyrate-producing bacteria alongside dynamic Enterococcus surveillance. Such an approach may overcome the limitations of conventional antimicrobial therapies and contribute to personalized microbiota-directed interventions.
Data availability
Raw 16 S amplicon metagenomic sequencing data are available at the National Center for Biotechnology Information (NCBI), under BioProject PRJNA1242409 with SRA accession numbers from SRR32883272 to SRR32883303. The data can be accessed via the following link:https://www.ncbi.nlm.nih.gov/bioproject/PRJNA1242409. We have uploaded the analytical code to the GitHub repository. Access link: https://github.com/jingxianhan/Gut-Microbiota-and-Perioperative-Infection-in-Heart-Transplant.
References
Abe, R. et al. Factors related to the severity of early postoperative infection after heart transplantation in patients surviving prolonged mechanical support periods: experience at a single university. J. Cardiothorac. Vasc Anesth. 32, 53–59. https://doi.org/10.1053/j.jvca.2017.07.004 (2018).
Colvin, M. et al. OPTN/SRTR 2018 annual data report: heart. Am. J. Transplant. 20(Suppl s1), 340–426. https://doi.org/10.1111/ajt.15676 (2020).
Shultes, K. C. et al. Outcomes and predictors of early infection after heart transplantation. Surg. Infect. (Larchmt). 19, 516–522. https://doi.org/10.1089/sur.2017.295 (2018).
Wilmes, P. et al. The gut microbiome molecular complex in human health and disease. Cell. Host Microbe. 30, 1201–1206. https://doi.org/10.1016/j.chom.2022.08.016 (2022).
Dethlefsen, L., Huse, S., Sogin, M. L. & Relman, D. A. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 6, e280. https://doi.org/10.1371/journal.pbio.0060280 (2008).
Palleja, A. et al. Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat. Microbiol. 3, 1255–1265. https://doi.org/10.1038/s41564-018-0257-9 (2018).
Yuzefpolskaya, M. et al. Gut microbiota, endotoxemia, inflammation, and oxidative stress in patients with heart failure, left ventricular assist device, and transplant. J. Heart Lung Transpl. 39, 880–890. https://doi.org/10.1016/j.healun.2020.02.004 (2020).
Olek, K. et al. Gut Microbiome in patients after heart Transplantation-Current state of knowledge. Biomedicines 11 https://doi.org/10.3390/biomedicines11061588 (2023).
Mallick, H. et al. Multivariable association discovery in population-scale meta-omics studies. PLoS Comput. Biol. 17, e1009442. https://doi.org/10.1371/journal.pcbi.1009442 (2021).
Langille, M. G. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. https://doi.org/10.1038/nbt.2676 (2013).
Chinese Society of Organ Transplantation & Chinese Medical Association. Technical guidelines for preoperative evaluation and preparation of cardiac transplant recipients (2019 Edition). Chin. J. Transplant. (Electronic Edition). 13, 1–7 (2019).
Ministry of Health of the People’s Republic of China. Diagnostic criteria for hospital infections (trial version). Chin. J. Med. 81, 314–320 (2001).
Louca, S., Parfrey, L. W. & Doebeli, M. Decoupling function and taxonomy in the global ocean microbiome. Science 353, 1272–1277. https://doi.org/10.1126/science.aaf4507 (2016).
Gabarre, P. et al. Immunosuppressive therapy after solid organ transplantation and the gut microbiota: bidirectional interactions with clinical consequences. Am. J. Transpl. 22, 1014–1030. https://doi.org/10.1111/ajt.16836 (2022).
Tang, W. H., Kitai, T. & Hazen, S. L. Gut microbiota in cardiovascular health and disease. Circ. Res. 120, 1183–1196. https://doi.org/10.1161/circresaha.117.309715 (2017).
Pasini, E. et al. Pathogenic gut flora in patients with chronic heart failure. JACC Heart Fail. 4, 220–227. https://doi.org/10.1016/j.jchf.2015.10.009 (2016).
Zafar, H. & Saier Jr, M. H. Gut bacteroides species in health and disease. Gut Microbes. 13, 1–20. https://doi.org/10.1080/19490976.2020.1848158 (2021).
Troy, E. B. & Kasper, D. L. Beneficial effects of bacteroides fragilis polysaccharides on the immune system. Front. Biosci. (Landmark Ed). 15, 25–34. https://doi.org/10.2741/3603 (2010).
Ramakrishna, C. et al. Bacteroides fragilis polysaccharide A induces IL-10 secreting B and T cells that prevent viral encephalitis. Nat. Commun. 10, 2153. https://doi.org/10.1038/s41467-019-09884-6 (2019).
Shen, Y. et al. Outer membrane vesicles of a human commensal mediate immune regulation and disease protection. Cell. Host Microbe. 12, 509–520. https://doi.org/10.1016/j.chom.2012.08.004 (2012).
Yao, Y. et al. The role of short-chain fatty acids in immunity, inflammation and metabolism. Crit. Rev. Food Sci. Nutr. 62, 1–12. https://doi.org/10.1080/10408398.2020.1854675 (2022).
Kim, C. H. Immune regulation by microbiome metabolites. Immunology 154, 220–229. https://doi.org/10.1111/imm.12930 (2018).
Liu, X. et al. Blautia-a new functional genus with potential probiotic properties? Gut Microbes. 13, 1–21. https://doi.org/10.1080/19490976.2021.1875796 (2021).
Akshaya, B. S. et al. Cinnamaldehyde inhibits Enterococcus faecalis biofilm formation and promotes clearance of its colonization by modulation of phagocytes in vitro. Microb. Pathog. 181, 106157. https://doi.org/10.1016/j.micpath.2023.106157 (2023).
Duan, Y. et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 575, 505–511. https://doi.org/10.1038/s41586-019-1742-x (2019).
Al Bshabshe, A. et al. Characterization and antimicrobial susceptibility patterns of Enterococcus species isolated from nosocomial infections in a Saudi Tertiary Care Hospital over a Ten-Year Period (2012–2021). Diagnostics (Basel) 14. https://doi.org/10.3390/diagnostics14111190 (2024).
Annavajhala, M. K. et al. Colonizing multidrug-resistant bacteria and the longitudinal evolution of the intestinal microbiome after liver transplantation. Nat. Commun. 10, 4715. https://doi.org/10.1038/s41467-019-12633-4 (2019).
Agudelo-Ochoa, G. M. et al. Gut microbiota profiles in critically ill patients, potential biomarkers and risk variables for sepsis. Gut Microbes. 12, 1707610. https://doi.org/10.1080/19490976.2019.1707610 (2020).
Patrier, J. et al. Oropharyngeal and intestinal concentrations of opportunistic pathogens are independently associated with death of SARS-CoV-2 critically ill adults. Crit. Care 26, 300. https://doi.org/10.1186/s13054-022-04164-0 (2022).
Lee, A. H., Jimenez, R., Meisel, M. & D. M. & Limosilactobacillus reuteri: a probiotic gut commensal with contextual impact on immunity. Gut Microbes 17, 2451088. https://doi.org/10.1080/19490976.2025.2451088 (2025).
Chen, Z., Chang, X., Ye, Q., Gao, Y. & Deng, R. Kidney transplantation and gut microbiota. Clin. Kidney J. 17, sfae214. https://doi.org/10.1093/ckj/sfae214 (2024).
Kato, K. et al. Longitudinal analysis of the intestinal microbiota in liver transplantation. Transpl. Direct 3, e144. https://doi.org/10.1097/txd.0000000000000661 (2017).
Fricke, W. F., Maddox, C., Song, Y. & Bromberg, J. S. Human microbiota characterization in the course of renal transplantation. Am. J. Transpl. 14, 416–427. https://doi.org/10.1111/ajt.12588 (2014).
Guo, T. et al. Oxygen-Inducible conversion of lactate to acetate in heterofermentative Lactobacillus brevis ATCC 367. Appl. Environ. Microbiol. 83 https://doi.org/10.1128/aem.01659-17 (2017).
Curiel, J. A. et al. Identification of target genes to control acetate yield during aerobic fermentation with Saccharomyces cerevisiae. Microb. Cell. Fact. 15, 156. https://doi.org/10.1186/s12934-016-0555-y (2016).
Rosenfeld, B. & Simon, E. The mechanism of the butanol-acetone fermentation. II. Phosphoenolpyruvate as a new intermediate. J. Biol. Chem. 186, 405–410 (1950).
Trischler, R. et al. 3-Hydroxypropionate production from myo-inositol by the gut acetogen blautia schinkii. Environ. Microbiol. 26, e16692. https://doi.org/10.1111/1462-2920.16692 (2024).
Reichardt, N. et al. Specific substrate-driven changes in human faecal microbiota composition contrast with functional redundancy in short-chain fatty acid production. ISME J. 12, 610–622. https://doi.org/10.1038/ismej.2017.196 (2018).
Acknowledgements
The authors would like to express their gratitude to EditSprings (https://www.editsprings.com) for the expert linguistic services provided.
Funding
This work was supported by the Zhengzhou Municipal Science and Technology Innovation Guidance and Planning Project in the Healthcare Field [grant no. 2024YLZDJH232]; the Joint Construction Project of Medical Science and Technology Research Plan of Henan Province [grant no. LHGJ20240900 and LHGJ20230736] and Henan Province Key Research and Development and Promotion Special Project (Scientific and Technological Research) [grant no. 242102310226].
Author information
Authors and Affiliations
Contributions
JXH and LH were responsible for conceptualization, methodology, validation, drafting the initial manuscript. SQZ and HQ were responsible for data curation. ZBZ and LC were responsible for visualization. XGM designed the study and was in charge of the formal analysis of bioinformatics data. ZYL confirmed the authenticity of all raw data and provided supervision for the paper. All authors have read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
Not applicable.
Ethics approval
This study is a retrospective data analysis conducted in accordance with the ethical guidelines of the Declaration of Helsinki (2000) and the Istanbul Declaration (2008). It was approved by the Ethics Committee of the Seventh People’s Hospital of Zhengzhou (Approval No.: 2024[ky-018]). We hereby declare that: all transplant organs/tissues originated from voluntary and unpaid donations by Chinese citizens, NO organs/tissues were procured from prisoners. Organ allocation was conducted through the “China Organ Transplant Response System” (COTRS). Written informed consent was obtained from all patients and their immediate family members, and all patient data were de-identified to ensure privacy protection.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Han, J., Hua, L., Yang, B. et al. Gut microbiota and metabolic pathway profiles in infected and non-infected heart transplant patients before and after surgery. Sci Rep 16, 7697 (2026). https://doi.org/10.1038/s41598-026-38911-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-38911-y
