
Abstract
Pramipexole, a dopamine receptor agonist, is clinically employed in the treatment of Parkinson’s disease. Accumulating evidence has demonstrated that it can enhance neurological function following cerebral ischemia–reperfusion via multiple mechanisms. This study is designed to explore whether pramipexole exerts a protective effect on neurons subjected to oxygen–glucose deprivation/reperfusion by inhibiting the ferroptosis pathway. Firstly, CCK8 assay was employed to evaluate cell viability to determine the optimal intervention time and dosage during oxygen–glucose deprivation/reperfusion injury. Mitochondrial function was evaluated via transmission electron microscopy and JC-1 fluorescent probe. Intracellular iron, glutathione, and glutathione peroxidase 4 enzyme activity were measured using ELISA. Ferroptosis-related proteins and genes were analyzed by immunofluorescence, real-time qPCR, and western blot. And then, a ferroptosis inducer (Erastin) was used as a positive control to validate the link between pramipexole’s effects and ferroptosis regulation. Oxygen–glucose deprivation/reperfusion exposure reduced cell viability, impaired mitochondrial membrane potential, elevated intracellular iron and reactive oxygen species, and suppressed antioxidant capacity. Pramipexole treatment reversed these pathological changes and activated the nuclear factor E2-related factor 2/solute carrier family 7 member 11/glutathione peroxidase 4 signaling axis. Erastin recapitulated oxygen–glucose deprivation/reperfusion-induced cellular dysfunction, which was also mitigated by pramipexole. Pramipexole alleviates oxygen–glucose deprivation/reperfusion-induced neuronal damage and oxidative stress by activating the nuclear factor E2-related factor 2/solute carrier family 7 member 11/glutathione peroxidase 4 pathway to inhibit ferroptosis, highlighting its potential as a therapeutic agent for cerebral ischemia–reperfusion injury.
Similar content being viewed by others


Introduction
Stroke persists as one of the most formidable global health challenges. According to the Global Burden of Disease data1, the number of stroke cases in China reached 28.76 million in 2019, with a continued upward trend. Notably, the increase in ischemic stroke cases (195.2%) has been far more prominent when compared to hemorrhagic stroke. The American Heart Association2 reported that the global prevalence of stroke reached 93.82 million in 2021, with ischemic stroke accounting for 69.94 million cases. Characterized by its high recurrence rate and substantial disability rate, ischemic stroke significantly impacts the quality of life and life expectancy across the global population.
During the onset of ischemic stroke, the anaerobic metabolism and lactate accumulation lead to a reduction in ATP levels and intracellular pH, thereby disrupting ATPase-dependent ion transport mechanisms. This disruption results in intracellular calcium overload, cellular swelling, rupture, and ultimately cell death3. Furthermore, when thrombolysis or thrombectomy is performed to restore blood flow (reperfusion), rather than alleviating the damage, it can paradoxically exacerbate brain injury. This phenomenon is referred to as ischemia–reperfusion (I/R) injury. The underlying mechanisms of I/R injury are complex and may be associated with the accumulation of local reactive oxygen species, infiltration of inflammatory cells, and the opening of mitochondrial membrane permeability transition pores, among other factors4.
Ferroptosis has been recognized to assume a crucial role in cerebral ischemia—reperfusion injury. It is intricately linked to the dysregulation of intracellular iron metabolism, lipid metabolism, and amino acid metabolism5. Glutathione peroxidase 4 (GPX4) is a key regulator of ferroptosis, which inhibits ferroptosis by consuming glutathione (GSH) to convert lipid peroxides into lipid alcohols. Experimental evidence has demonstrated that the activity of GPX4 is modulated by nuclear factor erythroid 2-related factor 2 (Nrf2)6, protein kinase B7, microRNAs such as miR-25-3p8, among other regulatory factors. When GPX4 is inactivated, it disrupts the normal oxidative stress balance, energy metabolism, and inflammatory responses within the brain, leading to lipid peroxide accumulation and ultimately triggering ferroptosis. The cystine/glutamate antiporter (system Xc-) is another important component in the context of ferroptosis. It transports extracellular cystine into cells, where it is used to generate GSH to participate in ferroptosis inhibition. Research has revealed that after cerebral ischemia, the Solute Carrier Family 7 Member 11 (SLC7A11) subunit of system Xc- is suppressed, resulting in decreased intracellular GSH levels and the accumulation of lethal lipid peroxides that promote ferroptosis9. Nrf2, a key transcription factor in the regulation of cellular oxidative stress, is involved in modulating mitochondrial function and lipid metabolism. Activation of Nrf2 can restore GSH/GPX4 activity and reduce the expression of long-chain acyl-CoA synthetase 4, thereby ameliorating neuronal ferroptosis after cerebral I/R10. Nrf2 also directly regulates SLC7A11 transcription. By increasing the expression of SLC7A11 and GPX4, Nrf2 inhibits the accumulation of lipid peroxides. This suppresses neuronal ferroptosis and improves post-ischemic cognitive dysfunction in turn11. Collectively, these findings highlight the Nrf2/SLC7A11/GPX4 signaling pathway as a potentially promising therapeutic target for the treatment of ischemic stroke, as specifically shown in Scheme 1.

The Role of the Nrf2/SLC7A11/GPX4 in the Ferroptosis Regulatory Pathway.
Pramipexole (PPX) is a dopamine receptor agonist clinically utilized in the treatment of Parkinson’s disease. Emerging evidence indicates that PPX might confer neuroprotective effects against brain injury via multiple mechanisms, including antioxidation, anti-apoptosis, inhibition of excitotoxicity, and the maintenance of mitochondrial function12. Notwithstanding these findings, whether PPX can ameliorate cerebral ischemia–reperfusion injury and whether this protective effect is associated with targeting ferroptosis (particularly through the Nrf2/SLC7A11/GPX4 pathway) remains to be elucidated. Preliminary research by our team has shown that pramipexole enhances cognitive and neurological function in a rat model of global cerebral ischemia–reperfusion injury and exerts certain inhibitory effects on ferroptosis13,14. Building on this previous work, the present study employs an oxygen–glucose deprivation/reoxygenation (OGD/R) cell model to mimic the in vivo.
This scheme illustrates the mechanism of ferroptosis, a regulated cell death pathway driven by iron-dependent lipid peroxidation, and describes how iron ions, ROS, GSH, Nrf2, SLC7A11, and GPX4 are interrelated microenvironment of ischemia–reperfusion. By investigating the effects of pramipexole on ferroptosis-related parameters (such as iron accumulation, lipid peroxidation, and mitochondrial morphological alterations) and the Nrf2/SLC7A11/GPX4 signaling pathway, this research endeavors to clarify the role of ferroptosis in OGD/R injury. Moreover, it aims to evaluate the therapeutic potential of pramipexole in the treatment of ischemic stroke and the mitigation of cerebral ischemia–reperfusion injury.
Materials and methods
Cell culture
HT22 cells (Guangzhou Jiniou Biotechnology Co., Ltd. Guangzhou, China) are a murine hippocampal neuronal cell line derived from the hippocampus of C57BL/6 mice. They were maintained in Dulbecco’s Modified Eagle Medium (DMEM) (11995065, Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS) (G8003, Gibco, Grand Island, NY, USA) and 1% penicillin–streptomycin (15140122, Gibco, Grand Island, NY, USA). The glucose concentration in the medium was 22.5 mM. Cells were cultured at 37 ℃ in a humidified incubator with 5% CO2/95% air and passaged when reaching 80–90% confluence using trypsin (25200056, Gibco, Grand Island, NY, USA). For each experiment, 5 × 103 cells were seeded in 96-well plates (for cell viability, ROS, Iron, GSH, and GPX4 activity assays), and 1.5 × 105 cells were seeded in 60-mm dishes (for TEM, JC-1 assays, Western blot, Immunofluorescence, and RT-qPCR) Two types of blanks were included: (1) Medium blank: Only medium without cells, used to subtract background fluorescence; (2) Vehicle blank: Medium containing the same concentration of DMSO as the Erastin treated groups, used to exclude solvent effects.
Experimental materials
HRP conjugated Goat Anti-Rabbit IgG (GB23303) and HRP conjugated Goat Anti-Mouse IgG (GB23301) were purchased from Wuhan Servicebio Technology (Wuhan, China); Cell Counting Kit-8 (C0038), Enhanced BCA Protein Assay Kit (P10010) and Antifade Mounting Medium with DAPI (P0131) were obtained from Biyuntian Biotechnology (Shanghai, China); Pramipexole dihydrochloride powder (HY-17355), Erastin, JC-1 and H2DCFDA were provided by MedChemExpress (Monmouth Junction, NJ, USA); Tissue iron assay kit (A039-2-1) and GSH assay kit (A006-2-1) were purchased from Nanjing Jiancheng Bioengineering Institute (Nanjing, China); Glutathione peroxidase 4 (GPX4) colorimetric assay kit (E-BC-K883-M) was obtained from Elabscience Biotechnology (Wuhan, China); Ultra-fast single-column animal cell total RNA extraction kit (RC102-01), HiScript IV All-in-One Ultra RT SuperMix for qPCR (R433-01), SupRealQ Ultra Hunter SYBR qPCR Master Mix (Q713-02) were provided by Nanjing Vazyme Biotech (Nanjing, China); The primary antibodies used in this study were GPX4 (ab125066, Abcam, Cambridge, MA, UK) and X-CT (ab175186, Abcam, Cambridge, MA, UK); Nrf2 (16396-1-AP, Proteintech, Wuhan, China) and p53 (10442-1-AP, Proteintech, Wuhan, China).
Experimental grouping
Firstly, cells were divided into the Control group, OGD/R group, and OGD/R + PPX group.
The Control group was cultured in complete DMEM medium containing 22.5 mM glucose, 10% FBS, and 1% penicillin–streptomycin. The gas environment was 5% CO2/95% air at 37 °C incubator (Thermo Scientific Forma 371 CO₂ incubator).
For the OGD/R group, the culture medium was replaced with glucose-free DMEM (11966025, Gibco, Grand Island, NY, USA) on the following day after cell plating. The cells were then placed in a 37 °C incubator (Thermo Scientific HERAcell VIOS 160i three-gas incubator) containing a mixed gas of 1% O2, 5% CO2, and 94% N2 for 4 h to induce OGD injury. After OGD, the medium was replaced with complete DMEM medium containing 22.5 mM glucose, 10% FBS, and 1% penicillin–streptomycin, and the cells were cultured in a 37 °C incubator with 5% CO2/95% air for 24 h to simulate reperfusion15.
For the OGD/R + PPX group, different drug intervention time points were established at four distinct stages: 2 h before OGD (pre-hypoxia, 2 h), during OGD (hypoxia, 4 h), during the reoxygenation period (reoxygenation, 24 h), and throughout the ischemia–reperfusion process (hypoxia and reoxygenation, 28 h). Pramipexole (PPX) was treated at concentrations of 25 μM, 50 μM, 100 μM, 150 μM, and 200 μM. Only during the OGD process do the cells experience a lack of glucose and oxygen.
For ferroptosis inducing study, the cells were divided into the Control group as described earlier (22.5 mM glucose, 5% CO2/95% air), Erastin group (ferroptosis inducer, with 22.5 mM glucose and 5% CO2/95% air), and Erastin + PPX group (22.5 mM glucose, 5% CO2/95% air).
For the Erastin group, cells were treated with 1 μM Erastin (0.1% DMSO) for 24 h to induce ferroptosis, followed by replacement with drug-free complete medium16.
For the Erastin + PPX group, after Erastin treatment, cells were treated with complete medium containing 50 μM pramipexole (dissolved in PBS) for 6 h.
CCK-8 Assay
Cell viability was determined using the CCK-8 assay according to the manufacturer’s protocol. Briefly, 10 μl of CCK-8 solution was added to each well of a 96-well plate, followed by incubation in a cell culture incubator for 30 min. The absorbance was measured at 490 nm using a microplate reader. The cell survival rate was calculated using the following formula: (OD value of experimental group − OD value of blank group)/(OD value of control group − OD value of blank group) × 100%. Based on the assay results, the optimal intervention time point and drug concentration were determined.
Transmission electron microscopy (TEM)
Cells were collected and fixed in 2.5% glutaraldehyde solution at 4 ℃ for 2 h, then washed with PBS, and post-fixed in 1% osmium tetroxide solution for 2 h, followed by another PBS wash. The cells were dehydrated through a graded series of ethanol and acetone, and embedded in resin blocks using acetone and 812 embedding medium. Ultrathin sections were stained with uranyl acetate and lead citrate, and observed on the TEM. Qualitative analysis of membrane morphological features was conducted on the TEM images by comparing the staining intensity and structural characteristics.
Mitochondrial Membrane Potential (JC-1) Assay
The changes in mitochondrial membrane potential were assessed using the JC-1 fluorescent probe17. Cells were incubated with 5 μg/ml JC-1 solution at 37 °C for 20–30 min, followed by two washes with PBS (5 min each). Imaging was performed using a long-term dynamic live-cell imaging and functional analysis system. For quantification of JC-1 images, ImageJ software was used to calculate the red (aggregates) and green (monomers) fluorescence intensities. The red/green fluorescence ratio was computed and averaged for each group to obtain the data.
Intracellular iron concentration
Following the instructions provided in the iron assay kit, cells were collected using a cell scraper and centrifuged at 1000 rpm for 10 min to obtain cell pellets. The cells were washed 1–2 times with isotonic PBS buffer (0.1 M, pH 7.4), resuspended in isotonic PBS buffer, and subjected to ultrasonic disruption in an ice-water bath. Complete cell disruption was confirmed under microscopic observation. Subsequently, 0.5 ml of double-distilled water/iron standard solution/cell suspension was mixed with 1.5 ml of iron chromogenic reagent, incubated in a metal bath at 95 °C for 5 min, and cooled. The mixture was then centrifuged at 3500 rpm for 10 min, and the supernatant was analyzed using a microplate reader at 520 nm. The intracellular iron content (mg(μmol)/g protein) was calculated using the following formula: (OD value of experimental group − OD value of blank group)/(OD value of standard group − OD value of blank group) × standard solution concentration × 2 ÷ protein concentration of experimental group.
Intracellular Reactive Oxygen Species (ROS)
The relative intracellular ROS content was determined using the H2DCFDA fluorescent probe. Cells were stained with 10 μM H2DCFDA at 37 °C in the dark for 20 min, followed by two washes with PBS. Fluorescence intensity was measured using a multifunctional fluorescence microplate reader (Ex/Em = 488/525 nm).
GPX4 Enzyme Activity Assay
GPX4 enzyme activity in cells was determined using a colorimetric method. According to the kit instructions, cells were collected using a diluted stabilizer and lysed by sonication on ice, followed by centrifugation at 10,000 × g for 10 min. The supernatant was collected, and 20 µL of the sample was added to the microplate wells. The assay wells received 40 µL of buffer, while the control wells received 40 µL of buffer containing GPX4 enzyme inhibitor. The plates were incubated at 37 °C for 30 min. Subsequently, 140 µL of reaction working solution and 40 µL of buffer-diluted oxidant were added to each well. The absorbance (A1) at 340 nm was immediately measured using a microplate reader within 15 s. After incubation at room temperature (25 °C) for 15 min, the absorbance (A2) was measured again. GPX4 activity (U/gprot) was calculated as follows: (ΔA of the assay − ΔA of the control) ÷ (molar extinction coefficient × optical path length) × (total reaction volume ÷ volume of supernatant in the reaction) ÷ reaction time of the cell sample × dilution factor ÷ protein concentration.
GSH content
Following the instructions provided by the GSH assay kit, cells were collected and washed 1–2 times with PBS. Subsequently, the cells were resuspended in isotonic PBS buffer (0.1 M, pH 7.4) and subjected to ultrasonic homogenization for cell lysis. The lysate was mixed with Reagent 1 in a 1:1 ratio and centrifuged at 3500 rpm for 10 min. The supernatant was collected for chromogenic reaction, and the absorbance at 405 nm was measured using a microplate reader. Additionally, a portion of the lysate was used to determine protein concentration using the BCA protein assay kit. The GSH content in cells was calculated as follows: (OD value of the experimental group − OD value of the blank group)/(OD value of the standard group − OD value of the blank group) × standard solution concentration × 2 ÷ protein concentration of the experimental group.
Western Blot (WB) Analysis
After cell treatment, cells were collected and lysed in 1 × RIPA lysis buffer containing protease inhibitors. Protein concentration was determined using the BCA method. Samples were separated by 10% SDS-PAGE and transferred to 0.45 μm PVDF membranes. The membranes were blocked with a protein-free rapid blocking solution at room temperature for 10 min, followed by overnight incubation at 4 °C with primary antibodies against Nrf2, SLC7A11, GPX4, and p53. After TBST washing, the membranes were incubated with secondary antibodies at room temperature for 1 h, and signals were detected using an ECL chemiluminescence kit. Western blot band intensities were quantified with ImageJ software. The optical density of each target protein band was normalized against that of the internal reference protein (β-actin). The relative expression level of the target protein was calculated as the ratio of the normalized intensity in the treatment group to that in the control group.
Real-Time Quantitative PCR (RT-qPCR)
After cell treatment, total RNA was extracted using an RNA extraction kit, and the OD260/OD280 ratio was measured. Total RNA was diluted tenfold, and reverse transcription was performed using the extracted RNA as a template, along with reverse transcriptase, DNase, and dNTPs, following the kit instructions. The cDNA template, fluorescent dye, and primers were added to an eight-tube strip, and the PCR reaction was initiated. Gene expression differences between samples were compared based on CT values. Relative gene expression was calculated as 2^(-ΔΔCt). All primer sequences are provided in Table 1.
Immunofluorescence (ICC)
The culture medium was removed, and cells were fixed with 4% paraformaldehyde at room temperature for 15 min, followed by permeabilization with Triton X-100 for 5–10 min. After washing, cells were blocked with 5% BSA for 30 min and incubated with primary antibodies (Nrf2, SLC7A11, or GPX4) diluted 1:500 at 4 °C overnight. After PBS washing, cells were incubated with Alexa Fluor 488-conjugated secondary antibodies at room temperature for 1 h. A drop of antifade mounting medium containing DAPI (10–20 µL) was applied to the slides, and images were captured using a confocal or fluorescence microscope.
Statistical analysis
Statistical analysis was performed using SPSS 27.0 software, and graphs were generated using GraphPad Prism 7. Technical replicates within each independent experiment were averaged to obtain a single data point, and these independent data points were used for statistical analysis. Data are presented as mean ± standard deviation (SD). For normally distributed data, one-way ANOVA was used for comparisons between groups at the same time point. If homogeneity of variance was met, post hoc tests were performed using the Bonferroni’s test; otherwise, Dunn’s test was applied. Comparisons across different drug concentrations and time points were analyzed using two-way ANOVA. A test value of p < 0.05 was considered statistically significant. Each experiment was performed in triplicate wells per group, and the entire experiment was repeated independently 3 times. This replication size applies to experiments including cell viability, JC-1, ROS, Iron, GSH, GPX4 activity, Western blot and RT-qPCR. For TEM, 3 independent samples per group were analyzed, with 5 fields of view per sample. For Immunofluorescence, 3 independent samples per group were analyzed, with 6 fields of view per sample, n = 18 fields of view total.
Software and computational environment
Our experiments were conducted on the Microsoft Windows 10 operating system. The code was implemented in Python 3.8.16 using the PyTorch 1.7.0 deep learning framework (URL: https://pytorch.org). Model training and inference were accelerated on an NVIDIA GPU with CUDA 12.0 and cuDNN 8.8.0 libraries (URL: https://developer.nvidia.com/cuda-toolkit and https://developer.nvidia.com/cudnn, respectively).
Results
The impact of pramipexole on cell viability following OGD/R
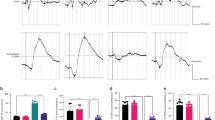
Earlier reports indicated that levodopa exisits controversial cytotoxic effects18, and pramipexole exhibits a superior safety profile compared with levodopa19. In this study, the results of cell viability indicate that pramipexole did not exhibit any cytotoxic effects at any concentration or time point, which partially confirmed the findings of previous research. Among the four drug intervention time points (pre-OGD, during OGD, during reoxygenation, full-phase), treatment during full-phase resulted in more pronounced protection against OGD/R-induced cell death, as reflected by higher overall cell viability compared to treatment during hypoxia (an increase of 13.1%, P = 0.0063). The efficacy of PPX treatment during the reoxygenation phase was statistically comparable to that of the full-phase treatment. Among all treatments with concentrations of 25, 50, 100, 150, and 200 μM, 50 μM was identified as the optimal concentration (P = 0.0138). Compared to the OGD/R group, treatment with 50 μM PPX during the reoxygenation phase resulted in the most significant improvement in cell viability (a 32.7% increase, P = 0.0318). Consequently, all subsequent experiments employed the intervention method of 50 μM pramipexole treatment during the reoxygenation phase (Fig. 1 a-b).

The impact of pramipexole on cell viability, mitochondrial-related indicators and ferroptosis-related indicators of HT22 cells subjected to OGD/R. (a) The influence of different drug concentrations on the viability of HT22 cells after OGD/R, definitions of the four drug intervention groups are provided in the Methods section. Pre-hypoxia means the Pre-OGD group refers to PPx treatment 2 h before the onset of OGD (hypoxia phase). (b) The influence of administering 50 μM pramipexole at different time intervals on cellular viability. (c) Representative TEM images depicting mitochondrial morphology across experimental groups, the red arrow points to the representative mitochondrion, with a scale bar of 500 nm. (d) Representative images of JC-1 fluorescent probe staining acquired via a long-term dynamic live-cell imaging and functional analysis system, with a scale bar of 400 μm.Magnification: 10×. (e) Changes in mitochondrial membrane potential. (f) Changes in Iron concentration. (g) Changes in relative ROS levels. (h) Changes in GPX4 enzyme activity. (i) Changes in GSH concentration. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Effects of pramipexole on mitochondrial morphology and function following OGD/R
Transmission electron microscopy analysis revealed significant alterations in mitochondrial morphology in the OGD/R group, characterized by reduced mitochondrial volume, diminished cristae, and increased membrane density. The treatment of PPX partially reversed these OGD/R-induced morphological changes (Fig. 1 c). Furthermore, mitochondrial membrane potential was assessed using the JC-1 fluorescent probe, with the red-to-green fluorescence ratio serving as an indicator of mitochondrial membrane potential. The results demonstrated a significant decrease of 28.2% compared to the control group in mitochondrial membrane potential following OGD/R treatment (P < 0.0001). Notably, pramipexole treatment effectively ameliorated 33.5% the mitochondrial membrane potential levels (P < 0.0001 vs OGD/R group) (Fig. 1d,e).
Effects of pramipexole on intracellular iron content, ROS levels, GPX4 enzyme activity and GSH levels following OGD/R
This study investigated the effects of pramipexole on intracellular iron content, relative ROS levels, GPX4 enzyme activity, and GSH levels in cells subjected to OGD/R. The results demonstrated a significant increase in intracellular iron content in the OGD/R group compared to the control group. The mean iron concentration in the Control group was 2.8 μmol/g, whereas the OGD/R group exhibited a mean iron concentration of 5.2 μmol/g (P < 0.0001). Treatment of pramipexole significantly reduced intracellular iron concentration to a mean value of 3.6 μmol/g (P = 0.0008 vs OGD/R group) (Fig. 1f).
ROS, as a product of iron-mediated Fenton reactions, showed a consistent trend with iron content variations. OGD/R treatment resulted a 7.7% increase relative ROS levels (P = 0.0084 vs Control group), while pramipexole effectively reduced by 5.5% intracellular ROS (P = 0.0289 vs OGD/R group) (Fig. 1g). GPX4 enzyme activity and GSH levels were quantified using ELISA. The control group demonstrated a mean GPX4 enzyme activity of 11.6 U/gprot, which significantly decreased to 3.5 U/gprot following OGD/R treatment (P = 0.0001 vs Control group). Pramipexole treatment enhanced GPX4 enzyme activity to 8.8 U/gprot (P = 0.0039 vs OGD/R group) (Fig. 1h). Compared to the control group, OGD/R treatment reduced intracellular GSH levels by 32% (P = 0.0312 cs Control), while pramipexole significantly elevated 45.2% intracellular GSH levels (P = 0.0351 vs OGD/R) (Fig. 1i).
The impact of pramipexole on the Nrf2/SLC7A11/GPX4 protein pathway in cells following OGD/R
Western blot analysis revealed that, compared to the control group, intracellular Nrf2 levels slightly increased post-OGD/R (P = 0.4936 vs Control group), while SLC7A11 and GPX4 expression significantly decreased (P = 0.0088 and P = 0.0034 vs Control group). Upon treatment of pramipexole, Nrf2 protein was markedly activated, with a 77.3% increase in Nrf2 protein content in the OGD/R + PPX group compared to the control (P = 0.0005). Additionally, SLC7A11 protein content increased 2.5 times (P = 0.0125) and GPX4 protein content increased 1.6 times (P = 0.0105) compared to the OGD/R group. Post-OGD/R injury, HT22 cells exhibited a 52.2% increase in p53 protein expression (P = 0.0083 vs Control), which was significantly suppressed by 39.2% when treating pramipexole (P = 0.0025 vs OGD/R) (Fig. 2 a–e).

Effect of pramipexole on Nrf2/SLC7A11/GPX4 proteins and genes in cells subjected to OGD/R. (a–e) Protein expression of Nrf2, SLC7A11, GPX4, p53 in HT22 cells after OGD/R. (f–i) Gene expression of Nrf2, SLC7A11, GPX4, p53 in HT22 cells after OGD/R. (j–o) Immunofluorescence staining and detection of Nrf2, SLC7A11, GPX4 in HT22 cells after OGD/R. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
RT-qPCR results indicated that, compared to the control group, Nrf2 gene expression had a insignificant increase in cells treated with OGD/R, while SLC7A11 and GPX4 gene expression significantly decreased by 30.3% and 36.1% (P = 0.0203 and P = 0.0015 vs OGD/R group). In the OGD/R + PPX group, Nrf2 gene expression was significantly higher than in the control group (an increase of 20.7%, P = 0.0459), and SLC7A11 and GPX4 gene expression were notably higher than in the OGD/R group (increase by 39.2% and 39.3%, P = 0.0349 and P = 0.0234). The expression of p53 gene was 1.1 times higher in the OGD/R group compared to the control (P = 0.0331), and pramipexole significantly inhibited p53 gene expression by 55.1% (P = 0.0242 vs OGD/R group) (Fig. 2 f–i).
Immunofluorescence assays showed that, compared to the control group, Nrf2 fluorescence intensity insignificantly increased in cells treated with OGD/R, while SLC7A11 and GPX4 fluorescence intensity decreased by 19.6% and 21.8% (P = 0.0007 and P = 0.0005 vs Control group). In the OGD/R + PPX group, Nrf2 fluorescence intensity was insignificantly stronger than in the OGD/R group and had an increase of 19.7% than in the control group (P = 0.0291). Furthermore, SLC7A11 and GPX4 fluorescence expression was pronounced in the OGD/R + PPX group compared to the OGD/R group ,reaching increases of 10.8% and 14.0% (P = 0.0389 and P = 0.0398) (Fig. 2 j–o).
Effects of pramipexole on cell viability and mitochondrial morphology and function after erastin treatment
The results of the CCK8 assay indicated that compared with the control group, the cell viability in the Erastin group decreased by 37.8% (P < 0.0001), while 50 μM pramipexole significantly increased the cell viability by 29.7% after 1 μM Erastin treatment (P = 0.0005) (Fig. 3 a,b). In addition, we used transmission electron microscopy to observe the morphological changes of the cells. The results showed that compared with the control group, the mitochondrial morphology in the Erastin group changed significantly, mainly manifested as reduced mitochondrial volume, decreased mitochondrial cristae, and increased membrane density, while the addition of pramipexole maintained the normal morphology of mitochondria (Fig. 3 c). The JC-1 probe was used to stain the mitochondria of cells to investigate the changes in mitochondrial membrane potential in the Erastin group and the Erastin + PPX group (Fig. 3 d,e). The results showed that the mitochondrial membrane potential decreased by 28.3% compared to the Control group after Erastin treatment (P < 0.0001), while the addition of pramipexole alleviated mitochondrial membrane dysfunction, reaching a 29.2% increase compared to Erastin group. (P < 0.0001).

Effect of pramipexole on cell viability, mitochondrial-related indicators and ferroptosis-related indicators in erastin-treated cells. (a) Changes in cell viability after 24-h erastin-treatment. (b) Effect of pramipexole on cell viability in 1 μM erastin-treated cells. (c) Representative TEM images depicting mitochondrial morphology across experimental groups, the red arrow points to the representative mitochondrion, with a scale bar of 500 nm. (d) Representative images of JC-1 fluorescent probe staining acquired via a long-term dynamic live-cell imaging and functional analysis system, with a scale bar of 400 μm. Magnification: 10×. (e) Changes in mitochondrial membrane potential. (f) Changes in Iron concentration. (g) Changes in relative ROS levels. (h) Changes in GPX4 enzyme activity. i Changes in GSH concentration. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Effects of pramipexole on intracellular iron content, ROS level, GPX4 enzyme activity, and GSH content after erastin treatment
The results of iron ion content detection showed that the average iron content in the control group cells was 2.8 μmol/g, while the average iron ion content in the Erastin group cells was 5.0 μmol/g (P < 0.0001). After adding pramipexole, the average intracellular iron ion content was 3.2 μmol/g (P = 0.0003 vs the Erastin group)(Fig. 3 f).
The relative content of intracellular ROS was detected by DCFH-DA fluorescent probe. The results showed that compared with the control group, the relative content of ROS in the Erastin group had a 11.5% increase (P = 0.0004), and pramipexole reduced the ROS level by 4.5% (P = 0.0286 vs Erastin) (Fig. 3 g). The intracellular GPX4 enzyme activity and GSH content were detected by ELISA. The results showed that the GPX4 enzyme activity was high and the GSH content was sufficient in the control group. In contrast, after Erastin treatment, the intracellular GPX4 enzyme activity decreased from 10.1U/gprot to 3.2U/gprot (P < 0.0001 vs Control group), and the phenomenon of intracellular GSH depletion occurred with a decrease of 74.3% (P = 0.0014 vs Control). After adding pramipexole, it significantly increased the intracellular GPX4 enzyme activity to 7.8U/gprot (P = 0.0008 vs Erastin group) and increased the GSH content by 2.4 times (P = 0.0131 vs Erastin) in Erastin-damaged cells (Fig. 3 h-i).
Effects of pramipexole on the Nrf2/SLC7A11/GPX4 signaling pathway in cells after erastin treatment
The results of WB experiments showed that compared with the control group, the expression of Nrf2 protein in the Erastin group had an insignificant change (P = 0.2584), the expression of p53 protein increased by 52.6% (P = 0.0031). The expression of SLC7A11 protein had a decrease of 56.5% (P = 0.0011), and the expression of GPX4 protein decreased significantly by 54.8% (P < 0.0001). Pramipexole activated Nrf2 protein by 2.0 times (P = 0.0032 vs Control group), increased the expression levels of intracellular SLC7A11 by 1.4 times and GPX4 by 96.3% (P = 0.0006 and P < 0.0001 vs Erastin group), and inhibited the expression of p53 by 44.7% (P = 0.0004 vs Erastin group) (Fig. 4 a–e). The data of RT-qPCR showed that compared with the control group, the expression level of Nrf2 gene in cells showed an insignificant increase after Erastin treatment, the expression of p53 gene increased by 54.4% (P = 0.0071), and the expression of SLC7A11 and GPX4 was inhibited, showed a decreased of 64.3% and 12.5% (P = 0.0443 and P = 0.0317). After adding pramipexole, it significantly activated the expression of Nrf2 by 59.3% (P = 0.0328 vs Control), promoted the expression of SLC7A11 gene by 2.0 times and GPX4 gene by 17.2% (P = 0.0172 and P = 0.0092 vs Erastin group), and inhibited the expression of p53 gene by 27.1% (P = 0.0313 vs Erastin group) (Fig. 4 f–i). The results of immunofluorescence experiments proved that compared with the control group, the expression of Nrf2 protein in the Erastin group had an insignificant change, the expression level of SLC7A11 protein decreased by 25.5% (P < 0.0001), and the expression of GPX4 protein decreased by 25.0% (P < 0.0001). After adding pramipexole, the expression of Nrf2 further increased by 31.4% (P = 0.0135 vs Control group), and the expression of SLC7A11 protein and GPX4 protein also significantly increased by 8.4% and 16.0% (P = 0.0117 and P = 0.0075 vs Erastin group) (Fig. 4 j–o).

Effect of pramipexole on Nrf2/SLC7A11/GPX4 proteins and genes in cells after erastin treatment. (a–e) Protein expression of Nrf2, SLC7A11, GPX4, p53 in HT22 cells after Erastin treatment. (f–i) Gene expression of Nrf2, SLC7A11, GPX4, p53 in HT22 cells after Erastin treatment. (j–o) Immunofluorescence staining and detection of Nrf2, SLC7A11, GPX4 in HT22 cells after Erastin treatment. Data are presented as mean ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Discussion
Current emergency treatment modalities for ischemic stroke primarily involve pharmacological thrombolysis or mechanical thrombectomy20. Nevertheless, the abrupt restoration of blood flow to ischemic cerebral regions can trigger localized inflammation and the accumulation of reactive oxygen species, potentially exacerbating tissue damage. Therefore, the alleviation of ischemia–reperfusion injury is of paramount importance for enhancing the prognosis of patients suffering from ischemic stroke. Ferroptosis, a regulated form of programmed cell death, is distinguished by the iron-dependent buildup of lipid peroxides. Significantly, ferroptosis is intricately linked to the development and advancement of cerebral ischemia–reperfusion injury. As far back as 1988, Dietrich et al.21 documented iron deposition in the basal ganglia, thalamus, periventricular areas, and subcortical white matter regions during the recovery stage of ischemic brain injury.
Pramipexole has emerged as a promising neuroprotective agent capable of exerting neuroprotective effects independent of cell death modalities and its dopamine receptor agonist properties22. Clinical investigations have provided compelling evidence that pramipexole can significantly expedite the restoration of consciousness in patients suffering from severe brain disorders23,24. Furthermore, pramipexole mitigates brain injury through multiple mechanisms. It can induce a state of mild hypothermia, inhibit necroptosis25,26, maintain the integrity of the blood—brain barrier, and attenuate the inflammatory responses27. Our research group’s earlier studies have revealed that pramipexole can enhance cognitive function in Sprague-Dawley (SD) rats subjected to global cerebral ischemia—reperfusion injury. It accomplishes this by reducing intracranial lipid peroxidation and inhibiting ferroptosis related pathways13,14. Therefore, the present study endeavors to conduct in vitro experiments to validate the mechanisms underlying pramipexole’s actions. By using Erastin as a positive control, this study aims to rule out the possibility that pramipexole’s neuroprotective effects are solely attributed to its ability to inhibit oxidative stress, thereby further elucidating whether pramipexole improves functional status in OGD/R injury models by specifically suppressing ferroptosis.
The CCK-8 assay was employed to assess cellular viability changes subsequent to OGD/R in order to identify the optimal concentration and treatment timing. The outcomes indicated that interventions using different concentrations of pramipexole (PPX) at various time points during OGD/R had distinct impacts on cellular viability. Specifically, treatment during hypoxia proved detrimental to cells, whereas both reoxygenation and continuous treatment demonstrated partial improvements in cellular viability, and 50 μM was determined to be the optimal concentration. Although prior research has indicated that administering agents before ischemia–reperfusion injury can yield significant therapeutic effects28, our findings suggest that pramipexole treatment during reoxygenation or throughout the process is more effective in enhancing cellular viability. Additionally, existing research has demonstrated that intervening after ischemia–reperfusion injury can significantly reduce the cerebral infarction volume and improve neurological deficits in mice models of middle cerebral artery occlusion29. Compared to pre-injury treatment, post-injury treatment may hold greater clinical relevance and applicability.
Transmission electron microscopy was employed to observe the cells subsequent to OGD/R treatment. It was found that certain cells exhibited ferroptosis-related mitochondrial morphological changes following OGD/R, while the mitochondrial morphology in the OGD/R + PPX group remained relatively intact. As the “energy factory” of cells, mitochondrial dysfunction is a key link in the occurrence and development of brain injury. Dysfunction of mitochondria can trigger oxidative stress, inflammatory responses, and calcium imbalance, thereby impairing brain plasticity and function30. To assess the relative level of mitochondrial membrane potential, the JC-1 fluorescent probe was utilized. The results showed that OGD/R injury led to a notable reduction in the mitochondrial membrane potential within cells and the addition of pramipexole effectively reversed these changes in the mitochondrial membrane potential. Collectively, these results suggest that OGD/R injury exerts a substantial influence on both mitochondrial morphology and function, while pramipexole can preserve normal mitochondrial morphology and function. Previous research31 has indicated that safeguarding mitochondrial function can mitigate the severity of ischemia–reperfusion-induced brain injury. Additionally, it has been reported30 that PPX can enhance the mitochondrial membrane potential following traumatic brain injury and alleviate the degree of mitochondrial dysfunction. The results of this study confirm that pramipexole can maintain mitochondrial morphology and function after OGD/R injury, thereby exerting a cytoprotective effect.
After cerebral ischemia–reperfusion injury, the integrity of ferritin is disrupted, leading to the release of a substantial amount of free iron. Fe2+ in the cytoplasm generates ROS through the Fenton reaction, increasing the cell’s sensitivity to ferroptosis. At the same time, the intracellular antioxidant defense system is inhibited, creating a conducive environment for the occurrence of ferroptosis. The results showed that following OGD/R treatment, both the intracellular iron content and ROS levels increased significantly. This elevation in oxidative stress within the cell greatly augmented the likelihood of ferroptosis. However, the addition of PPX effectively mitigated the intracellular iron and ROS contents. GPX4, a pivotal antioxidant peroxidase, uses GSH as a substrate to counteract the intracellular lipid peroxidation process, directly reducing lipid hydroperoxides or indirectly reducing cholesterol hydroperoxides and thymidine hydroperoxides to inhibit ferroptosis32. The research results demonstrated that after OGD/R injury, the activity of GPX4 in cells was inhibited, and the GSH content decreased substantially. Conversely, when pramipexole was introduced, the activity of the GPX4 enzyme was enhanced, and the GSH content increased significantly. These results clearly indicated a marked attenuation of the cell’s antioxidant capacity after OGD/R and pramipexole can effectively ameliorate the intracellular oxidative stress induced by OGD/R and enhance the cell’s resistance to ferroptosis.
Oxidative stress represents a crucial factor contributing to cell damage during cerebral ischemia—reperfusion injury. Nrf2, an essential antioxidant factor, is degraded by the KEAP1 and other E3 ubiquitin ligase complexes under basal conditions and translocates into the nucleus, where it activates target genes harboring antioxidant response elements upon exposure to stress conditions. This activation participates in multiple physiological processes, including iron metabolism, lipid metabolism, and the regulation of mitochondrial function33. Research findings indicate that following OGD/R treatment, the expression of Nrf2 in neurons experiences a modest increase. This observation suggests that Nrf2 is activated under stress conditions, potentially serving as an endogenous protective mechanism. The addition of pramipexole can significantly increase the expression of Nrf2 protein and gene in neurons, proving that pramipexole can further promote the expression of Nrf2 subsequent to OGD/R injury. Activated Nrf2 plays a pivotal role in promoting the expression of downstream antioxidant genes. This, in turn, helps to mitigate oxidative stress and mitochondrial dysfunction34,35. GPX4 holds a unique position as the sole enzyme capable of directly reducing phospholipid hydroperoxides and is involved in the regulation of ferroptosis across various diseases36. Activation of GPX4 has been shown to alleviate the severity of brain injury in animal models of different brain diseases, such as subarachnoid hemorrhage37 and Alzheimer’s disease38. In the present study, OGD/R treatment was found to significantly reduce the protein and gene expression of GPX4 in HT22 cells. Conversely, the addition of pramipexole was able to reverse this trend, leading to a significant enhancement of antioxidant capacity. In addition, SLC7A11 mediates cystine uptake and glutamate release. Dysfunction of SLC7A11 can result in a decline in intracellular cysteine and depletion of glutathione (GSH) synthesis39. Downregulation of SLC7A11 indirectly inhibits the activity of GPX4 by impeding the cysteine metabolic pathway, leading to the accumulation of lipid peroxides and ultimately inducing ferroptosis40,41. The results of this study revealed that OGD/R treatment significantly inhibits the expression of both the SLC7A11 protein and gene in cells. However, the addition of pramipexole can effectively increase the levels of SLC7A11 protein and gene. Additionally, p53, an important tumor suppressor protein, undergoes serine phosphorylation and activation after cerebral ischemia–reperfusion injury42. Inhibiting the expression of p53 has been shown to improve mitochondrial function and exert neuroprotective effects in rats with transient middle cerebral artery occlusion43. Through WB and RT—qPCR experiments, this study investigated the expression changes of p53. The results showed that the expression level of p53 increased after OGD/R treatment, while pramipexole inhibited the activation of p53. Collectively, these findings suggest that the protective effect of pramipexole on OGD/R—injured cells is closely associated with the activation of the Nrf2/SLC7A11/GPX4 protein pathway. Additionally, pramipexole’s ability to inhibit p53 activation may also contribute to its neuroprotective effects in the context of cerebral ischemia—reperfusion injury.
In fact, a significant oxidative stress process is evident during ischemia–reperfusion injury, which is closely related to the occurrence of ferroptosis. Our research findings indicate that pramipexole can effectively lower intracellular iron ion and ROS levels subsequent to OGD/R injury. Moreover, it enhances the activity of GPX4 and increases the content of GSH. Thus, the protective effect of pramipexole against OGD/R injury may be attributed to its inhibition of ferroptosis. To verify this hypothesis, we established a positive control group using the ferroptosis inducer Erastin to determine whether pramipexole indeed exerts an inhibitory effect on ferroptosis. As anticipated, cell viability significantly decreased after Erastin treatment, while 50 μM pramipexole significantly improved cell viability. Furthermore, Erastin treatment induced distinct ferroptosis-related morphological alterations in mitochondria, accompanied by a significant reduction in mitochondrial membrane potential. This indicates a disruption of normal mitochondrial morphology and function. The addition of pramipexole, however, reversed these mitochondrial morphological changes and preserved normal mitochondrial function. The core mechanism of ferroptosis involves intracellular iron overload and lipid peroxidation accumulation44. Excess iron reacts with H2O2 to generate ROS, which further oxidizes polyunsaturated fatty acids to form lipid peroxides, damaging cell membrane structures and attacking DNA and proteins. In this process, GPX4 and GSH, as pivotal antioxidants, convert toxic phospholipid peroxides into non-toxic phospholipid alcohols, thereby alleviating oxidative stress45. When compared to the control group, the Erastin group showed significantly reduced expression of ferroptosis-related biomarkers, including iron ions, ROS, and GSH, along with inhibited GPX4 enzyme activity. The addition of pramipexole alleviated intracellular iron overload, reduced ROS content, enhanced GPX4 enzyme activity, and increased intracellular GSH content following Erastin treatment. The decline in mitochondrial membrane potential is one of the important characteristics of ferroptosis, and mitochondrial dysfunction plays a crucial role in the regulation of programmed cell death, such as ferroptosis46. Iron ions, ROS, GSH, and GPX4 enzyme are all closely associated indicators in the progression of ferroptosis. This study further confirmed the inhibitory effect of pramipexole on ferroptosis by detecting the differential characteristics between the Erastin group and the Erastin + PPX group. This confirms that the protective effect of pramipexole on OGD/R-injured cells is achieved through the inhibition of ferroptosis.
Research findings indicate that Nrf2 is closely associated with ferroptosis, capable of inhibiting lipid peroxidation and modulating the progression of ferroptosis through multiple signaling pathways35. GPX4 has been experimentally demonstrated to alleviate cerebral ischemia—reperfusion injury through inhibiting ferroptosis6. Similarly, SLC7A11 plays a crucial role in the occurrence of ferroptosis by regulating the cysteine metabolic pathway40. Investigations into intracellular protein expression following ferroptosis reveal that, comparable to the OGD/R group, the Erastin group exhibits increased Nrf2 protein and gene expression, while SLC7A11 and GPX4 protein and gene expression levels are significantly reduced. When pramipexole was introduced, it further activated Nrf2 and significantly enhanced the protein and gene expression of GPX4 and SLC7A11. Additionally, p53 exerts a dual regulatory effect on ferroptosis. On one hand, p53 can trigger ferroptosis by suppressing SLC7A11 expression or enhancing the expression of SAT1 (spermidine/spermine N1-acetyltransferase 1) and GLS2 (glutaminase 2). On the other hand, p53 can also inhibit ferroptosis through the direct suppression of dipeptidyl peptidase 447. The results of the present study show that Erastin treatment leads to an upregulation of p53 expression, while pramipexole effectively inhibits p53 activation. These findings strongly suggest that pramipexole inhibits the onset of ferroptosis by promoting the expression of Nrf2/SLC7A11/GPX4 proteins and genes, thereby enhancing cellular antioxidant stress capacity.
In summary, ferroptosis occurs during OGD/R-induced injury, marked by several key features, including intracellular iron overload, the accumulation of ROS, the decline of GPX4 enzyme activity, the depletion of glutathione (GSH), and mitochondrial dysfunction. Our research demonstrates that pramipexole exhibits multifaceted neuroprotective effects. It effectively maintains the normal mitochondrial membrane potential and morphology, ameliorates intracellular iron overload, scavenges excessive ROS, and preserves the activity of GPX4 enzyme and the levels of GSH. Specifically, pramipexole mitigates OGD/R-induced ferroptosis through the Nrf2/SLC7A11/GPX4 pathway, exerting protective effects against cerebral ischemia–reperfusion injury, thereby demonstrating its potential as a neuroprotective pharmaceutical agent.
Nevertheless, this study has certain limitations. Regarding the research subjects, HT22 cells are dopaminergic neurons, and our findings confirm that pramipexole helps maintain their normal physiological functions. Nonetheless, other studies have shown that pramipexole can inhibit astrocyte activation and suppress neuroinflammation48, and may possess extra-cerebral protective effects49. These findings suggest that the protective mechanisms of pramipexole might be more widespread. Thus, future research should explore the cytoprotective potential of pramipexole across diverse cell types. Moreover, this study solely focused on ferroptosis as the cell death mechanism, without analyzing other ferroptotic pathways or alternative cell death types. Given the intricate interplay between various cell death modalities50, future studies should concurrently examine multiple cell death mechanisms to further elucidate the underlying mechanisms of pramipexole’s neuroprotective effects.
Data availability
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request. [The original Western Blot bands have been uploaded as supplementary material.]
References
Ma, Q. et al. Temporal trend and attributable risk factors of stroke burden in China, 1990-2019: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 6(12), e897–e906 (2021).
Martin, S. S. et al. 2025 Heart Disease and Stroke Statistics: A Report of US and Global Data From the American Heart Association[J]. Circulation 151(8), e41–e660 (2025).
Siesjö, B. K. Pathophysiology and treatment of focal cerebral ischemia. Part I: Pathophysiology[J]. J. Neurosurg. 77(2), 169–184 (1992).
Kalogeris, T., Baines, C. P., Krenz, M. & Korthuis, R. Ischemia/Reperfusion. Compr. Physiol. 7(1), 113–170 (2016).
Zhou, P., Luo, K. & Shi, Y. Research progress on ferroptosis signaling pathways in ischemic brain injury. Chin. J. Neurol. 54(7), 6 (2021).
Liu, H. et al. Rhein attenuates cerebral ischemia-reperfusion injury via inhibition of ferroptosis through NRF2/SLC7A11/GPX4 pathway. Exp. Neurol. 369, 114541 (2023).
Bai, X. et al. Angong Niuhuang Wan inhibit ferroptosis on ischemic and hemorrhagic stroke by activating PPARγ/AKT/GPX4 pathway. J. Ethnopharmacol. 321, 117438 (2024).
Yang, W. et al. Exosomes from young healthy human plasma promote functional recovery from intracerebral hemorrhage via counteracting ferroptotic injury[J]. Bioact. Mater. 27, 1–14 (2023).
Yuan, Y., Zhai, Y., Chen, J., Xu, X., Wang, H. Kaempferol ameliorates oxygen-glucose deprivation/reoxygenation-induced neuronal ferroptosis by activating Nrf2/SLC7A11/GPX4 axis. Biomolecules. 11(7) (2021).
Hu, Q. et al. β-Caryophyllene suppresses ferroptosis induced by cerebral ischemia reperfusion via activation of the NRF2/HO-1 signaling pathway in MCAO/R rats[J]. Phytomedicine 102, 154112 (2022).
Fu, C. et al. Rehmannioside A improves cognitive impairment and alleviates ferroptosis via activating PI3K/AKT/Nrf2 and SLC7A11/GPX4 signaling pathway after ischemia. J. Ethnopharmacol. 289, 115021 (2022).
Wilson, S. M., Wurst, M. G., Whatley, M. F. & Daniels, R. N. Classics in chemical neuroscience: Pramipexole. ACS Chem. Neurosci. 11(17), 2506–2512 (2020).
Kang, X., Liu, L., Wang, W. & Wang, Y. Effects of pramipexole combined with levodopa on cognitive function and mitochondrial function in rats with global cerebral ischemia-reperfusion injury. Chin. J. Rehabil. Theory Pract. 29(05), 533–540 (2023).
Kang, X. et al. Dopamine receptor agonist pramipexole exerts neuroprotection on global cerebral ischemia/reperfusion injury by inhibiting ferroptosis. J. Stroke Cerebrovasc. Dis. 34(1), 108101 (2025).
Xie, K., Mo, Y., Yue, E., Shi, N. & Liu, K. Exosomes derived from M2-type microglia ameliorate oxygen-glucose deprivation/reoxygenation-induced HT22 cell injury by regulating miR-124-3p/NCOA4-mediated ferroptosis. Heliyon 9(7), e17592 (2023).
Fang, Y. et al. Inhibiting ferroptosis through disrupting the NCOA4-FTH1 interaction: A new mechanism of action. ACS Cent. Sci. 7(6), 980–989 (2021).
Perelman, A. et al. JC-1: Alternative excitation wavelengths facilitate mitochondrial membrane potential cytometry. Cell Death Dis. 3(11), e430 (2012).
Melamed, E. et al. Levodopa toxicity and apoptosis. Ann. Neurol. 44(3 Suppl 1), S149–S154 (1998).
Clarke, C. E. & Guttman, M. Dopamine agonist monotherapy in Parkinson’s disease. Lancet 360(9347), 1767–1769 (2002).
Association, C. S. & Stroke, W. G. O. C. Guidelines of the Chinese Stroke Association for Reperfusion Therapy in Acute Ischemic Stroke (2024)[J]. Chin. J. Stroke 19(12), 1460–1478 (2024).
Dietrich, R. B. & Bradley, W. G. J. Iron accumulation in the basal ganglia following severe ischemic-anoxic insults in children. Radiology 168(1), 203–206 (1988).
Huang, F., Vemuri, M. C. & Schneider, J. S. Modulation of ATP levels alters the mode of hydrogen peroxide-induced cell death in primary cortical cultures: Effects of putative neuroprotective agents. Brain Res. 997(1), 79–88 (2004).
Patrick, P. D., Buck, M. L., Conaway, M. R. & Blackman, J. A. The use of dopamine enhancing medications with children in low response states following brain injury. Brain Inj. 17(6), 497–506 (2003).
Liu, L., Dong, W. & Shi, R. New approaches to clinical management of cerebral resuscitation. Chin. Med. J. 29, 2069–2071 (2006).
Zhang, Q. et al. Reassessment of subacute MPTP-treated mice as animal model of Parkinson’s disease[J]. Acta Pharmacol. Sin. 38(10), 1317–1328 (2017).
Liu, C. et al. Pramipexole alleviates traumatic brain injury in rats through inhibiting necroptosis[J]. Neurosci. Lett. 791, 136911 (2022).
Hall, E. D., Andrus, P. K., Oostveen, J. A., Althaus, J. S. & VonVoigtlander, P. F. Neuroprotective effects of the dopamine D2/D3 agonist pramipexole against postischemic or methamphetamine-induced degeneration of nigrostriatal neurons. Brain Res. 742(1–2), 80–88 (1996).
Yang, H. et al. Cerebral ischemia/reperfusion injury and pharmacologic preconditioning as a means to reduce stroke-induced inflammation and damage. Neurochem. Res. 47(12), 3598–3614 (2022).
Wang, Y. et al. Anti-CHAC1 exosomes for nose-to-brain delivery of miR-760-3p in cerebral ischemia/reperfusion injury mice inhibiting neuron ferroptosis. J. Nanobiotechnology 21(1), 109 (2023).
Thapak, P. & Gomez-Pinilla, F. The bioenergetics of traumatic brain injury and its long-term impact for brain plasticity and function. Pharmacol. Res. 208, 107389 (2024).
Zhao, Y. et al. Synergistic interaction between zinc and reactive oxygen species amplifies ischemic brain injury in rats[J]. Stroke 49(9), 2200–2210 (2018).
Li, M. et al. Baicalein ameliorates cerebral ischemia-reperfusion injury by inhibiting ferroptosis via regulating GPX4/ACSL4/ACSL3 axis. Chem. Biol. Interact. 366, 110137 (2022).
Li, J. et al. The crosstalk between ferroptosis and mitochondrial dynamic regulatory networks. Int. J. Biol. Sci. 19(9), 2756–2771 (2023).
Lan, X. et al. Isoliquiritigenin alleviates cerebral ischemia-reperfusion injury by reducing oxidative stress and ameliorating mitochondrial dysfunction via activating the Nrf2 pathway[J]. Redox Biol. 77, 103406 (2024).
Dodson, M., Castro-Portuguez, R. & Zhang, D. D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol. 23, 101107 (2019).
Huang, B. et al. Palmitoylation-dependent regulation of GPX4 suppresses ferroptosis. Nat. Commun. 16(1), 867 (2025).
Chen, J. et al. Oroxin A alleviates early brain injury after subarachnoid hemorrhage by regulating ferroptosis and neuroinflammation. J. Neuroinflammation 21(1), 116 (2024).
Li, X. et al. Berberine ameliorates iron levels and ferroptosis in the brain of 3 × Tg-AD mice. Phytomedicine 118, 154962 (2023).
Wang, X., Kong, X., Feng, X. & Jiang, D. Effects of DNA, RNA, and protein methylation on the regulation of ferroptosis. Int. J. Biol. Sci. 19(11), 3558–3575 (2023).
Koppula, P., Zhuang, L. & Gan, B. Cystine transporter SLC7A11/xCT in cancer: Ferroptosis, nutrient dependency, and cancer therapy. Protein Cell 12(8), 599–620 (2021).
Iida, Y. et al. Effective ferroptotic small-cell lung cancer cell death from SLC7A11 inhibition by sulforaphane. Oncol. Lett. 21(1), 71 (2021).
Kmet, T. I. & Tkachuk, S. S. [Dynamics of changes of p53-positive cells morphofunctional condition in the cerebral cortex of the temporal lobe in rats affected by carotid ischemia-reperfusion]. Patol. Fiziol. Eksp. Ter. 59(2), 19–22 (2015).
Shang, J. et al. Naoxintong capsule accelerates mitophagy in cerebral ischemia-reperfusion injury via TP53/PINK1/PRKN pathway based on network pharmacology analysis and experimental validation. J. Ethnopharmacol. 336, 118721 (2025).
Rochette, L., Dogon, G., Rigal, E., Zeller, M., Cottin, Y., Vergely, C. Lipid peroxidation and iron metabolism: Two corner stones in the homeostasis control of ferroptosis. Int. J. Mol. Sci. 24(1) (2022).
Ru, Q. et al. Iron homeostasis and ferroptosis in human diseases: Mechanisms and therapeutic prospects. Signal Transduct. Target. Ther. 9(1), 271 (2024).
She, R. et al. Mitochondrial dysfunctions induce PANoptosis and ferroptosis in cerebral ischemia/reperfusion injury: From pathology to therapeutic potential. Front. Cell Neurosci. 17, 1191629 (2023).
Kang, R., Kroemer, G. & Tang, D. The tumor suppressor protein p53 and the ferroptosis network. Free Radic. Biol. Med. 133, 162–168 (2019).
Dong, A. et al. Pramipexole inhibits astrocytic NLRP3 inflammasome activation via Drd3-dependent autophagy in a mouse model of Parkinson’s disease. Acta Pharmacol. Sin. 44(1), 32–43 (2023).
Salminen, A. V. et al. Disconnection between periodic leg movements and cortical arousals in spinal cord injury. J. Clin. Sleep Med. 9(11), 1207–1209 (2013).
Galluzzi, L. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018[J]. Cell Death Differ. 25(3), 486–541 (2018).
Funding
The present study was supported by the National Natural Science Foundation of China (82271427), the National Key R&D Program of China (2022YFC3602802) and the Scientific Research Foundation of China Rehabilitation Research Center (2022ZX-08).
Author information
Authors and Affiliations
Contributions
Lixu Liu made substantial contributions to the conception and design of the work and acquisition of funds and project supervision. Lixu Liu, Xiaoyu Kang and Linyao Zhang revised it critically for important intellectual content. Linyao Zhang made substantial contributions to the acquisition, analysis, or interpretation of data and drafted the initial work. Qianhui Wang, Zijie Cao and Ruixin Gao participated in the acquisition and analysis of data. Yan Yu and Ting Li provided technology support and made indispensable contributions to the design of the work.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
The cell line used in this study (HT22) was purchased from the Guangzhou Jiniou Biotechnology Co., Ltd. and is commercially available and de-identified. The study only involved in vitro cell culture and did not include any in vivo animal experiments or human clinical trials. According to the regulations of our institution’s ethics committee, studies using commercialized, non-primary cell lines do not require additional animal or human ethics approval.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Zhang, L., Kang, X., Wang, Q. et al. Pramipexole alleviates ferroptosis in HT22 cells induced by oxygen–glucose deprivation/reoxygenation via the Nrf2/SLC7A11/GPX4 pathway. Sci Rep 16, 11134 (2026). https://doi.org/10.1038/s41598-026-40289-w
Received:
Accepted:
Published:
Version of record:
DOI: https://doi.org/10.1038/s41598-026-40289-w
