
Abstract
Fish allergy is a significant global health concern and often leads to persistent and potentially severe hypersensitivity reactions. The molecular heterogeneity of fish allergens poses challenges for developing broadly applicable immunotherapies. In this study, an integrated immunoinformatics pipeline was used to design a multi-epitope vaccine construct targeting three major fish allergens: parvalbumin, enolase, and aldolase. Predicted B-cell and T-cell epitopes were screened for immunogenicity, non-allergenicity, and lack of toxicity, and the selected peptides were assembled into a 432-amino acid chimeric construct using rationally chosen linkers to facilitate antigen processing. Computational analyses predicted the construct to exhibit favorable physicochemical properties, including stability, solubility, and a broad theoretical population coverage. Docking and molecular dynamics simulations suggested a potential interaction with Toll-like receptor 4, offering preliminary insight into possible innate immune engagement. As these findings are entirely in silico, they represent a theoretical framework; experimental validation is necessary to determine the construct’s biological behavior, safety, and immunological relevance.
Similar content being viewed by others


Introduction
Fish allergy is a significant global health issue that affects populations worldwide, with variable prevalence depending on dietary habits and geographic regions. Globally, it is estimated that up to 1% of the general population and higher in fish-heavy diets might suffer from this type of food allergy, causing severe reactions that can be life-threatening. In the United States alone, fish allergies affect approximately 0.4% of the population, underscoring the pressing need for effective management strategies1,2.
Management of fish allergy currently involves strict dietary avoidance and the preparedness to treat accidental ingestions with interventions such as epinephrine. However, the hidden presence of fish proteins in food and issues with food labeling complicate these avoidance strategies, often leading to accidental exposure3. Diagnostic challenges further exacerbate this problem, as standard testing methods such as skin prick tests and specific IgE measurements may not always accurately diagnose fish allergies or predict the severity of allergic reactions4.
Among the distinct proteins identified in fish, parvalbumin is recognized as the major allergen responsible for widespread cross-reactivity across distantly related fish species. Other significant allergens include aldolase and enolase, which vary in their allergenic potential and prevalence across different fish types1,5. Component-resolved diagnostics and curated databases such as the WHO/IUIS Allergen Nomenclature have revealed a broader allergen repertoire that includes collagen and some minor allergens such as tropomyosin, creatine kinase, triosephosphate isomerase, and pyruvate kinase, with variable prevalence and clinical relevance depending on species and population. To date, 44 fish allergens from 22 species spanning 13 protein families have been officially registered, highlighting the molecular heterogeneity of fish allergy5,6.
IgE-mediated fish allergy develops through a biphasic immunological process. During the sensitization phase, allergen proteins are internalized by antigen-presenting cells, processed, and presented as peptide-MHC complexes to T cells. In genetically susceptible individuals, this leads to Th2 polarization and the production of allergen-specific IgE7,8. The elicitation phase occurs upon re-exposure, when the allergen cross-links IgE bound to FcεRI receptors on mast cells and basophils, triggering degranulation and anaphylaxis. Importantly, a structural dichotomy exists in this recognition: T cells recognize short linear peptide epitopes presented by MHC molecules, whereas IgE antibodies predominantly recognize three-dimensional conformational epitopes on the intact allergen surface, such as the calcium-loaded EF-hand domains of parvalbumin. This mechanistic divergence provides the fundamental rationale for the multi-epitope peptide vaccine design proposed here. By selectively presenting promiscuous, immunodominant CD4⁺ T-cell epitopes in the absence of conformational B-cell epitopes, such constructs are designed to induce regulatory T cells (Tregs) and shift the immune response from a Th2-dominated allergic phenotype toward a protective Th1 or Treg-mediated profile9,10. This shift is characterized by the production of IL-10 and IFN-γ, which promote class switching to ‘blocking’ IgG4 antibodies that compete with IgE for allergen binding9,11. Critically, because the short linear peptides in the vaccine lack the tertiary structure required to recreate the native EF-hand domains, the risk of IgE cross-linking and anaphylaxis during treatment is significantly reduced10,12. Furthermore, computational screening allows for the systematic exclusion of sequences that overlap with experimentally validated IgE-binding regions, thereby operationalizing these immunological principles into a rational, safety-optimized design13.
In this context, computational approaches have become increasingly valuable tools in allergy research. Bioinformatics and molecular modeling enable the identification and characterization of T- and B-cell epitopes, providing a framework for designing multi-epitope constructs and for pre-clinical hypothesis generation. Furthermore, the integration of artificial intelligence (AI) has accelerated this landscape, offering more accurate prediction of immunogenic epitopes and more efficient prioritization of vaccine candidates14. Such in silico analyses help prioritize candidates that merit further experimental evaluation by focusing on predicted immunogenic regions while excluding sequences associated with allergenicity or toxicity15.
The present study applies a computational workflow to design a multi-epitope construct targeting major fish allergens. By integrating epitope prediction, filtering, structural modeling, and receptor interaction analyses, we aimed to generate a preliminary in silico vaccine prototype that can serve as a basis for subsequent experimental investigation. While the findings provide useful insights for candidate selection, the proposed construct remains a computational design that requires extensive in vitro and in vivo validation before any conclusions regarding safety, immunogenicity, or clinical applicability can be drawn.
Methods
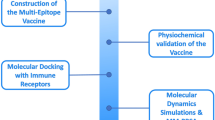
The computational workflow for designing the multi-epitope fish allergy vaccine is summarized in Fig. 1.

Workflow of multi epitope vaccine development for fish allergy.
Sequence retrieval
To develop a multi-epitope vaccine for fish allergy, we focused on the selection of major fish allergens, specifically parvalbumin, aldolase, and enolase. These allergenic sequences were identified through searches in Allergome (https://www.allergome.org/) and the WHO/IUIS Allergen Nomenclature Database (https://allergen.org/). Subsequently, the corresponding amino acid sequences were obtained in FASTA format from the Universal Protein Resource (UniProt) and National Center for Biotechnology Information (NCBI).
Sequence alignment
To perform multiple sequence alignment and identify conserved sequences crucial for our study, we gathered parvalbumins from a diverse array of fish species, specifically Clu h 1, Sar sa 1, Cyp c 1, Gad c 1, Gad m 1, Lat c 1, Lep w 1, Onc m 1, Sal s 1, Seb m 1, Thu a 1, Cten i 1, Pan h 1, and Xip g 1. To investigate cross-reactivity, the sequence of chicken parvalbumin (Gal d 8) was also included. The sequences of aldolases (Gad m 3, Pan h 3, Sal s 3, and Thu a 3) and enolases (Sal s 2, Gad m 2, Pan h 2, and Thu a 2) were compiled for further analysis. These sequences, obtained in FASTA format (S1 file), were imported into the Jalview software and aligned using the Clustal Omega algorithm, enabling the identification of conserved regions across these allergens. A consensus sequence was selected.
T-cell, B-cell epitopes prediction
To identify T-cell epitopes within the selected proteins, we employed two distinct computational approaches. The NETCTL (https://services.healthtech.dtu.dk/services/NetCTL-1.2/) server which integrates MHC class I binding prediction, proteasomal cleavage, and TAP transport efficiency through artificial neural networks. Predictions were performed using the A1 supertype reference set, with all parameters maintained at their default thresholds (combined score threshold = 0.75) and only those epitopes that were deemed to be strong binders were chosen16, along with the IEDB MHC Class I binding prediction tool (https://tools.iedb.org/mhci/) based on the 27 reference HLA class I alleles, was used to predict cytotoxic T lymphocyte (CTL) epitopes, with epitopes showing a lower percentile rank were chosen17. Conversely, helper T lymphocyte (HTL) epitopes were predicted using the IEDB MHC Class II binding prediction tool (https://tools.iedb.org/mhcii/) selecting the NetMHCIIpan 4.1 EL prediction method. Predictions were conducted using the 7-allele HLA-DR reference set (HLA-DRB1*03:01, HLA-DRB1*07:01, HLA-DRB1*15:01, HLA-DRB3*01:01, HLA-DRB3*02:02, HLA-DRB4*01:01, and HLA-DRB5*01:01), The default IEDB selection criterion (percentile rank ≤ 10%) was applied, whereby epitopes with lower percentile ranks are indicative of higher-affinity binders18. B-cell epitope prediction was conducted using the ABCpred server (https://webs.iiitd.edu.in/raghava/abcpred/) which is based on a recurrent neural network (RNN) algorithm. Predictions were performed using a fixed epitope length of 16 amino acids and the default prediction threshold of 0.5119, which allowed for the identification of linear B-cell epitopes across the allergenic sequences.
Antigenicity allergenicity and toxicity evaluation
For antigenicity prediction, the VaxiJen V2 (https://www.ddg-pharmfac.net/vaxijen/VaxiJen/VaxiJen.html) server was used with a standard threshold of 0.4, and only epitopes classified as antigenic were retained20. For allergenicity prediction, AllerTOP v2.0 (https://www.ddg-pharmfac.net/allertop_test/) server was employed, and epitopes predicted as non-allergenic were selected21. Toxicity prediction was further assessed using the ToxinPred (https://crdd.osdd.net/raghava/toxinpred/) server to ensure that the selected epitopes were non-toxic22. These tools were chosen to facilitate the identification of candidates suitable for inclusion in multi-epitope vaccine design.
IgE-epitope screening and population coverage
Given that fish allergy is primarily mediated by IgE antibodies, the predicted epitopes were screened against experimentally validated IgE-binding epitopes from fish allergens. A total of 323 IgE epitopes (predominantly from parvalbumin) were retrieved from the Immune Epitope Database (https://www.iedb.org/). Each vaccine candidate epitope was aligned against this reference dataset to identify shared sequence segments of ≥ 5 consecutive amino acid residues, which were considered indicative of potential IgE cross-reactivity. Epitopes sharing such segments with experimentally validated IgE epitopes were classified as “overlapping” and flagged for potential IgE cross-reactivity risk, while epitopes with no detectable overlap were classified as “non-overlapping”. Population coverage analysis was performed to evaluate the distribution of HLA allele frequencies corresponding to predicted epitope-binding specificities, utilizing the IEDB population coverage calculation tool (http://tools.iedb.org/population)23.
Design of the multi-epitope vaccine
In our vaccine construct design, epitopes from parvalbumins, enolases, and aldolases, corresponding to HTL, CTL, and B-cell responses, respectively, were concatenated using specific linkers: GPGPG for HTL epitopes, AYY for CTL epitopes, and KK linkers to separate B-cell epitopes and intersperse the different protein-derived epitopes. This configuration has led to the development of a multi-epitope vaccine construct, RS09. To augment immunogenicity, the construct was further enhanced by integrating the universal memory T helper peptide (TpD) and the Pan HLA-DR reactive epitope (PADRE) attached through strategic linker placement. A PorB adjuvant (TLR2 ligand) was affixed to the C-terminus, whereas the N-terminus was modified with RS09, a synthetic adjuvant recognized for its potent TLR4 activation capability. A hexahistidine (6×His) tag was added at the C-terminus of the construct to facilitate purification in future experimental work. The physicochemical characteristics of the multi-epitope vaccine construct were assessed using the ProtParam tool (https://web.expasy.org/protparam/) in the ExPASy database server24.
Secondary structure and 3D modeling validation of protein constructs
Secondary structure prediction was performed using the PSIPRED (https://bioinf.cs.ucl.ac.uk/psipred/) server, followed by 3D structure prediction using the AlphaFold v3 (https://alphafoldserver.com/) server25. The best model was selected and refined using the GalaxyRefine Web tool (https://galaxy.seoklab.org/cgi-bin/submit.cgi?type=REFINE) to identify any errors in the predicted tertiary structures26. To validate the quality of the model, the ProSA-Web tool (https://prosa.services.came.sbg.ac.at/prosa.php), ERRAT tool, and Ramachandran plot generated by the PROCHECK (https://saves.mbi.ucla.edu/) web tool were used27. The PyMOL software (version 3.1.3) was used to visualize and edit the protein data.
Molecular Docking
Molecular docking was performed using the ClusPro (https://cluspro.org/) protein-protein docking server28,29,30. Docking involved interactions between the multi-epitope vaccine construct and Toll-like receptor 2 (TLR2, PDB ID: 2z7x) and Toll-like receptor 4 (TLR4, PDB ID: 3fxi). The model with the best score was selected for further analysis.
Molecular dynamics simulation of TLR2 and TLR4 complexed with vaccine construct
Molecular Dynamics (MD) simulations were performed using a multi-epitope vaccine complexed with TLR2 and TLR4. System preparation was conducted using the CHARMM-GUI (https://www.charmm-gui.org/)31with the CHARMM36 force field32. The system was solvated using the TIP3P water model within a cubic box, ensuring a 10 Å buffer zone. Sodium chloride (NaCl) was added at a concentration of 0.15 M. The simulation workflow, executed via GROMACS 2024.133, included energy minimization, equilibration, and production phases. Energy minimization was performed using the steepest descent method with positional restraints on heavy atoms over 50,000 steps. Equilibration was performed in the NPT ensemble for 125 ps, maintaining isotropic pressure of 1 bar. The production run was extended to 100 ns under the NPT ensemble, utilizing the leapfrog integrator and the Verlet cutoff scheme, with a 1.2 nm cutoff for van der Waals and electrostatic interactions. Long-range electrostatics were handled using Particle Mesh Ewald (PME), while all hydrogen bonds were constrained via the LINCS algorithm. The system stability was reinforced by removing the center-of-mass motion every 100 steps for both the solute and solvent groups. Trajectory data were recorded at 100 ps intervals and subsequently analyzed using VMD 1.9.434 (Visual Molecular Dynamics) to assess structural and dynamic properties. MD simulation trajectories were visualized and further analyzed using QtGrace v0.2.6.
Immune simulation and in Silico cloning
Immune simulations were conducted using the C-IMMSIM server (https://kraken.iac.rm.cnr.it/C-IMMSIM/) using the following settings: simulation volume = 10, 100 simulation steps, random seed = 12,345, and an LPS-free vaccine formulation35. The cloning process was carried out using SnapGene software. Prior to cloning, the vaccine construct was subjected to codon optimization using the JCat web tool (https://www.jcat.de/) to enhance expression efficiency36. Subsequently, the optimized sequence was cloned into the pET28a (+) plasmid using the XhoI and BamHI restriction sites.
Results
Analysis of conserved regions and physicochemical characteristics of major fish allergens
The physicochemical and immunogenic properties of the consensus sequences for parvalbumin, aldolase, and enolase were determined and are summarized in Table 1. The analysis revealed significant differences among the three major allergens.
Parvalbumin has the lowest molecular weight (11.9 kDa, is classified as an allergen, and displays the highest stability (instability index: 17.39) among the three proteins. Despite its stability, it was predicted to be a non-antigen (antigenicity: 0.3595). Both aldolase and enolase have higher molecular weights (39.6 kDa and 47.2 kDa, respectively) and are predicted as probable non-allergens. However, their antigenicity scores (0.4173 for aldolase and 0.4499 for enolase) suggest probable antigenic potential.
T cell and B cell epitope prediction
We designed a multi-epitope vaccine by selecting cytotoxic T-lymphocyte (CTL), helper T-lymphocyte (HTL), and B-cell epitopes from parvalbumin, aldolase, and enolase proteins. Computational assessment indicated that all chosen epitopes were antigenic but not allergenic or toxic. High-scoring epitopes, which are predictive of a higher likelihood of natural processing and presentation by MHC-I molecules, were selected for further analysis. For parvalbumin, initial predictions identified only two epitopes, both of which were non-antigenic and therefore excluded. Additional screening subsequently yielded five high-affinity epitopes predicted to be non-allergenic, antigenic, and non-toxic. The selected epitopes, including KAADSFNHK, EFAALVKAR, and DSFNHKAFF, are summarized in Table 2. For enolase and aldolase, six and nine epitopes, respectively, were identified from the larger prediction pools (356 candidates for enolase and 426 for aldolase). These epitopes were selected based on their high combined prediction scores and their compliance with the filtering criteria for non-toxicity, antigenicity, and non-allergenicity (Table 3).
HTL epitope identification yielded a limited set of high-affinity candidates based on the NetMHCIIpan 4.1 EL predictions. Among the predicted peptides, only the top-ranked epitopes with the lowest percentile and adjusted ranks across the seven HLA-DR reference alleles were retained. These epitopes also satisfied the filtering criteria for predicted antigenicity, non-allergenicity, and non-toxicity. The final selected HTL epitopes, derived from parvalbumin, aldolase, and enolase, are presented in Table 4.
For B-cell epitopes, the predicted peptides were screened for antigenicity, non-allergenicity, and non-toxicity. Only the highest-scoring candidates that met all filtering criteria were retained for inclusion in the MEV construct. The final selected linear B-cell epitopes, derived from parvalbumin, aldolase, and enolase, are presented in Table 5.
Table 6 summarizes the immunological filtering outcomes for the final selected epitopes. Each CTL, HTL, and B-cell epitope displayed positive antigenicity scores and was predicted to be non-allergenic and non-toxic. These findings provided the basis for their inclusion in the MEV construct.
IgE epitope screening and population coverage
Screening of the 16 predicted epitopes against 323 experimentally validated IgE epitopes from fish allergens retrieved from the IEDB revealed that 10 epitopes (62.5%) showed no sequence overlap with known IgE-binding regions. In contrast, six epitopes (37.5%) demonstrated sequence overlap with validated IgE epitopes, sharing conserved parvalbumin-associated motifs including FAALVKA, AADSFNHK, HKAFF, KVGLA, and SADDVKKAF (Table 7).
Population coverage was estimated using the IEDB population coverage tool to evaluate the recognition potential of the selected epitopes and their corresponding HLA-DR, -DQ, and -DP supertypes across different geographic regions. In Africa, the calculated coverage values were 92.82%, 96.50%, 89.83%, and 94.10% for East, West, Central, and North Africa, respectively. In the Americas, the coverage was 99.59%, 25.10%, and 90.91% for North, Central, and South America, respectively. Oceania showed a coverage of 95.58%. In Asia, the calculated values were 98.60%, 96.90%, 97.60%, 95.85%, and 94.31% for East Asia, Northeast Asia, South Asia, Southeast Asia, and Southwest Asia, respectively. Europe demonstrated a coverage of 99.86%. The overall global population coverage was 99.26%.
Design of a multi-epitope vaccine construct
The multi-epitope vaccine was constructed using high-affinity epitopes derived from three source proteins. Seven HTL and seven CTL epitopes were selected based on their HLA binding scores, alongside three non-allergenic, non-toxic, and immunogenic B-cell epitopes. These antigenic regions were fused using GPGPG, AAY, EAAAK, and KK linkers to facilitate efficient intracellular processing and prevent epitope folding interference. To potentiate the immune response, the construct was adjuvanted with the TLR2 agonist PorB at the N-terminus and the TLR4 agonist RS09 at the C-terminus, both attached via EAAAK linkers. Additionally, the T-cell helper peptide (TpD) and Pan HLA-DR reactive epitope (PADRE) were incorporated to further boost broad-spectrum immunogenicity. The overall architecture of the MEV construct is illustrated in Fig. 2.

Construction and structural features of the MEV vaccine for fish allergy.
Physiochemical properties, antigenicity and allergenicity of multi-epitope vaccine construct
Utilizing the ProtParam tool, the multi-epitope vaccine construct was characterized to have a molecular weight of 44.81 kDa and comprised 432 amino acid residues. The charge distribution included 44 negatively charged (Asp + Glu) and 52 positively charged residues (Arg + Lys). A stability index of 15.94 suggested a stable configuration, with a grand average of hydropathicity (GRAVY) at -0.099 indicating a hydrophilic nature. The aliphatic index stood at 84.86, hinting at good thermostability. The theoretical isoelectric point (pI) was calculated to be 9.15. Antigenic potential, predicted using VaxiJen, yielded a score of 0.6309, indicating that the designed MEV is predicted to elicit an immune response. The vaccine was also identified as non-allergenic, with the closest protein match in AllerTOPv2.0, assigned UniProtKB accession number Q75I13. BlastP analysis against human proteome suggested that there was no sequence similarity to the human proteome.
Secondary and tertiary structure prediction and validation
PSIPRED server analysis revealed that the secondary structure of the multi-epitope vaccine comprised 168 α-helices (39%), 76 extended strands (17.5%), and 188 random coils (43.5%) out of a total of 432 amino acid residues (Fig. 3).

Secondary structure distribution of the vaccine construct predicted by the PSIPRED server: alpha helices (39%) in pink, beta strands (17.5%) in yellow, and random coils (43.5%) in grey.
The tertiary structure of the MEV construct was generated using AlphaFold, which produced a low predicted template modeling (pTM) score of 0.19, indicating limited confidence in the global fold. The model was refined using GalaxyRefine, and the resulting output was evaluated using standard quality assessment tools. ProSA-web Z-scores changed from − 3.23 (before-refinement) to − 3.97 (after-refinement). Ramachandran plot analysis showed an increase from 84.1% to 97.5% of residues in the favored region after refinement, with no residues in disallowed regions in either model. ERRAT quality scores also increased from 93.08 to 97.53. These parameters reflect improvements in local stereochemistry, although the low AlphaFold pTM score indicates that the global tertiary fold remains uncertain. The predicted solubility estimated by ProteinSol was 0.464, with an isoelectric point (pI) of 9.520. The pre- and post-refinement models are shown in Fig. 4.

Tertiary structure prediction and optimization of the MEV. (a) Z-score pre-optimization. (b) Z-score post-optimization. (c) Ramachandran plot pre-optimization. (d) Ramachandran plot post-optimization. (e) Superposition of initial AlphaFold model (violet) and GalaxyRefine-optimized model (cyan). (f) ProteinSol solubility score.
Molecular Docking of multi epitope vaccine construct with receptors
To elucidate the interactions between the multi-epitope vaccine (MEV) and TLR2 and TLR4, we employed the ClusPro2.0 server to generate 30 model complexes for each receptor. The models were evaluated based on their binding energies, and complexes with the lowest energies were selected for analysis. For the MEV-TLR4 interaction, the model that demonstrated optimal binding affinity had a central energy-weighted score of -896.5 kcal/mol and a minimum energy-weighted score of -1079.0 kcal/mol. LigPlot + analysis showed that this complex formed 12 hydrogen bonds, indicating a strong and stable interaction. Conversely, the best-performing MEV-TLR2 model showed a central energy-weighted score of -1099.8 kcal/mol and a minimum energy-weighted score of -1282.9 kcal/mol, with LigPlot+ revealing 3 hydrogen bonds (Fig. 5). These docking outputs provide computational predictions of potential interaction patterns between the MEV construct and the TLR2/TLR4 receptors.

3D structures and 2D interaction diagrams of MEV–TLR complexes. (a, b) Docked MEV peptide (cyan) within the binding sites of TLR2 and TLR4 (green). (c, d) 2D interaction diagrams of MEV with TLR2 and TLR4 generated using LigPlot+, showing key interacting residues.
Molecular dynamics simulation
The molecular stability of the interactions between the multi-epitope vaccine and TLR-2, as well as TLR-4, was analyzed using molecular dynamics (MD) simulations. A 100ns MD simulation was performed for the multi-epitope vaccine, TLR-2, and TLR-4 complexes. Important trajectory-derived parameters, such as radius of gyration (Rg), root mean square deviation (RMSD), root mean square fluctuation (RMSF), solvent accessible surface area (SASA), and the number of hydrogen bonds, were used to obtain a better understanding of the binding dynamics and structural flexibility of our systems. Trajectory-derived parameters, presented as mean ± standard deviation (SD), are summarized in Table 8.
The stability of the backbone atoms was assessed using RMSD (Fig. 6a). The TLR4-MEV complex demonstrated superior stability, equilibrating at a lower mean RMSD of 14.81 Å, whereas the TLR2-MEV complex exhibited a higher mean deviation of 20.25 Å. These values indicate that while both systems reached equilibrium, the vaccine maintained a trajectory closer to its initial docked conformation with TLR4, suggesting a more stable complex. Local structural flexibility was characterized by RMSF (Fig. 6b, c). The TLR4 complex displayed a mean RMSF of 7.53 ± 3.53 Å, which was comparable to the 7.15 Å observed for the TLR2 complex. The fluctuations observed in both systems are consistent with the presence of flexible linker regions in the vaccine construct, which are essential for effective antigen presentation. To determine the structural compactness of the complexes, Rg was analyzed (Fig. 6d). The TLR4 complex maintained a more expanded conformation with a mean Rg of 56.35 Å, in contrast to the more compact TLR2 complex, which had a mean Rg of 42.14 Å. This structural expansion in the TLR4 complex was further corroborated by SASA analysis (Fig. 6e). The TLR4 complex exhibited a significantly higher accessible surface area (87,325.95 Ų) compared to the TLR2 complex (56,571.94 Ų), suggesting that the vaccine adopts an extended binding mode on the TLR4 dimer interface, thereby exposing a larger surface for potential solvent and co-receptor interactions.
The strength and consistency of the binding interface were evaluated by monitoring intermolecular hydrogen bond formation over the simulation time (Fig. 6f). The TLR4-MEV complex established a dense and stable interaction network, maintaining an average of 10.29 hydrogen bonds. This is significantly higher than the TLR2-MEV complex, which formed an average of 2.86 hydrogen bonds. The sustained high number of hydrogen bonds in the TLR4 complex provides strong evidence for high-affinity binding and suggests TLR4 as the most likely primary receptor for the designed vaccine candidate.

Molecular dynamics simulation analysis of the MEV–TLR complexes. (a) RMSD, (b, c) RMSF, (d) Rg, (e) SASA, and (f) hydrogen-bond profiles for MEV–TLR2 (colored in blue) and MEV–TLR4 (colored in red) over 100 ns.
Immune responses induced by the multi epitope vaccine in Silico
The C-IMMSIM simulation was used to evaluate the predicted immune response profile of the multi-epitope vaccine (MEV) construct. The simulation showed a clear primary response following antigen exposure, characterized by an early rise in IgM levels. Subsequent cycles demonstrated increases in IgG1, IgG2, IgM, and total immunoglobulin levels, accompanied by expansion of the corresponding B-cell populations (Fig. 7a–c). The progressive increase in memory B-cell counts across simulation time points suggests the potential for sustained immune recall. A secondary and tertiary response pattern was also observed, with increases in helper (CD4⁺) and cytotoxic (CD8⁺) T-cell populations after repeated antigen exposures (Fig. 7d–f). These trends indicate predicted activation and engagement of both arms of the adaptive immune response. Cytokine profiling revealed elevated levels of IFN-γ and IL-2 following immunization (Fig. 7g–i). These immune simulation predicts coordinated activation of humoral and cellular components following MEV administration.

Simulation of immune responses for the multi-epitope vaccine construct using the C-IMMSIM server. (a) Antigen and immunoglobulin response, (b) Total B-cell population, (c) B-cell population per state, (d) Total Helper T-cell (TH) population, (e) TH-cell population per state, (f) Cytotoxic T-cell (TC) population per state, (g) Macrophage (MA) population per state, (h) Dendritic cell (DC) population per state, (i) Cytokine and interleukin profiles.
Codon optimization and in silico cloning
Before designing of the multi-epitope vaccine (MEV), the codon sequence was optimized using the JCat server36, with Escherichia coli (strain K12) chosen as the optimal host for expression. The optimization process sought to improve translational efficiency while reducing the occurrence of rho-independent transcription terminators and prokaryotic ribosome-binding sites. The codon adaptation index (CAI) for the modified sequence was 0.993, indicating potential compatibility with the E. coli expression system. The GC content was adjusted to 51.92%, aligned closely with the native GC content of E. coli (50.73%), thereby supporting stable and efficient expression. Analysis of restriction enzyme sites showed that the optimized vaccine construct lacked recognition sites for XhoI and BamHI, supporting their suitability for in silico subcloning. The vaccine sequence was successfully inserted into the pET28a(+) expression vector at designated restriction sites (Fig. 8).

In silico cloning using restriction enzymes of the multi-epitope vaccine by using pET28a (+) expression vector. The black circle indicates the vector and the red section indicates where the MEV is inserted.
Discussion
Conventional vaccines, inactivated or attenuated, produce a good immunity but pose risks of allergenicity and toxicity owing to residual pathogenic components37.Subunit, toxoid, and conjugate vaccines, although more specific have diminished efficacy and side effects38. To overcome such challenges, Immunoinformatics has emerged as a pivotal tool in modern vaccine design, leveraging reverse vaccinology to identify safe and immunogenic epitopes with high precision. By utilizing these in silico approaches, researchers can significantly accelerate the development pipeline, matching the accuracy of experimental methods while substantially reducing the need for extensive laboratory screening14,39,40. This method accelerates vaccine design by reducing cost and enhancing antigen selection. Epitope-based peptide vaccines, built upon the precise selection of B-cell and T-cell epitopes, harness the specificity of the immune system to induce a targeted and controlled response41. Here, utilizing computational tools, we constructed a multi-epitope vaccine candidate for fish allergy, selecting epitopes based on predicted immunogenicity, safety, and limited allergenic risk.
Why fish allergy? Despite its lower prevalence relative to peanut or tree nut allergies, fish allergies pose a similar risk of severe reactions, rendering it a notable concern in food allergy management2,42. Parvalbumin, enolase, and aldolase were identified as the target allergens for our multi-epitope vaccine because of their established involvement in fish allergy, with parvalbumin serving as the principal allergen responsible for cross-reactivity among various fish species, while enolase and aldolase facilitate IgE-mediated hypersensitivity reactions7,43,44. Recent advances in component-resolved diagnostics, proteomic profiling, and curated databases like the WHO/IUIS Allergen Nomenclature have characterized additional clinically relevant allergens, including collagen, tropomyosin, creatine kinase, triosephosphate isomerase, and pyruvate kinase5. Although these minor allergens contribute to the broader sensitization profile, this study strategically prioritized the major targets to maximize serological coverage for most patients while ensuring the vaccine construct remains of a manageable molecular size for stability and efficient expression. Several studies have previously explored potential treatments for fish allergy, notably Laurian Zuidmeer-Jongejan et al. reported the first-in-human subcutaneous immunotherapy trial using a hypoallergenic recombinant parvalbumin (mCyp c 1) designed to significantly reduce allergenic activity while retaining immunogenicity44. In alignment with this approach, our research leveraged computational immunoinformatics to construct a multi-epitope vaccine compiling all major fish allergens to develop a broader immunotherapeutic strategy capable of simultaneously addressing multiple allergenic components.
Promiscuous, highly immunogenic, nontoxic, and non-allergenic B-cell and T-cell epitopes (both MHC-I and MHC-II) were identified through integrated computational predictions. Subsequently, these selected epitopes were strategically combined using appropriate linkers, to generate a vaccine construct, which was further enhanced by the incorporation of immunogenic adjuvants45,46.
However, safety is paramount in allergy vaccine design. A critical step in our study was the IgE-epitope screening to assess the risk of IgE-mediated anaphylaxis. IgE epitope screening against experimentally validated IEDB data identified six overlapping regions, including several T-cell epitopes and one linear B-cell epitope. Because T-cell epitopes require intracellular processing and presentation via MHC, their linear overlap with IgE-binding regions does not necessarily translate into functional IgE recognition, and the altered structural context within the multi-epitope construct further reduces this likelihood47. Moreover, although most native B-cell epitopes are conformational, the vaccine’s architecture incorporates flexible (GPGPG) and rigid (EAAAK) linkers, which are expected to limit the formation of native globular folds and favor the exposure of linear peptide segments48. Given this structural configuration, linear motif overlap serves as the most pertinent metric for evaluating the potential for IgE cross-reactivity.
Native Parvalbumin has two calcium-binding EF-hands that form the primary IgE binding sites12. Our vaccine contains only linear fragments. It physically lacks the tertiary structure required to form these sites. Should experimental validation confirm IgE reactivity, sequence optimization strategies such as site-directed mutagenesis can be employed to disrupt IgE-binding residues while preserving T-cell efficacy44. This approach aligns with findings by Sircar et al., who demonstrated that modified epitopes designed to induce blocking IgG antibodies achieved up to 70% inhibition of IgE binding and a 10-fold suppression of histamine release in mold allergy models49.
While evaluating the physicochemical features of our potential vaccine, we discovered that its molecular weight was 44.81 kDa, which aligns with the acceptable range for simple synthesis and purification, given that proteins should normally have a molecular weight of ≤ 100 kDa50. The theoretical isoelectric point (pI) of our vaccine was 9.15, showing an alkaline nature. In standard applications, the vaccine exhibited a high degree of stability, as indicated by a stability index of 15.94, suggesting a stable configuration. Additionally, the elevated aliphatic index denotes exceptional thermostability, whereas a low GRAVY value reflects its hydrophilic properties51,52.
Structural assessments provided meaningful insights into the conformational characteristics of the vaccine construct. After refinement, the model demonstrated improved local stereochemistry, with 97.5% of residues located in favored regions of the Ramachandran plot. The model also showed a low AlphaFold-predicted Template Modeling (pTM) score (0.19), which is commonly observed in multi-epitope constructs that lack natural homologous templates53. Although low pTM values generally indicate limited confidence in global folding, multi-epitope constructs are not expected to adopt a stable tertiary structure. Their flexible and linker-rich architecture is intended to facilitate intracellular degradation and subsequent MHC presentation. Thus, the structural flexibility reflected by the low pTM score is consistent with the expected behavior of a vaccine construct designed for efficient antigen processing13,54,55.
Molecular docking studies indicated favorable binding between the MEV construct and Toll-like receptors. The most energetically favorable docking model for the MEV-TLR4 complex exhibited a substantial binding affinity (minimum energy-weighted score of -1079.0 kcal/mol) and formed 12 hydrogen bonds, indicative of robust and specific interactions. Interestingly, the initial docking analysis for TLR2 indicated only three hydrogen bonds. However, subsequent molecular dynamics simulations were necessary to validate these static poses and capture the true interaction dynamics56.
We conducted 100 ns molecular dynamics simulations to validate static docking poses and analyze vaccine-receptor interaction dynamics56,57. The MEV-TLR2 and MEV-TLR4 complexes demonstrated distinct stability profiles. The MEV-TLR4 complex exhibited superior stability, equilibrating at a lower mean RMSD of 14.81 Å, compared to the MEV-TLR2 complex, which displayed a higher mean deviation of 20.25 Å. Structural analysis revealed that the vaccine adopts a significantly more expanded conformation when bound to TLR4, as evidenced by a higher Radius of Gyration (Rg 56.35 Å) and a larger Solvent Accessible Surface Area (SASA 87,325.95 Ų). In contrast, the TLR2 complex remained more compact (Rg 42.14 Å; SASA 56,571.94 Ų). Crucially, despite the expanded conformation, the MEV-TLR4 complex maintained a much stronger interaction network, averaging 10.29 hydrogen bonds throughout the simulation, whereas the TLR2 complex sustained only 2.86 hydrogen bonds. This indicates that the MEV-TLR4 interaction is dynamic but highly specific; the extended binding mode likely allows the vaccine to “wrap” across the receptor interface, maximizing contact points and facilitating the robust signaling required for potent receptor activation58. The significant difference in hydrogen bonding suggests that while the vaccine can engage both receptors, TLR4 is likely the primary driver of the innate immune response for this candidate.
The in silico immune simulations generated response patterns consistent with expected adaptive immune dynamics following repeated antigen exposure. Successive administrations of the construct were associated with increased levels of simulated IgM and IgG responses, accompanied by a rise in memory B-cell populations. The model also predicted expansion of helper T-cell subsets, suggesting sustained activation within the simulated immune environment59. Finally, in silico cloning analysis revealed that the vaccine could be cloned and expressed in E. coli for further in vitro and in vivo experiments.
The computational vaccine design presented in this study provides an initial framework for exploring multi-epitope–based approaches for fish allergy. By incorporating epitopes from several clinically relevant allergens, the construct was designed to broaden theoretical antigenic coverage while reducing reliance on whole-allergen preparations. The immunoinformatics workflow enabled the selection of epitopes predicted to be antigenic, non-allergenic, and non-toxic, and the subsequent structural, docking, and immune-simulation analyses offered a preliminary characterization of the construct from a computational standpoint. Nevertheless, these findings remain purely predictive. All structural, immunological, and receptor-interaction outcomes described here are based on in silico models, which do not capture the full complexity of IgE-mediated sensitization or immune signaling in vivo. The behavior of the construct in biological systems, its processing, MHC presentation, IgE reactivity, and overall immune profile, cannot be inferred from computational analysis alone. Experimental evaluation is therefore essential. Future work should include in vitro assays, such as basophil activation tests and IgE-binding studies, to assess allergenic potential, followed by in vivo immunogenicity studies to determine biological relevance. Within these limitations, the present work should be viewed as a preliminary computational exploration that may inform future experimental efforts aimed at developing allergen-specific immunotherapies.
Conclusion
We proposed a computational multi-epitope vaccine construct as an initial framework for addressing fish allergy through an epitope-focused approach. Through reverse vaccinology, we identified epitopes from parvalbumin, enolase, and aldolase that are predicted to be immunogenic yet reduced in allergenic potential. Molecular dynamics simulations point to TLR4 as a likely receptor for this construct, suggesting a pathway for immune activation. However, as these results are derived solely from computational models, they represent a theoretical blueprint. Future experimental studies are essential to verify the safety, stability, and biological efficacy of this candidate before it can be considered for clinical application.
Data availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
References
Sharp, M. F. & Lopata, A. L. Fish allergy: in review. Clin. Rev. Allergy Immunol. 46, 258–271. https://doi.org/10.1007/s12016-013-8363-1 (2014).
Sicherer, S. H. & Sampson, H. A. Food allergy: A review and update on epidemiology, pathogenesis, diagnosis, prevention, and management. J. Allergy Clin. Immunol. 141, 41–58. https://doi.org/10.1016/j.jaci.2017.11.003 (2018).
Taylor, S. L. & Hefle, S. L. Food allergen labeling in the USA and Europe. Curr. Opin. Allergy Clin. Immunol. 3, 186–190. https://doi.org/10.1097/01.all.0000225158.75521.ad (2006).
Sampson, H. A. Update on food allergy. J. Allergy Clin. Immunol. 113, 805–819. https://doi.org/10.1016/j.jaci.2004.03.014 (2004).
Yang, Y. et al. An overview of seafood allergens: Structure–Allergenicity relationship and allergenicity elimination processing techniques. Foods 14, 2241. https://doi.org/10.3390/FOODS14132241 (2025).
Sudharson, S., Kalic, T., Hafner, C. & Breiteneder, H. Newly defined allergens in the WHO/IUIS allergen nomenclature database during 01/2019-03/2021. Allergy 76, 3359–3373. https://doi.org/10.1111/ALL.15021 (2021).
Sun, Y. et al. Fish allergy: A review of clinical Characteristics, Mechanism, Allergens, Epitopes, and Cross-Reactivity. ACS Food Sci. Technol. 4, 304–315. https://doi.org/10.1021/acsfoodscitech.3c00572 (2024).
Zieglmayer, P. et al. Mechanisms, safety and efficacy of a B cell epitope-based vaccine for immunotherapy of grass pollen allergy. EBioMedicine 11, 43–57. https://doi.org/10.1016/J.EBIOM.2016.08.022 (2016).
Cezmi, A. & Akdis Mübeccel Akdis. Advances in allergen immunotherapy: aiming for complete tolerance to allergens. Sci. Transl Med. https://doi.org/10.1126/scitranslmed.aaa7390 (2015).
Verhoef, A., Alexander, C., Kay, A. B. & Larché, M. T cell epitope immunotherapy induces a CD4 + T cell population with regulatory activity. PLoS Med. 2, 0253–0261. https://doi.org/10.1371/JOURNAL.PMED.0020078 (2005).
Noriega, D. B., Teodorowicz, M., Savelkoul, H. & Ruinemans-Koerts, J. The basophil activation test for clinical management of food allergies: recent advances and future directions. J. Asthma Allergy. 14, 1335–1348. https://doi.org/10.2147/JAA.S237759 (2021).
Kuehn, A., Swoboda, I., Arumugam, K., Hilger, C. & Hentges, F. Fish allergens at a glance: variable allergenicity of parvalbumins, the major fish allergens. Front. Immunol. 5 (APR), 79259. https://doi.org/10.3389/FIMMU.2014.00179/XML (2014).
Rawat, S. S., Keshri, A. K., Kaur, R. & Prasad, A. Immunoinformatics approaches for vaccine design: A fast and secure strategy for successful vaccine development. Vaccines 11. https://doi.org/10.3390/vaccines11020221 (2023).
Villanueva-Flores, F., Sanchez-Villamil, J. I. & Garcia-Atutxa, I. AI-driven epitope prediction: a systematic review, comparative analysis, and practical guide for vaccine development. Npj Vaccines 2025. 10, 1. https://doi.org/10.1038/s41541-025-01258-y (2025).
De Groot, A. S. & Martin, W. Reducing risk, improving outcomes: bioengineering less Immunogenic protein therapeutics. Clin. Immunol. 131, 189–201. https://doi.org/10.1016/j.clim.2009.01.009 (2009).
Larsen, M. V. et al. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinform. 8, 1–12. https://doi.org/10.1186/1471-2105-8-424/TABLES/3 (2007).
Stranzl, T., Larsen, M. V., Lundegaard, C., Nielsen, M. & NetCTLpan Pan-specific MHC class I pathway epitope predictions. Immunogenetics 62, 357–368. https://doi.org/10.1007/S00251-010-0441-4 (2010).
Kaabinejadian, S. et al. Contribution of DRB3, 4 and 5 to the Total DR Immunopeptidome. Front. Immunol. 13. https://doi.org/10.3389/FIMMU.2022.835454/PDF (2022). MHC Motif Deconvolution of Immunopeptidomics Data Reveals a Significant.
Saha, S. & Raghava, G. P. S. Prediction of continuous B-cell epitopes in an antigen using recurrent neural network. Proteins: Struct. Function Genet. 65, 40–48. https://doi.org/10.1002/PROT.21078 (2006).
Doytchinova, I. A. & Flower, D. R. VaxiJen: A server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinform. 8. https://doi.org/10.1186/1471-2105-8-4 (2007).
Dimitrov, I., Bangov, I., Flower, D. R. & Doytchinova, I. AllerTOP v.2 - A server for in Silico prediction of allergens. J. Mol. Model. 20. https://doi.org/10.1007/s00894-014-2278-5 (2014).
Rathore, A. S., Choudhury, S., Arora, A., Tijare, P. & Raghava, G. P. S. ToxinPred 3.0: an improved method for predicting the toxicity of peptides. Comput. Biol. Med. 179. https://doi.org/10.1016/j.compbiomed.2024.108926 (2024).
Bui, H. H. et al. Predicting population coverage of T-cell epitope-based diagnostics and vaccines. BMC Bioinform. 7, 153. https://doi.org/10.1186/1471-2105-7-153 (2006).
Gasteiger, E. et al. The proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res. 31, 3784–3788. https://doi.org/10.1093/nar/gkg563 (2003).
Abramson, J. et al. Accurate structure prediction of biomolecular interactions with alphafold 3. Nature 630, 493–500. https://doi.org/10.1038/s41586-024-07487-w (2024).
Heo, L., Park, H., Seok, C. & GalaxyRefine Protein structure refinement driven by side-chain repacking. Nucleic Acids Res. 41. https://doi.org/10.1093/nar/gkt458 (2013).
Laskowski, R. A., MacArthur, M. W., Moss, D. S. & Thornton, J. M. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 26, 283–291. https://doi.org/10.1107/s0021889892009944 (1993).
Vajda, S. et al. New additions to the ClusPro server motivated by CAPRI. Proteins: Structure, Function and Bioinformatics. ;85:435–44. (2017). https://doi.org/10.1002/prot.25219
Desta, I. T., Porter, K. A., Xia, B., Kozakov, D. & Vajda, S. Performance and its limits in rigid body Protein-Protein Docking. Structure 28, 1071–1081e3. https://doi.org/10.1016/j.str.2020.06.006 (2020).
Jones, G. et al. Elucidation of protein function using computational Docking and hotspot analysis by cluspro and ftmap. Acta Crystallogr. D Struct. Biol. 78, 690–697. https://doi.org/10.1107/S2059798322002741 (2022).
Jo, S., Kim, T., Iyer, V. G. & Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 29, 1859–1865. https://doi.org/10.1002/jcc.20945 (2008).
Huang, J. & Mackerell, A. D. CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. J. Comput. Chem. 34, 2135–2145. https://doi.org/10.1002/jcc.23354 (2013).
Abraham, M. J. et al. Gromacs: high performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25. https://doi.org/10.1016/j.softx.2015.06.001 (2015).
Humphrey, W., Dalke, A. & Schulten, K. VMD: visual molecular dynamics. J. Mol. Graph. 14, 33–38. https://doi.org/10.1016/0263-7855(96)00018-5 (1996).
Rapin, N., Lund, O., Bernaschi, M. & Castiglione, F. Computational immunology Meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS One. 5. https://doi.org/10.1371/journal.pone.0009862 (2010).
Grote, A. et al. JCat: A novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 33. https://doi.org/10.1093/nar/gki376 (2005).
Elliott, S. L. et al. Phase I trial of a CD8 + T-cell peptide epitope-based vaccine for infectious mononucleosis. J. Virol. 82, 1448–1457. https://doi.org/10.1128/JVI.01409-07 (2008).
Rappuoli, R. & Aderem, A. A 2020 vision for vaccines against HIV, tuberculosis and malaria. Nature 473, 463–469. https://doi.org/10.1038/NATURE10124 (2011).
Mora, M., Veggi, D., Santini, L., Pizza, M. & Rappuoli, R. Reverse vaccinology. Drug Discov Today. 8, 459–464. https://doi.org/10.1016/S1359-6446(03)02689-8 (2003).
Kanampalliwar, A. M. Reverse vaccinology and its applications. Methods Mol. Biol. 2131, 1–16. https://doi.org/10.1007/978-1-0716-0389-5_1 (2020).
Meloen, R. H., Langeveld, J. P. M., Schaaper, W. M. M. & Slootstra, J. W. Synthetic peptide vaccines: Unexpected fulfillment of discarded hope? In: Biologicals. Academic Press; pp. 233–6. (2001). https://doi.org/10.1006/biol.2001.0298
Pereira, B. et al. Prevalence of sensitization to food allergens, reported adverse reaction to foods, food avoidance, and food hypersensitivity among teenagers. J. Allergy Clin. Immunol. 116, 884–892. https://doi.org/10.1016/j.jaci.2005.05.047 (2005).
Jędrychowski, L. & Wichers, H. J. Chemical and biological properties of food allergens. Chem. Biol. Prop. Food Allergens. 1–429. https://doi.org/10.1201/9781420058574 (2009).
Zuidmeer-Jongejan, L. et al. Development of a hypoallergenic Recombinant parvalbumin for First-in-Man subcutaneous immunotherapy of fish allergy. Int. Arch. Allergy Immunol. 166, 41–51. https://doi.org/10.1159/000371657 (2015).
Yuen, R., Kuniholm, J., Lisk, C. & Wetzler, L. M. Neisserial PorB immune enhancing activity and use as a vaccine adjuvant. Hum. Vaccin Immunother. 15, 2778–2781. https://doi.org/10.1080/21645515.2019.1609852 (2019).
Li, M., Jiang, Y., Gong, T., Zhang, Z. & Sun, X. Intranasal vaccination against HIV-1 with adenoviral Vector-Based nanocomplex using synthetic TLR-4 agonist peptide as adjuvant. Mol. Pharm. 13, 885–894. https://doi.org/10.1021/acs.molpharmaceut.5b00802 (2016).
Prickett, S. R., Rolland, J. M. & O’Hehir, R. E. Immunoregulatory T cell epitope peptides: the new frontier in allergy therapy. Clin. Exp. Allergy. 45, 1015–1026. https://doi.org/10.1111/CEA.12554 (2015).
Chen, X., Zaro, J. L. & Shen, W. C. Fusion protein linkers: Property, design and functionality. Adv. Drug Deliv Rev. 65, 1357–1369. https://doi.org/10.1016/j.addr.2012.09.039 (2013).
Sircar, G., Jana, K., Dasgupta, A., Saha, S. & Bhattacharya, S. G. Epitope mapping of Rhi o 1 and generation of a hypoallergenic variant: A CANDIDATE MOLECULE FOR FUNGAL ALLERGY VACCINES. J. Biol. Chem. 291, 18016. https://doi.org/10.1074/JBC.M116.732032 (2016).
Solomon, J. S. et al. Expression, purification, and human antibody response to a 67 kDa vaccine candidate for schistosomiasis Japonica. Protein Expr Purif. 36, 226–231. https://doi.org/10.1016/j.pep.2004.04.011 (2004).
Gasteiger, E. et al. Protein identification and analysis tools on the expasy server. In: The Proteomics Protocols Handbook. Humana; 571–607. https://doi.org/10.1385/1-59259-890-0:571. (2005).
Karp, C. L. et al. Evaluating the value proposition for improving vaccine thermostability to increase vaccine impact in low and middle-income countries. Vaccine 33, 3471–3479. https://doi.org/10.1016/j.vaccine.2015.05.071 (2015).
Mandal, S., Chanu, W. P. & Natarajaseenivasan, K. Development of a multi-epitope vaccine candidate to combat SARS-CoV-2 and dengue virus co-infection through an immunoinformatic approach. Front. Immunol. 16, 1442101. https://doi.org/10.3389/FIMMU.2025.1442101/BIBTEX (2025).
Nezafat, N. et al. Designing an efficient multi-epitope peptide vaccine against vibrio cholerae via combined immunoinformatics and protein interaction based approaches. Comput. Biol. Chem. 62, 82–95. https://doi.org/10.1016/j.compbiolchem.2016.04.006 (2016).
Tsvetkov, P., Myers, N., Adler, J. & Shaul, Y. Degradation of intrinsically disordered proteins by the NADH 26S proteasome. Biomolecules 10, 1–10. https://doi.org/10.3390/BIOM10121642 (2020).
Hollingsworth, S. A. & Dror, R. O. Molecular dynamics simulation for all. Neuron 99, 1129. https://doi.org/10.1016/J.NEURON.2018.08.011 (2018).
Mallik, B. & Morikis, D. Applications of molecular dynamics simulations in immunology: A useful computational method in aiding vaccine design. Curr. Proteom. 3, 259–270. https://doi.org/10.2174/157016406780655568 (2007).
Troutman, T. D., Bazan, J. F. & Pasare, C. Toll-like receptors, signaling adapters and regulation of the pro-inflammatory response by PI3K. Cell. Cycle. 11, 3559. https://doi.org/10.4161/CC.21572 (2012).
Peters, B., Nielsen, M., Sette, A. T. & Cell Epitope Predictions Annu. Rev. Immunol. ;38:123. https://doi.org/10.1146/ANNUREV-IMMUNOL-082119-124838. (2020).
Acknowledgements
The authors would like to express their sincere gratitude to the Pediatric Translational Clinical Research Unit for kindly providing access to the workstation used for conducting molecular dynamics simulations and trajectory analysis.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Resources, Visualization, and Writing – original draft: Abdelfateh Chourir ; Formal analysis, Visualization: Hafs Essaadi; Methodology **,** Writing – review & editing: Farah Makhloufi; Resources, Writing – review & editing: Aya Abidou; Resources, Writing – review & editing: Ouissal Hachlaf; Writing – review & editing: Zineb Lafi; Writing – review & editing: Boutaina Elgharbaoui; Investigation, Methodology, Resources, Validation: Mohammed Hakmi; Writing – review & editing: Rachid Eljaoudi; Conceptualization, Project administration, Supervision, Writing – review & editing: Naima El Hafidi.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Chourir, A., Essaadi, H., Makhloufi, F. et al. Design of a multiepitope immunotherapy for fish allergy. Sci Rep 16, 8838 (2026). https://doi.org/10.1038/s41598-026-39519-y
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-39519-y
